
Induced Necroptosis and Its Role in Cancer Immunotherapy


 and
and
Abstract
1. Introduction
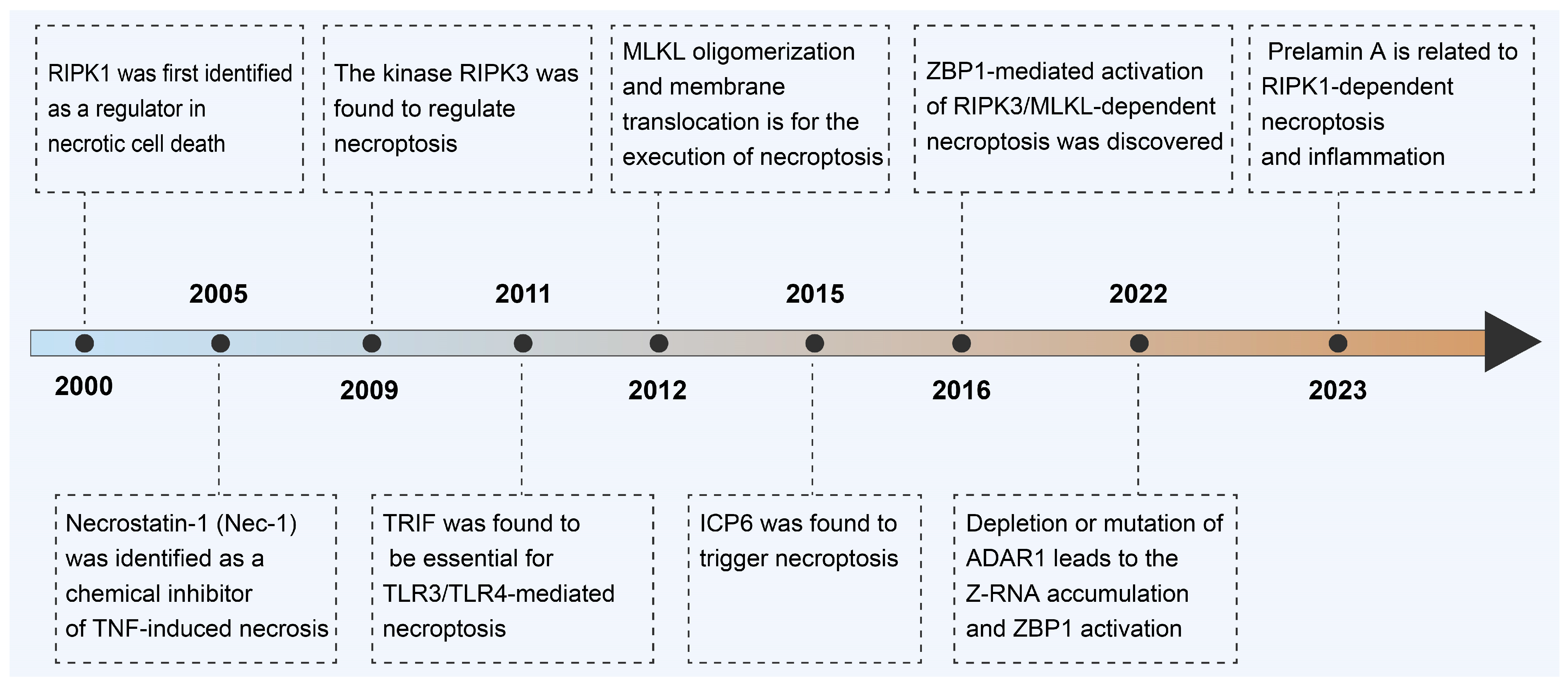
2. Milestone on Discovery of Necroptosis
3. Classic Signaling Pathway of Necroptosis
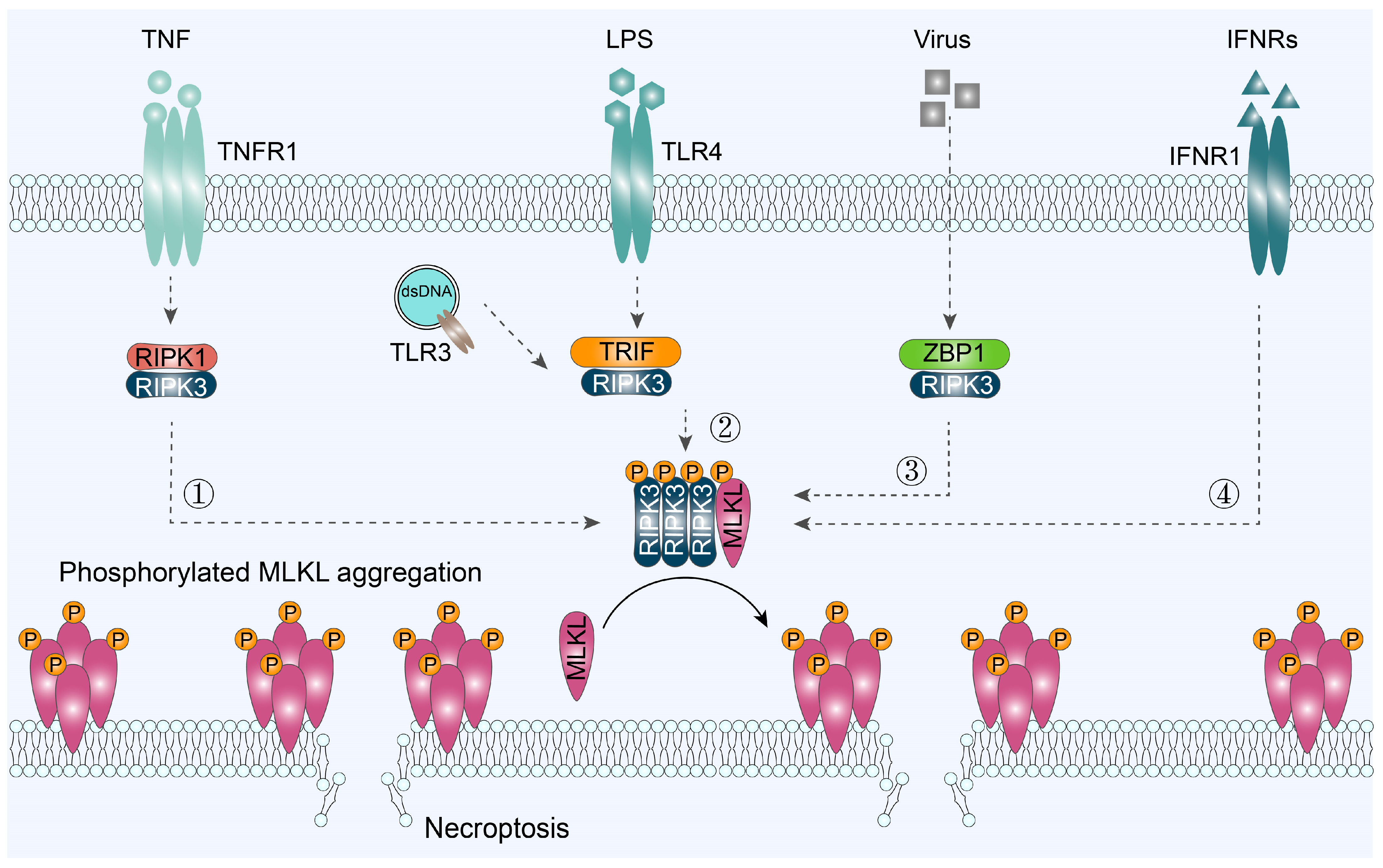
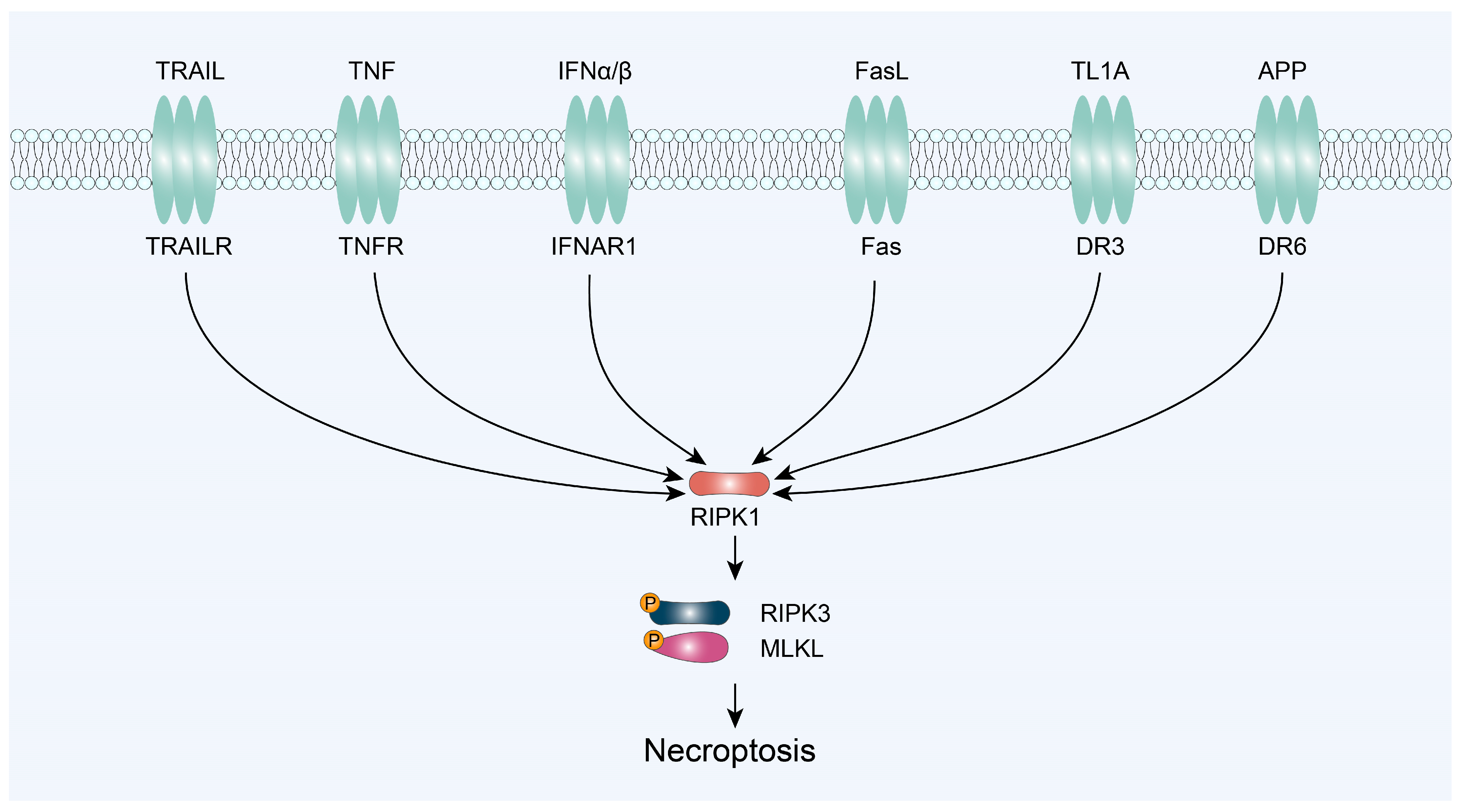
3.1. RIPK1/RIPK3/MLKL-Mediated Necroptosis
3.2. TRIF/RIPK3/MLKL-Mediated Necroptosis
3.3. ZBP1/RIPK3/MLKL-Mediated Necroptosis
3.4. IFNR/MLKL-Mediated Necroptosis
4. Necroptosis and Cancer Immunotherapy
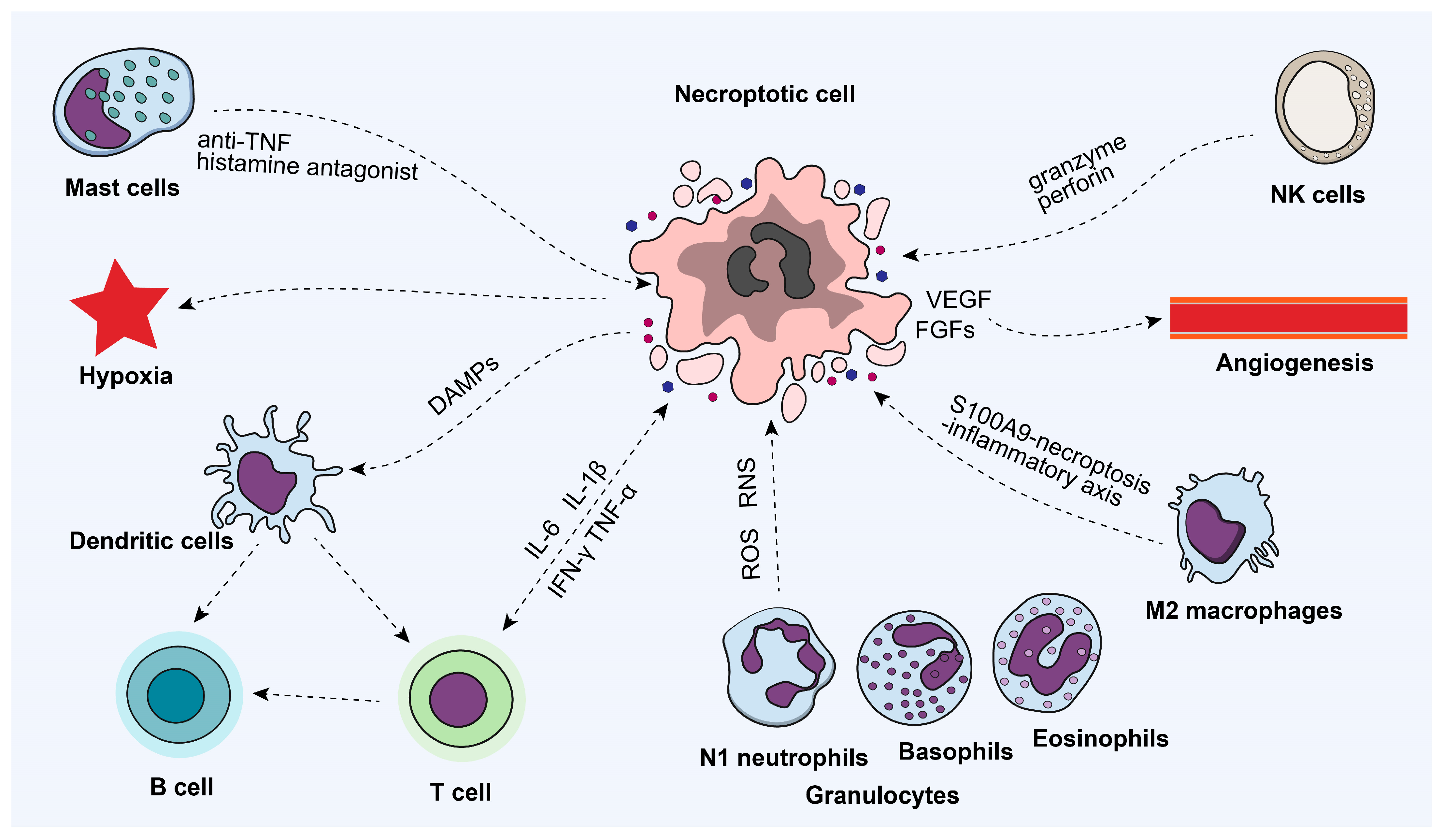
4.1. Necroptosis and Immune Microenvironment
4.2. Necroptosis and Cancer Immunosurveillance
4.3. Induced Necroptosis and Immunogenic Cell Death
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Yan, J.; Wan, P.; Choksi, S.; Liu, Z.-G. Necroptosis and Tumor Progression. Trends Cancer 2022, 8, 21–27. [Google Scholar] [CrossRef]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, Pyroptosis and Apoptosis: An Intricate Game of Cell Death. Cell Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef]
- Lin, J.; Kumari, S.; Kim, C.; Van, T.-M.; Wachsmuth, L.; Polykratis, A.; Pasparakis, M. RIPK1 Counteracts ZBP1-Mediated Necroptosis to Inhibit Inflammation. Nature 2016, 540, 124–128. [Google Scholar] [CrossRef]
- Someda, M.; Kuroki, S.; Miyachi, H.; Tachibana, M.; Yonehara, S. Caspase-8, Receptor-Interacting Protein Kinase 1 (RIPK1), and RIPK3 Regulate Retinoic Acid-Induced Cell Differentiation and Necroptosis. Cell Death Differ. 2020, 27, 1539–1553. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, D.A.; Quarato, G.; Liedmann, S.; Tummers, B.; Zhang, T.; Guy, C.; Crawford, J.C.; Palacios, G.; Pelletier, S.; Kalkavan, H.; et al. Caspase-8 and FADD Prevent Spontaneous ZBP1 Expression and Necroptosis. Proc. Natl. Acad. Sci. USA 2022, 119, e2207240119. [Google Scholar] [CrossRef]
- Victorelli, S.; Salmonowicz, H.; Chapman, J.; Martini, H.; Vizioli, M.G.; Riley, J.S.; Cloix, C.; Hall-Younger, E.; Machado Espindola-Netto, J.; Jurk, D.; et al. Apoptotic Stress Causes mtDNA Release during Senescence and Drives the SASP. Nature 2023, 622, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Chan, F.K.-M.; Luz, N.F.; Moriwaki, K. Programmed Necrosis in the Cross Talk of Cell Death and Inflammation. Annu. Rev. Immunol. 2015, 33, 79–106. [Google Scholar] [CrossRef]
- Newton, K.; Strasser, A.; Kayagaki, N.; Dixit, V.M. Cell Death. Cell 2024, 187, 235–256. [Google Scholar] [CrossRef]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.-L.; Schneider, P.; Seed, B.; Tschopp, J. Fas Triggers an Alternative, Caspase-8–Independent Cell Death Pathway Using the Kinase RIP as Effector Molecule. Nat. Immunol. 2000, 1, 489–495. [Google Scholar] [CrossRef]
- Chan, F.K.-M.; Shisler, J.; Bixby, J.G.; Felices, M.; Zheng, L.; Appel, M.; Orenstein, J.; Moss, B.; Lenardo, M.J. A Role for Tumor Necrosis Factor Receptor-2 and Receptor-Interacting Protein in Programmed Necrosis and Antiviral Responses. J. Biol. Chem. 2003, 278, 51613–51621. [Google Scholar] [CrossRef]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical Inhibitor of Nonapoptotic Cell Death with Therapeutic Potential for Ischemic Brain Injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Amin, P.; Ofengeim, D. Necroptosis and RIPK1-Mediated Neuroinflammation in CNS Diseases. Nat. Rev. Neurosci. 2019, 20, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.-W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.-J.; Lin, S.-C.; Dong, M.-Q.; Han, J. RIP3, an Energy Metabolism Regulator That Switches TNF-Induced Cell Death from Apoptosis to Necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef]
- Cho, Y.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K.-M. Phosphorylation-Driven Assembly of the RIP1-RIP3 Complex Regulates Programmed Necrosis and Virus-Induced Inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef]
- He, S.; Liang, Y.; Shao, F.; Wang, X. Toll-like Receptors Activate Programmed Necrosis in Macrophages through a Receptor-Interacting Kinase-3–Mediated Pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 20054–20059. [Google Scholar] [CrossRef]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed Lineage Kinase Domain-like Protein Mediates Necrosis Signaling Downstream of RIP3 Kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.-C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.; Liu, Z.-G. Plasma Membrane Translocation of Trimerized MLKL Protein Is Required for TNF-Induced Necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef]
- Huang, Z.; Wu, S.-Q.; Liang, Y.; Zhou, X.; Chen, W.; Li, L.; Wu, J.; Zhuang, Q.; Chen, C.; Li, J.; et al. RIP1/RIP3 Binding to HSV-1 ICP6 Initiates Necroptosis to Restrict Virus Propagation in Mice. Cell Host Microbe 2015, 17, 229–242. [Google Scholar] [CrossRef]
- Zhang, T.; Yin, C.; Fedorov, A.; Qiao, L.; Bao, H.; Beknazarov, N.; Wang, S.; Gautam, A.; Williams, R.M.; Crawford, J.C.; et al. ADAR1 Masks the Cancer Immunotherapeutic Promise of ZBP1-Driven Necroptosis. Nature 2022, 606, 594–602. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, J.; Lv, M.; Cui, N.; Shan, B.; Sun, Q.; Yan, L.; Zhang, M.; Zou, C.; Yuan, J.; et al. Defective Prelamin A Processing Promotes Unconventional Necroptosis Driven by Nuclear RIPK1. Nat. Cell Biol. 2024, 26, 567–580. [Google Scholar] [CrossRef]
- Wong, W.W.-L.; Vince, J.E.; Lalaoui, N.; Lawlor, K.E.; Chau, D.; Bankovacki, A.; Anderton, H.; Metcalf, D.; O’Reilly, L.; Jost, P.J.; et al. cIAPs and XIAP Regulate Myelopoiesis through Cytokine Production in an RIPK1- and RIPK3-Dependent Manner. Blood 2014, 123, 2562–2572. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, M.; Günther, S.D.; Schwarzer, R.; Albert, M.-C.; Schorn, F.; Werthenbach, J.P.; Schiffmann, L.M.; Stair, N.; Stocks, H.; Seeger, J.M.; et al. Caspase-8 Is the Molecular Switch for Apoptosis, Necroptosis and Pyroptosis. Nature 2019, 575, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhu, F.; Wei, J.; Xie, J.; He, J.; Wei, D.; Li, Y.; Lai, K.; Liu, L.; Su, Q.; et al. The Active Fraction of Polyrhachis Vicina Roger (AFPR) Activates ERK to Cause Necroptosis in Colorectal Cancer. J. Ethnopharmacol. 2023, 312, 116454. [Google Scholar] [CrossRef] [PubMed]
- Massacci, G.; Perfetto, L.; Sacco, F. The Cyclin-Dependent Kinase 1: More than a Cell Cycle Regulator. Br. J. Cancer 2023, 129, 1707–1716. [Google Scholar] [CrossRef]
- Shi, C.-S.; Kehrl, J.H. Bcl-2 Regulates Pyroptosis and Necroptosis by Targeting BH3-like Domains in GSDMD and MLKL. Cell Death Discov. 2019, 5, 151. [Google Scholar] [CrossRef]
- Zhou, J.; Qin, X.; Li, L.; Tian, D.; Zou, Z.; Gu, Z.; Su, L. Heat Stress-Induced Intestinal Epithelial Cells Necroptosis via TLR3-TRIF-RIP3 Pathway Was Dependent on P53. Int. Immunopharmacol. 2023, 122, 110574. [Google Scholar] [CrossRef]
- de Almagro, M.C.; Vucic, D. Necroptosis: Pathway Diversity and Characteristics. Semin. Cell Dev. Biol. 2015, 39, 56–62. [Google Scholar] [CrossRef]
- Wang, J.; Wang, H.; Gao, M.; Zhang, Y.; Zhang, L.; Huang, D.; Tu, K.; Xu, Q. The Regulation of Amino Acid Metabolism in Tumor Cell Death: From the Perspective of Physiological Functions. Apoptosis 2023, 28, 1304–1314. [Google Scholar] [CrossRef]
- Gong, Y.-N.; Guy, C.; Olauson, H.; Becker, J.U.; Yang, M.; Fitzgerald, P.; Linkermann, A.; Green, D.R. ESCRT-III Acts Downstream of MLKL to Regulate Necroptotic Cell Death and Its Consequences. Cell 2017, 169, 286–300. [Google Scholar] [CrossRef]
- Zhang, T.; Xu, D.; Trefts, E.; Lv, M.; Inuzuka, H.; Song, G.; Liu, M.; Lu, J.; Liu, J.; Chu, C.; et al. Metabolic Orchestration of Cell Death by AMPK-Mediated Phosphorylation of RIPK1. Science 2023, 380, 1372–1380. [Google Scholar] [CrossRef]
- Wu, X.; Nagy, L.E.; Gautheron, J. Mediators of Necroptosis: From Cell Death to Metabolic Regulation. EMBO Mol. Med. 2024, 16, 219–237. [Google Scholar] [CrossRef] [PubMed]
- Riebeling, T.; Kunzendorf, U.; Krautwald, S. The Role of RHIM in Necroptosis. Biochem. Soc. Trans. 2022, 50, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Martens, S.; Bridelance, J.; Roelandt, R.; Vandenabeele, P.; Takahashi, N. MLKL in Cancer: More than a Necroptosis Regulator. Cell Death Differ. 2021, 28, 1757–1772. [Google Scholar] [CrossRef] [PubMed]
- lim, J.; Park, H.; Heisler, J.; Maculins, T.; Roose-Girma, M.; Xu, M.; Mckenzie, B.; Campagne, M.L.; Newton, K.; Murthy, A. Autophagy regulates inflammatory programmed cell death via turnover of RHIM-domain proteins. eLife 2019, 8, e44452. [Google Scholar] [CrossRef] [PubMed]
- Stanger, B.Z.; Leder, P.; Lee, T.-H.; Kim, E.; Seed, B. RIP: A Novel Protein Containing a Death Domain That Interacts with Fas/APO-1 (CD95) in Yeast and Causes Cell Death. Cell 1995, 81, 513–523. [Google Scholar] [CrossRef]
- Degterev, A.; Ofengeim, D.; Yuan, J. Targeting RIPK1 for the Treatment of Human Diseases. Proc. Natl. Acad. Sci. USA 2019, 116, 9714–9722. [Google Scholar] [CrossRef]
- Holbrook, J.; Lara-Reyna, S.; Jarosz-Griffiths, H.; McDermott, M.F. Tumour Necrosis Factor Signalling in Health and Disease. F1000Research 2019, 8, 111. [Google Scholar] [CrossRef]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and Its Role in Inflammation. Nature 2015, 517, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Lin, C.; He, R.; Chen, H.; Teng, Z.; Yao, H.; Liu, S.; Hoffman, R.M.; Ye, J.; Zhu, G. TRAF6 Regulates the Abundance of RIPK1 and Inhibits the RIPK1/RIPK3/MLKL Necroptosis Signaling Pathway and Affects the Progression of Colorectal Cancer. Cell Death Dis. 2023, 14, 6. [Google Scholar] [CrossRef]
- Deng, X.-X.; Li, S.-S.; Sun, F.-Y. Necrostatin-1 Prevents Necroptosis in Brains after Ischemic Stroke via Inhibition of RIPK1-Mediated RIPK3/MLKL Signaling. Aging Dis. 2019, 10, 807–817. [Google Scholar] [CrossRef]
- Shi, F.; Yuan, L.; Wong, T.; Li, Q.; Li, Y.; Xu, R.; You, Y.; Yuan, T.; zhang, H.; Shi, Z.; et al. Dimethyl Fumarate Inhibits Necroptosis and Alleviates Systemic Inflammatory Response Syndrome by Blocking the RIPK1-RIPK3-MLKL Axis. Pharmacol. Res. 2023, 189, 106697. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen Recognition by the Innate Immune System. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, W.J.; Sridharan, H.; Huang, C.; Mandal, P.; Upton, J.W.; Gough, P.J.; Sehon, C.A.; Marquis, R.W.; Bertin, J.; Mocarski, E.S. Toll-like Receptor 3-Mediated Necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 2013, 288, 31268–31279. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, T.; Behlke, J.; Lowenhaupt, K.; Heinemann, U.; Rich, A. Structure of the DLM-1-Z-DNA Complex Reveals a Conserved Family of Z-DNA-Binding Proteins. Nat. Struct. Biol. 2001, 8, 761–765. [Google Scholar] [CrossRef]
- Rebsamen, M.; Heinz, L.X.; Meylan, E.; Michallet, M.; Schroder, K.; Hofmann, K.; Vazquez, J.; Benedict, C.A.; Tschopp, J. DAI/ZBP1 Recruits RIP1 and RIP3 through RIP Homotypic Interaction Motifs to Activate NF-κB. EMBO Rep. 2009, 10, 916–922. [Google Scholar] [CrossRef]
- McComb, S.; Cessford, E.; Alturki, N.A.; Joseph, J.; Shutinoski, B.; Startek, J.B.; Gamero, A.M.; Mossman, K.L.; Sad, S. Type-I Interferon Signaling through ISGF3 Complex Is Required for Sustained Rip3 Activation and Necroptosis in Macrophages. Proc. Natl. Acad. Sci. USA 2014, 111, E3206–E3213. [Google Scholar] [CrossRef]
- Thapa, R.J.; Nogusa, S.; Chen, P.; Maki, J.L.; Lerro, A.; Andrake, M.; Rall, G.F.; Degterev, A.; Balachandran, S. Interferon-Induced RIP1/RIP3-Mediated Necrosis Requires PKR and Is Licensed by FADD and Caspases. Proc. Natl. Acad. Sci. USA 2013, 110, E3109–E3118. [Google Scholar] [CrossRef]
- Yang, D.; Liang, Y.; Zhao, S.; Ding, Y.; Zhuang, Q.; Shi, Q.; Ai, T.; Wu, S.-Q.; Han, J. ZBP1 Mediates Interferon-Induced Necroptosis. Cell Mol. Immunol. 2020, 17, 356–368. [Google Scholar] [CrossRef]
- Ye, H.; Lu, M.; Tu, C.; Min, L. Necroptosis in the Sarcoma Immune Microenvironment: From Biology to Therapy. Int. Immunopharmacol. 2023, 122, 110603. [Google Scholar] [CrossRef]
- He, C.; Liu, Y.; Huang, Z.; Yang, Z.; Zhou, T.; Liu, S.; Hao, Z.; Wang, J.; Feng, Q.; Liu, Y.; et al. A Specific RIP3+ Subpopulation of Microglia Promotes Retinopathy through a Hypoxia-Triggered Necroptotic Mechanism. Proc. Natl. Acad. Sci. USA 2021, 118, e2023290118. [Google Scholar] [CrossRef]
- Ye, K.; Chen, Z.; Xu, Y. The Double-Edged Functions of Necroptosis. Cell Death Dis. 2023, 14, 163. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Yang, Z.; Xu, Y.; Chen, Y.; Yu, Q. Apoptosis, Autophagy, Necroptosis, and Cancer Metastasis. Mol. Cancer 2015, 14, 48. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ma, Y.; Ye, J.; Li, G.; Kang, X.; Xie, W.; Wang, X. Drug Delivery System for Enhanced Tumour Treatment by Eliminating Intra-Tumoral Bacteria. J. Mater. Chem. B 2024, 12, 1194–1207. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Fang, J.; Wang, J.; Shao, X.; Xu, S.; Liu, P.; Ye, W.; Liu, Z. Regulation of Toll-like Receptor (TLR) Signaling Pathways in Atherosclerosis: From Mechanisms to Targeted Therapeutics. Acta Pharmacol. Sin. 2023, 44, 2358–2375. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Li, Y.; Guo, Z.; Lu, J.; Li, G.; Zhang, Z.; Zhang, F.; Wei, Y.; Wang, X.; Zhao, L. FePd Nanozyme- and SKN-Encapsulated Functional Lipid Nanoparticles for Cancer Nanotherapy via ROS-Boosting Necroptosis. ACS Appl. Mater. Interfaces 2024, 16, 18411–18421. [Google Scholar] [CrossRef]
- Xie, W.; Lu, J.; Guo, Z.; Guo, X.; Chi, Y.; Ye, J.; Zhang, J.; Xu, W.; Zhao, L.; Wei, Y. Necroptosis-Elicited Host Immunity: GOx-Loaded MoS2 Nanocatalysts for Self-Amplified Chemodynamic Immunotherapy. Nano Res. 2022, 15, 2244–2253. [Google Scholar] [CrossRef]
- Jaillon, S.; Ponzetta, A.; Di Mitri, D.; Santoni, A.; Bonecchi, R.; Mantovani, A. Neutrophil Diversity and Plasticity in Tumour Progression and Therapy. Nat. Rev. Cancer 2020, 20, 485–503. [Google Scholar] [CrossRef]
- Bai, L.; Kong, M.; Duan, Z.; Liu, S.; Zheng, S.; Chen, Y. M2-like Macrophages Exert Hepatoprotection in Acute-on-Chronic Liver Failure through Inhibiting Necroptosis-S100A9-Necroinflammation Axis. Cell Death Dis. 2021, 12, 93. [Google Scholar] [CrossRef]
- Noto, C.N.; Hoft, S.G.; DiPaolo, R.J. Mast Cells as Important Regulators in Autoimmunity and Cancer Development. Front. Cell Dev. Biol. 2021, 9, 752350. [Google Scholar] [CrossRef]
- Ramírez-Labrada, A.; Pesini, C.; Santiago, L.; Hidalgo, S.; Calvo-Pérez, A.; Oñate, C.; Andrés-Tovar, A.; Garzón-Tituaña, M.; Uranga-Murillo, I.; Arias, M.A.; et al. All About (NK Cell-Mediated) Death in Two Acts and an Unexpected Encore: Initiation, Execution and Activation of Adaptive Immunity. Front. Immunol. 2022, 13, 896228. [Google Scholar] [CrossRef]
- Swann, J.B.; Smyth, M.J. Immune Surveillance of Tumors. J. Clin. Investig. 2007, 117, 1137–1146. [Google Scholar] [CrossRef]
- Smyth, M.J.; Thia, K.Y.T.; Street, S.E.A.; Cretney, E.; Trapani, J.A.; Taniguchi, M.; Kawano, T.; Pelikan, S.B.; Crowe, N.Y.; Godfrey, D.I. Differential Tumor Surveillance by Natural Killer (Nk) and Nkt Cells. J. Exp. Med. 2000, 191, 661–668. [Google Scholar] [CrossRef]
- Moriwaki, K.; Balaji, S.; McQuade, T.; Malhotra, N.; Kang, J.; Chan, F.K.-M. The Necroptosis Adaptor RIPK3 Promotes Injury-Induced Cytokine Expression and Tissue Repair. Immunity 2014, 41, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, W.J.; Upton, J.W.; Long, A.B.; Livingston-Rosanoff, D.; Daley-Bauer, L.P.; Hakem, R.; Caspary, T.; Mocarski, E.S. RIP3 Mediates the Embryonic Lethality of Caspase-8-Deficient Mice. Nature 2011, 471, 368–372. [Google Scholar] [CrossRef]
- Yatim, N.; Jusforgues-Saklani, H.; Orozco, S.; Schulz, O.; Barreira Da Silva, R.; Reis E Sousa, C.; Green, D.R.; Oberst, A.; Albert, M.L. RIPK1 and NF-κB Signaling in Dying Cells Determines Cross-Priming of CD8+ T Cells. Science 2015, 350, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Xie, J.; Shi, J. Rac1/ROCK-Driven Membrane Dynamics Promote Natural Killer Cell Cytotoxicity via Granzyme-Induced Necroptosis. BMC Biol. 2021, 19, 140. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.J.; Bang, B.-R.; Han, K.H.; Hong, L.; Shim, E.-J.; Ma, J.; Lerner, R.A.; Otsuka, M. Regulation of NKT Cell-Mediated Immune Responses to Tumours and Liver Inflammation by Mitochondrial PGAM5-Drp1 Signalling. Nat. Commun. 2015, 6, 8371. [Google Scholar] [CrossRef]
- Molnár, T.; Mázló, A.; Tslaf, V.; Szöllősi, A.G.; Emri, G.; Koncz, G. Current Translational Potential and Underlying Molecular Mechanisms of Necroptosis. Cell Death Dis. 2019, 10, 860. [Google Scholar] [CrossRef]
- Thomas, D.A.; Massagué, J. TGF-β Directly Targets Cytotoxic T Cell Functions during Tumor Evasion of Immune Surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef]
- Meier, P.; Legrand, A.J.; Adam, D.; Silke, J. Immunogenic Cell Death in Cancer: Targeting Necroptosis to Induce Antitumour Immunity. Nat. Rev. Cancer 2024, 24, 299–315. [Google Scholar] [CrossRef]
- Mai, Z.; Lin, Y.; Lin, P.; Zhao, X.; Cui, L. Modulating Extracellular Matrix Stiffness: A Strategic Approach to Boost Cancer Immunotherapy. Cell Death Dis. 2024, 15, 307. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Galluzzi, L.; Apetoh, L.; Baert, T.; Birge, R.B.; Bravo-San Pedro, J.M.; Breckpot, K.; Brough, D.; Chaurio, R.; Cirone, M.; et al. Molecular and Translational Classifications of DAMPs in Immunogenic Cell Death. Front. Immunol. 2015, 6, 588. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Manning, G. Necroptosis and Inflammation. Annu. Rev. Biochem. 2016, 85, 743–763. [Google Scholar] [CrossRef]
- Snyder, A.G.; Hubbard, N.W.; Messmer, M.N.; Kofman, S.B.; Hagan, C.E.; Orozco, S.L.; Chiang, K.; Daniels, B.P.; Baker, D.; Oberst, A. Intratumoral Activation of the Necroptotic Pathway Components RIPK1 and RIPK3 Potentiates Antitumor Immunity. Sci. Immunol. 2019, 4, eaaw2004. [Google Scholar] [CrossRef]
- Oliver Metzig, M.; Fuchs, D.; Tagscherer, K.E.; Gröne, H.-J.; Schirmacher, P.; Roth, W. Inhibition of Caspases Primes Colon Cancer Cells for 5-Fluorouracil-Induced TNF-α-Dependent Necroptosis Driven by RIP1 Kinase and NF-κB. Oncogene 2016, 35, 3399–3409. [Google Scholar] [CrossRef]
- Yang, H.; Ma, Y.; Chen, G.; Zhou, H.; Yamazaki, T.; Klein, C.; Pietrocola, F.; Vacchelli, E.; Souquere, S.; Sauvat, A.; et al. Contribution of RIP3 and MLKL to Immunogenic Cell Death Signaling in Cancer Chemotherapy. OncoImmunology 2016, 5, e1149673. [Google Scholar] [CrossRef]
- Takemura, R.; Takaki, H.; Okada, S.; Shime, H.; Akazawa, T.; Oshiumi, H.; Matsumoto, M.; Teshima, T.; Seya, T. PolyI:C–Induced, TLR3/RIP3-Dependent Necroptosis Backs Up Immune Effector–Mediated Tumor Elimination In Vivo. Cancer Immunol. Res. 2015, 3, 902–914. [Google Scholar] [CrossRef]
- Um, W.; Ko, H.; You, D.G.; Lim, S.; Kwak, G.; Shim, M.K.; Yang, S.; Lee, J.; Song, Y.; Kim, K.; et al. Necroptosis-Inducible Polymeric Nanobubbles for Enhanced Cancer Sonoimmunotherapy. Adv. Mater. 2020, 32, 1907953. [Google Scholar] [CrossRef] [PubMed]
- Van Hoecke, L.; Van Lint, S.; Roose, K.; Van Parys, A.; Vandenabeele, P.; Grooten, J.; Tavernier, J.; De Koker, S.; Saelens, X. Treatment with mRNA Coding for the Necroptosis Mediator MLKL Induces Antitumor Immunity Directed against Neo-Epitopes. Nat. Commun. 2018, 9, 3417. [Google Scholar] [CrossRef]
- Zhao, H.; Chen, H.; Guo, Z.; Zhang, W.; Yu, H.; Zhuang, Z.; Zhong, H.; Liu, Z. In Situ Photothermal Activation of Necroptosis Potentiates Black Phosphorus-Mediated Cancer Photo-Immunotherapy. Chem. Eng. J. 2020, 394, 124314. [Google Scholar] [CrossRef]
- Sun, W.; Yu, J.; Gao, H.; Wu, X.; Wang, S.; Hou, Y.; Lu, J.-J.; Chen, X. Inhibition of Lung Cancer by 2-Methoxy-6-Acetyl-7-Methyljuglone Through Induction of Necroptosis by Targeting Receptor-Interacting Protein 1. Antioxid. Redox Signal. 2019, 31, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Workenhe, S.T.; Nguyen, A.; Bakhshinyan, D.; Wei, J.; Hare, D.N.; MacNeill, K.L.; Wan, Y.; Oberst, A.; Bramson, J.L.; Nasir, J.A.; et al. De Novo Necroptosis Creates an Inflammatory Environment Mediating Tumor Susceptibility to Immune Checkpoint Inhibitors. Commun. Biol. 2020, 3, 645. [Google Scholar] [CrossRef] [PubMed]
- Otani, T.; Matsuda, M.; Mizokami, A.; Kitagawa, N.; Takeuchi, H.; Jimi, E.; Inai, T.; Hirata, M. Osteocalcin Triggers Fas/FasL-Mediated Necroptosis in Adipocytes via Activation of P300. Cell Death Dis. 2018, 9, 1194. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, H.; Yang, R.; Ji, D.; Xia, X. GSK872 and Necrostatin-1 Protect Retinal Ganglion Cells against Necroptosis through Inhibition of RIP1/RIP3/MLKL Pathway in Glutamate-Induced Retinal Excitotoxic Model of Glaucoma. J. Neuroinflamm. 2022, 19, 262. [Google Scholar] [CrossRef]
- Jouan-Lanhouet, S.; Arshad, M.I.; Piquet-Pellorce, C.; Martin-Chouly, C.; Le Moigne-Muller, G.; Van Herreweghe, F.; Takahashi, N.; Sergent, O.; Lagadic-Gossmann, D.; Vandenabeele, P.; et al. TRAIL Induces Necroptosis Involving RIPK1/RIPK3-Dependent PARP-1 Activation. Cell Death Differ. 2012, 19, 2003–2014. [Google Scholar] [CrossRef]
- van Loo, G.; Bertrand, M.J.M. Death by TNF: A Road to Inflammation. Nat. Rev. Immunol. 2023, 23, 289–303. [Google Scholar] [CrossRef]
- Liu, S.; Perez, P.; Sun, X.; Chen, K.; Fatirkhorani, R.; Mammadova, J.; Wang, Z. MLKL Polymerization-Induced Lysosomal Membrane Permeabilization Promotes Necroptosis. Cell Death Differ. 2024, 31, 40–52. [Google Scholar] [CrossRef]
- Samson, A.L.; Zhang, Y.; Geoghegan, N.D.; Gavin, X.J.; Davies, K.A.; Mlodzianoski, M.J.; Whitehead, L.W.; Frank, D.; Garnish, S.E.; Fitzgibbon, C.; et al. MLKL Trafficking and Accumulation at the Plasma Membrane Control the Kinetics and Threshold for Necroptosis. Nat. Commun. 2020, 11, 3151. [Google Scholar] [CrossRef]
- Zang, X.; Song, J.; Li, Y.; Han, Y. Targeting Necroptosis as an Alternative Strategy in Tumor Treatment: From Drugs to Nanoparticles. J. Control. Release 2022, 349, 213–226. [Google Scholar] [CrossRef]
- Su, Z.; Yang, Z.; Xie, L.; DeWitt, J.P.; Chen, Y. Cancer Therapy in the Necroptosis Era. Cell Death Differ. 2016, 23, 748–756. [Google Scholar] [CrossRef]
- Aaes, T.L.; Kaczmarek, A.; Delvaeye, T.; De Craene, B.; De Koker, S.; Heyndrickx, L.; Delrue, I.; Taminau, J.; Wiernicki, B.; De Groote, P.; et al. Vaccination with Necroptotic Cancer Cells Induces Efficient Anti-Tumor Immunity. Cell Rep. 2016, 15, 274–287. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Onyshchenko, K.; Wang, L.; Gaedicke, S.; Grosu, A.-L.; Firat, E.; Niedermann, G. Necroptosis-Dependent Immunogenicity of Cisplatin: Implications for Enhancing the Radiation-Induced Abscopal Effect. Clin. Cancer Res. 2023, 29, 667–683. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.; Huang, Y.; Zhu, Q.; Cheng, H.; Pei, Y.; Feng, J.; Xu, M.; Jiang, G.; Song, Q.; Jiang, T.; et al. Necroptotic Cancer Cells-Mimicry Nanovaccine Boosts Anti-Tumor Immunity with Tailored Immune-Stimulatory Modality. Biomaterials 2018, 164, 80–97. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Xu, J.; Zhang, B.; Liu, J.; Liang, C.; Hua, J.; Meng, Q.; Yu, X.; Shi, S. Ferroptosis, Necroptosis, and Pyroptosis in Anticancer Immunity. J. Hematol. Oncol. 2020, 13, 110. [Google Scholar] [CrossRef]
- Yu, L.; Huang, K.; Liao, Y.; Wang, L.; Sethi, G.; Ma, Z. Targeting Novel Regulated Cell Death: Ferroptosis, Pyroptosis and Necroptosis in anti-PD-1/PD-L1 Cancer Immunotherapy. Cell Prolif. 2024, e13644. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agents | Cancer Type | Pathway | Ref. |
|---|---|---|---|
| Lytic dead cell | B16F10 melanoma | BATF3+ cDC1- and CD8+ leukocyte-dependent | [74] |
| 5-FU/IDN-7314 | hHT-29 Colon cancer | NF-κB- and RIP1-mediated necroptosis | [75] |
| TSZ/mitoxantrone | mTC-1 Lung carcinoma | RIPK3/MLKL | [76] |
| Poly(l:C)/zVAD-fmk | mCT-26 Colon carcinoma | Modulating the tumoricidal microenvironment and dendritic cell-inducing antitumor immune system | [77] |
| Polymeric Nanobubbles | CT26 Colon carcinoma | Sonoimmunotherapy-mediated maturation of dendritic cells and activation of CD8+ cytotoxic T cells | [78] |
| MLKL- and tBid-mRNA | B16F10 melanoma | Type I interferon signaling and Batf3-dependent dendritic cells | [79] |
| BP-bPEI-PEG/CpG | 4T1 Breast cancer | Trigger the release of damage-associated molecular patterns to potentiate the immune response | [80] |
| FePd/SKN@Lip | 4T1 Breast cancer | Trigger the release of damage-associated molecular patterns to potentiate the immune response | [55] |
| 2-methoxy-6-acetyl-7-methyjuglone | HCT116, HT29, A549 cells and cisplatin-resistant A549 cells | Lysosomal membrane permeabilization, mitochondrial dysfunction, ROS production | [81] |
| Oncolytic virus/mitoxantrone | U2OS cells and TUBO cells osteosarcoma | Revealing pro-inflammatory cytokine production and myeloid cells and cytotoxic T cell influx in local and distant tumors | [82] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Zhang, F.; Xie, W.; Niu, Y.; Wang, H.; Li, G.; Zhao, L.; Wang, X.; Xie, W. Induced Necroptosis and Its Role in Cancer Immunotherapy. Int. J. Mol. Sci. 2024, 25, 10760. https://doi.org/10.3390/ijms251910760
Zhang Z, Zhang F, Xie W, Niu Y, Wang H, Li G, Zhao L, Wang X, Xie W. Induced Necroptosis and Its Role in Cancer Immunotherapy. International Journal of Molecular Sciences. 2024; 25(19):10760. https://doi.org/10.3390/ijms251910760
Chicago/Turabian StyleZhang, Ziyao, Fangming Zhang, Wenjing Xie, Yubo Niu, Haonan Wang, Guofeng Li, Lingyun Zhao, Xing Wang, and Wensheng Xie. 2024. "Induced Necroptosis and Its Role in Cancer Immunotherapy" International Journal of Molecular Sciences 25, no. 19: 10760. https://doi.org/10.3390/ijms251910760
APA StyleZhang, Z., Zhang, F., Xie, W., Niu, Y., Wang, H., Li, G., Zhao, L., Wang, X., & Xie, W. (2024). Induced Necroptosis and Its Role in Cancer Immunotherapy. International Journal of Molecular Sciences, 25(19), 10760. https://doi.org/10.3390/ijms251910760