1. Introduction
Following the tremendous success of mRNA vaccines in protecting against the disease caused by SARS-CoV-2 infection, mRNA-based drugs are being widely studied as candidates for various pharmaceutical products [
1]. Compared to conventional vaccines, which are relatively laborious and slow to produce, mRNA vaccines can be designed and scaled up more quickly and are versatile, easily adaptable, highly effective, and non-toxic since they do not integrate or alter genomic DNA [
2]. However, RNA is a molecule with high chemical instability and is rapidly degraded by ribonucleases in the extracellular environment, preventing its entry into target cells. This poses an important limitation for using RNA as a free molecule and underscores the need for an encapsulation system for the administration and delivery of RNA-based therapeutics. Among the encapsulation systems that have been studied, two stand out for their demonstrated effectiveness and robustness: lipoplexes and lipid nanoparticles (LNPs). Both systems are based on similar lipid mixtures, usually comprising an ionizable/cationic lipid, a helper lipid, and cholesterol. However, they differ in size, heterogeneity, and the location of the mRNA molecules they transport: while in lipoplexes the mRNA is adsorbed on their surface, in LNPs it is encapsulated inside. Although both systems work well in vitro, LNPs have proven to be more robust, safe, and effective for in vivo use [
3].
Another important aspect of mRNA therapeutics are the strict requirements to preserve mRNA integrity during manufacturing, storage, and transportation, which directly affects the logistics chain and the final product price [
4]. While conventional vaccines based on inactivated microorganisms can be stored under refrigeration conditions (2–8 °C) for at least 6 months, currently approved mRNA vaccines require ultra-low temperatures for storage, hampering their distribution, especially in developing countries with structural limitations. In this regard, numerous studies have been conducted aiming at finding optimal freezing conditions for LNPs to minimize chemical damage [
4]. To that end, cryoprotectants such as sucrose are added to mRNA-LNP formulations to preserve structural and functional properties during the freezing process. Thus, both mRNA-based vaccines authorized against COVID-19, SpikeVax
® and Comirnaty
®, contain a 10% sucrose content in the final product (prior to dilution). Notably, although sucrose is used as a cryoprotective agent in both cases, the initial storage temperature requirements were different, as SpikeVax
® required storage between −15 °C and −20 °C while Comirnaty
®/BNT162b2 necessitated storage between −60 °C and −80 °C [
5]. In 2021, the Comirnaty
® formulation was updated by replacing the originally used PBS (phosphate-buffered saline) with Tris (abbreviation of tris(hydroxymethyl)aminomethane) as the buffering agent, which improved its stability and allowed the approval by the European Medicines Agency (EMA) for storing the BioNTech/Pfizer vaccine between −15 °C and −25 °C for up to two weeks [
6]. Nevertheless, the mid- to long-term storage of mRNA-LNPs vaccines still requires infrastructure capabilities to ensure sub-zero temperatures along the entire chain of supply. While these requirements have been addressed from an engineering perspective by using special shipping containers and ultra-freezers, this still poses a logistical challenge that limits distribution and significantly increases the final cost, highlighting the need to develop biotechnological advancements to improve mRNA vaccine stability under non-freezing conditions to facilitate logistics and distribution [
4].
Lyophilization presents itself an effective alternative to prolong the shelf life of mRNA vaccines under refrigeration conditions, without the need for sub-zero temperatures. Lyophilization is a relatively mild drying method used in the pharmaceutical industry to improve the stability of biomolecules and drugs, based on removing water molecules from formulations through sublimation under a high vacuum and low temperature [
7]. There is currently an enormous interest in obtaining lyophilized formulations of mRNA-LNPs. However, the lyophilization of mRNA-LNPs is a complex process since freezing and dehydration impose mechanical stress and deformation on lipid structures, leading to LNP aggregation and the release of encapsulated mRNA [
8,
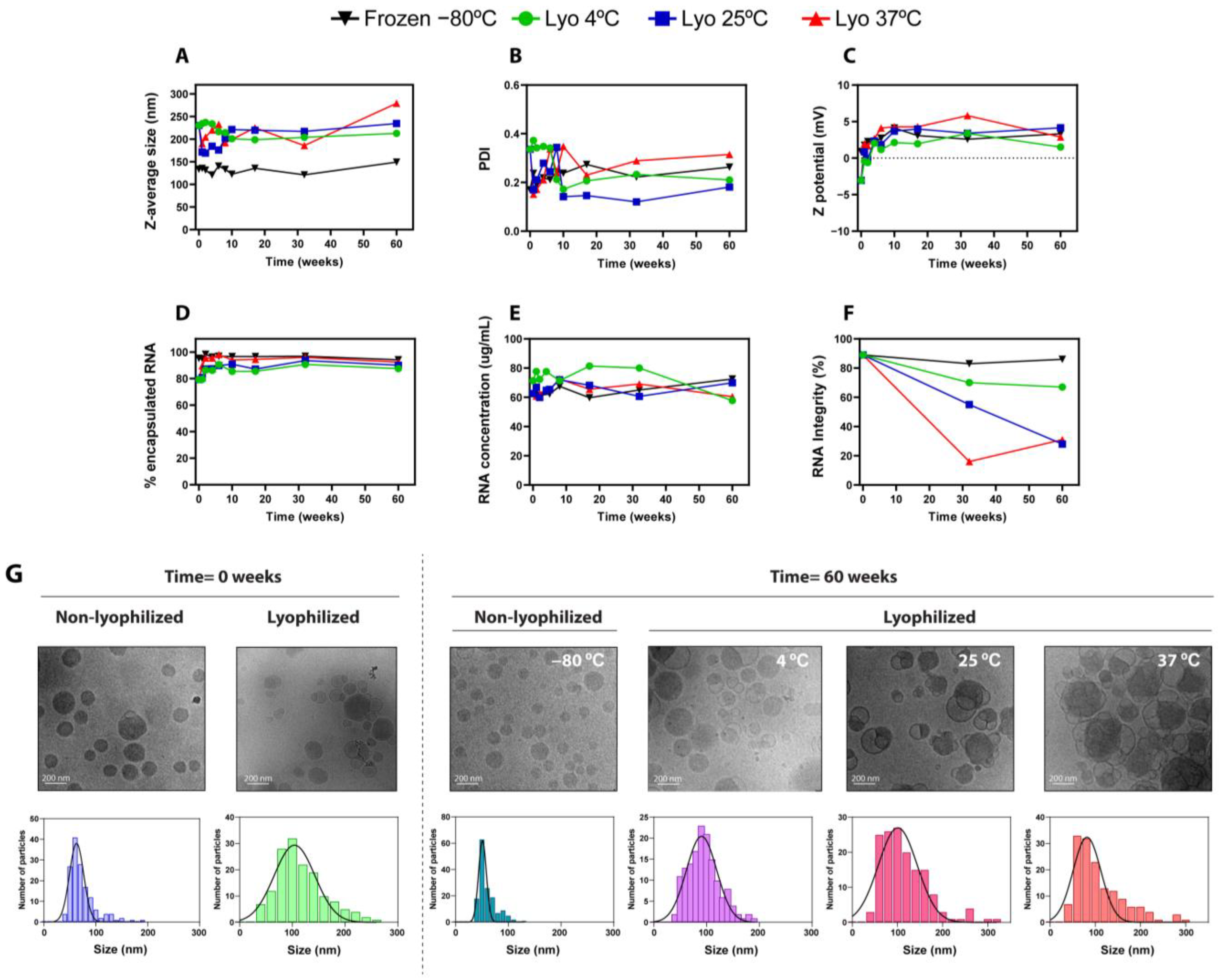
9]. In this regard, the physicochemical parameters of LNPs such as particle size, polydispersity, and encapsulation efficiency are critical for their bioactivity and efficacy and therefore must be preserved during the lyophilization process and subsequent storage. During the last few years, several studies have described lyophilized mRNA-LNPs maintaining their activity compared to liquid formulations [
10,
11,
12,
13]. However, only two studies show information about the long-term stability of lyophilized mRNA-LNPs [
11,
12]. Regarding commercial products, the GEMCOVAC-19 vaccine authorized for use in India consists of a lyophilized formulation based on mRNA encoding the SARS-CoV-2 S protein. This vaccine is composed of a lipoplex consisting of DOTAB and squalene, stabilized with polysorbates, and uses sucrose as a cryoprotectant; however, it does not use LNPs as a vehicle. According to the authors, this product can be stored under refrigeration and preserves its activity for 21 months at room temperature, although details on stability profile are not publicly available [
4].
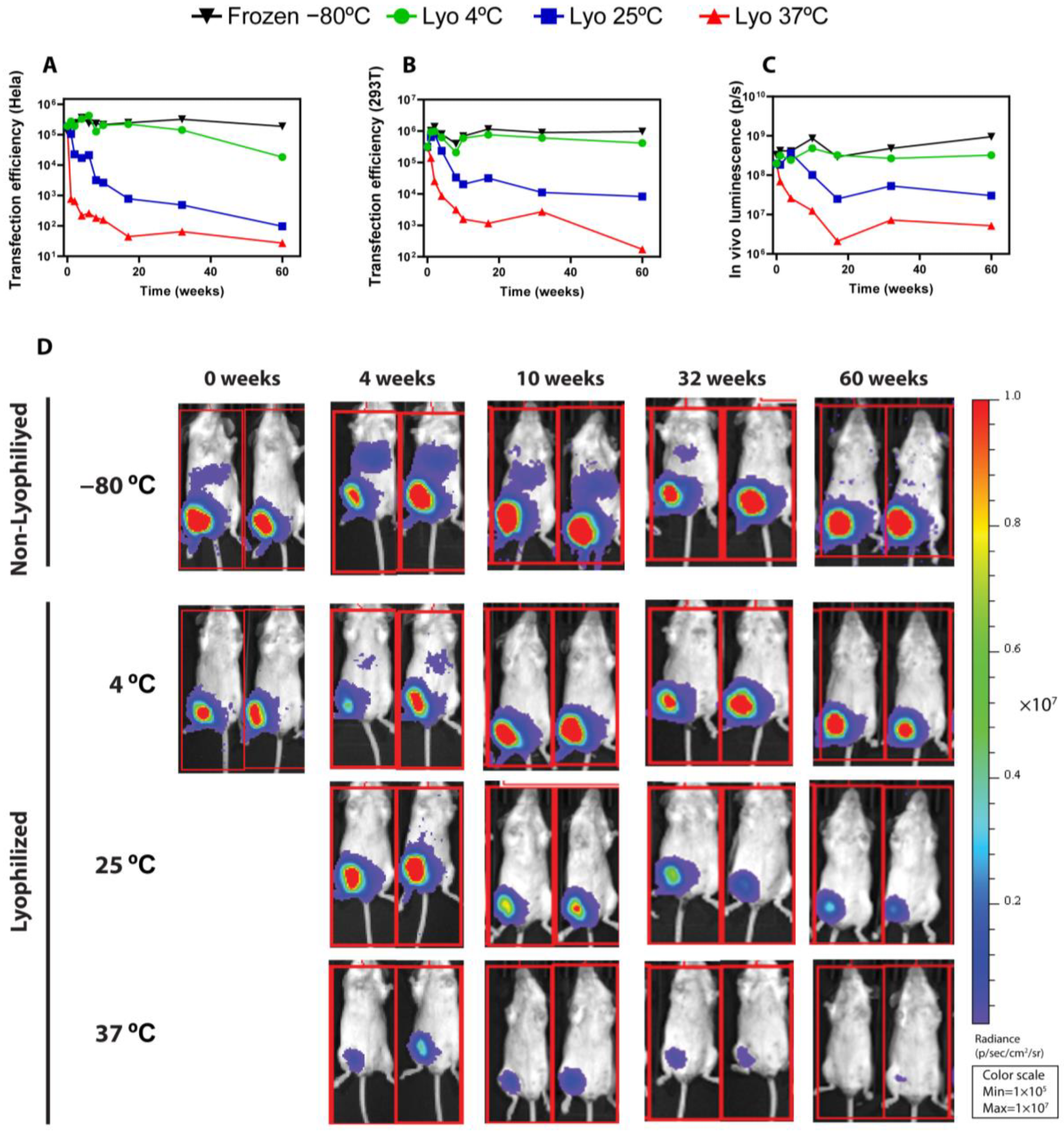
The evidence obtained so far underscores that, to obtain robust lyophilizable mRNA-LNPs formulations, it is imperative to comprehensively explore the various factors involved in the lyophilization process, such as the choice of buffers and cryoprotectants or the addition of excipients, as well as the intrinsic parameters of the lyophilization process, such as the temperature and pressure used. Thus, in the present study, we describe the systematic optimization process of a lyophilizable mRNA-LNP formulation, first paying attention to the type of buffer and lyoprotectant used and then adjusting the physical variables involved in the different stages of the freeze-drying process until obtaining the optimal conditions required to maintain the physicochemical and functional attributes of LNPs. Moreover, a long-term stability study was carried out for over one year, demonstrating that the lyophilization process managed to maintain the characteristics and in vivo functionality of LNPs under a refrigerated temperature throughout the studied period, without the need for freezing. Altogether, these results validate our freeze-drying process and pave the way for the systematic application of this methodology to obtain lyophilized mRNA-LNPs.
3. Materials and Methods
3.1. Materials
1-Octylnonyl 8-((2-hydroxyethyl) (6-oxo-6-(undecyloxy)hexyl)amino)octanoate (SM-102) ionizable lipid was purchase from BOC Sciences (NY, USA). The helper lipid 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) was obtained from Corden Pharma (Boulder, CO, USA), cholesterol was purchased from Merck (Darmstadt, Germany), and 1,2-dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 (DMG-PEG2000) was obtained from Cayman Chemical (Ann Arbor, MI, USA). Citrate buffer was purchased from Thermo Scientific (Waltham, MA, USA). Ethanol, maltose, sucrose, Tris (tris(hydroxymethyl)aminomethane), PBS (phosphate-buffered saline) buffers and other chemicals were obtained from PanReac AppliChem ITW Reagents (Barcelona, Spain).
3.2. mRNA Production
mRNA was produced by in vitro transcription (IVT) as previously reported [
28]. Prior to RNA IVT, pDNA encoding firefly luciferase (pTLuc) and green fluorescent protein (pTGFP) was linearized using the BspQI (NEB) enzyme, according to the manufacturer’s instructions. The linearization reaction was then purified with the Wizard
®SV Gel and PCR Clean-Up (Promega, Madison, WI, USA), in accordance with the manufacturer’s instructions.
The purified linear DNA was subsequently employed for mRNA production by in vitro transcription using T7 RNA polymerase, following the manufacturer’s instructions. Briefly, transcription reactions were performed at 37 °C for 3 h using the following materials: template linear DNA (50 μg/mL); T7 RNA polymerase (5000 U/mL; HONGENE, Shanghai, China); RNase inhibitor (1000 U/mL, HONGENE, ON-039); inorganic pyrophosphatase (2 U/mL, HONGENE, ON-025); ATP (5 μg/mL, HONGENE, R1331); GTP (5 μg/mL, HONGENE, R2331); CTP (5 μg/mL, HONGENE, R3331); N1-Methylpseudouridine (5 μg/mL, HONGENE, R5-027); CleanCap® AG (4 μg/mL, TRILINK®, San Diego, CA, USA); RNAse-free double-distilled water.
3.3. mRNA Purification
The generated mRNA transcripts were initially purified using DNaseI incubation (NEB, Ipswich, MA, USA) according to the manufacturer’s instructions. Following that, additional purification was carried out through affinity chromatography using POROS Oligo (dT) 25 columns (ThermoFisher, Waltham, MA, USA). Specifically, the buffers we employed were as follows: Buffer A, which contained 50 mM disodium phosphate, 0.5 M NaCl, and 5 mM EDTA, with pH = 7.0; and Buffer B, which contained 50 mM sodium dihydrogen phosphate and 5 mM EDTA, with pH = 7.0. The mRNA samples were initially half-diluted in Buffer A 2×. Following this, the column was equilibrated with 100% Buffer A, loaded with mRNA, washed with Buffer B, and ultimately eluted using double-deionized water. To completely eliminate Buffer B, mRNA was washed with a 30 KDa Amicon filter and then equilibrated through a one-tenth dilution in citrate buffer 10× with a pH of 6.5. The concentration of mRNA was determined by measuring the optical density at 260 nm, then adjusted to a final concentration of 1 mg/mL, aliquoted, and stored at −80 °C until needed. For quality assurance, all mRNAs underwent analysis through automated capillary electrophoresis (2100 Bioanalyzer G2938B, Agilent, Santa Clara, CA, USA). Subsequently, the mRNA samples were aliquoted and stored at −80 °C until needed.
3.4. Lipid Nanoparticle (LNP) Formulation
The microfluidic method was used for the controlled synthesis of LNPs. An ethanolic solution containing an ionizable lipid (SM-102), a helper lipid DOPE, cholesterol, and DMG-PEG2000 at a molar ratio of 50:10:38.5:1.5 was combined with an aqueous phase containing LUC-encoding mRNA diluted in 10 mM citrate buffer at pH 4, to achieve an ionizable lipid/RNA weight ratio of 10:1. A NanoAssemblr® Ignite microfluidic device (Precision Nanosystems, Vancouver, Canada) was set to operate at a total flow rate of 12 mL/min and a aqueous–ethanol volume ratio of 3. After this, ethanol was removed by dialysis (Pur-A-Lyzer™ Midi Dialysis Kit, Sigma, Sant Louis, MO, USA) using Tris or PBS and the different lyoprotectants for the buffer exchange. The resulting formulation was adjusted to 100 μg/mL mRNA concentration and lyophilized and/or stored as required for subsequent stability studies.
3.5. Lyophilization Process
Lyophilization was performed in a Virtis Genesis Pilot Freeze Dryer (SP, Warminster, PA, USA). The lyophilization process is based on three stages: firstly, samples undergo a freezing step, followed by a primary drying step and a subsequent secondary drying step. The experimental conditions of the initial lyophilization protocol and the modified protocol are depicted in
Tables S1 and S2, respectively. Vials were backfilled with pure nitrogen, capped, and transferred to various temperatures for stability assessments. To reconstitute lyophilized samples, 300 μL of RNase-free water was added to each vial and softly mixed until the solution turned into a homogeneous slightly white clear suspension.
3.6. LNP Characterization
LNP formulations were characterized to obtain the physicochemical characteristic parameters critical to their biological performance, such as particle size, polydispersity, and payload encapsulation. The average size, polydispersity (PDI), and zeta potential of LNPs were determined using a Malvern Zetasizer Advance Lab Blue Label (Malvern Instruments Ltd., Worcestershire, UK) by using a capillary cell (DTS1070) and diluting the sample (typically 1:100) in a filtered solution of 10 mM KCl.
The concentration and encapsulation efficiency of mRNA in LNPs were measured using a Quant-iT™ RiboGreen™ RNA Assay Kit from Thermo Fisher Scientific (Waltham, MA, USA), following the manufacturer’s protocols. Thus, the % of RNA encapsulated was calculated by comparing the total RNA obtained by the lysis of mRNA-LNPs using 0.5% Triton X-100 and the non-encapsulated RNA obtained when the LNPs are not lysed in the absence of detergent. Fluorescence was quantified in a Fluostar Omega microplate reader (BMG Labtech, Ortenberg, Germany). The EE% was calculated by the following equation:
Agarose gel electrophoresis was additionally used to determine the encapsulation of mRNA in LNPs. The quantification of encapsulated mRNA was determined by band densitometry using ImageJ software. Briefly, pixel densities of the upper, slow-migrating bands (corresponding to LNP-encapsulated mRNA) and of the lower, fast-migrating bands (corresponding to free mRNA) were measured separately, and the percentage of encapsulated mRNA was calculated by dividing the amount of encapsulated mRNA by the total amount of mRNA, corresponding to the sum of the upper and the lower bands within the same lane. Samples were loaded in a 1% agarose gel including SYBR-Safe, and electrophoresis was run at 120 V for 30 min. Gels were visualized with a UV transilluminator iBright™ CL750 imaging system (Thermo Fisher Scientific, Waltham, MA, USA), using adequate exposure times to avoid image saturation.
mRNA integrity was determined by capillary electrophoresis on the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA) using the Agilent RNA 6000 Nano Kit (Agilent, Santa Clara, CA, USA) [
29]. For the analysis of mRNA integrity, LNPs were disrupted by the addition of 0.5% Triton X-100 followed by heating at 70 °C during 2 min. As a reference of RNA integrity, the non-encapsulated RNA stock was treated and measured under the same conditions as the LNP samples, and the % of integrity was calculated relative to the integrity of this non-encapsulated RNA. Electropherograms obtained at each time point were analyzed using RNA 2100 Expert Software (version B.02.10).
3.7. Cryo-TEM Imaging of Fresh and Lyophilized LNPs
All CryoTEM analyses, including sample preparation and image collection, were performed using the CIC-bioGUNE Electron Microscopy Platform (Bilbao, Spain). For preparing the samples, L-polylysine was deposited in freshly glow-discharged carbon-only grids (EMResolutions, Newcastle, UK)), and then the grids were placed inside the chamber of the EM GP2 Automatic Plunge Freezing device (Leica, Wetzlar, Germany), which was maintained at 8 °C temperature and relative humidity close to saturation (90% RH). Four microliters of the sample was dropped onto the grid for 30 s. After incubation, most of the liquid on the grid was removed by blotting with absorbent standard filter paper (Ø55 mm, Grade 595, Hahnemühle, Dassel, Germany). After the blotting step, the grid was abruptly plunged into a liquid ethane bath, automatically set to −184 °C. Once the specimen was frozen, the vitrified grid was removed from the plunger and stored under liquid nitrogen inside a cryo-grid storage box.
The cryo-TEM data collection of the samples was performed on a JEM-1230 (JEOL Europe, Croissy, France) transmission electron microscope operated at 100 kV. This microscope has an UltraScan 4000 SP (4096 × 4096 pixels) cooled slow-scan CCD camera (GATAN, Leicester, UK). The images were recorded using DigitalMicrograph™ (Gatan Inc., Leicester, UK,
https://www.gatan.com/products/tem-analysis/gatan-microscopy-suite-software) software at nominal magnifications of 10.000× and 25.000× with a pixel size of 1.106 nm and 0.473 nm, respectively. TEM images were analyzed using the open-source image processing software ImageJ (version 1.52i) to obtain the size distributions of LNPs.
3.8. Cell Culture and mRNA Transfection
HeLa (ACC57, DSMZ GmbH, Berlin, Germany) cells were cultured on high-glucose DMEM (Merck D6429, Darmstadt, Germany) supplemented with fetal bovine serum 10% (Sigma F7524, Sant Louis, MO, USA), penicillin–streptomycin solution 1% (GibcoTM, 15140122, Waltham, MA, USA), and Glutamax 2 mM (Fisher 35050038, Waltham, MA, USA). HEK-293T (ATCC HB-8065) cells were cultured on RPMI 1640 (GibcoTM, 31870074, Waltham, MA, USA) supplemented with fetal bovine serum 10% (Sigma F7524, Sant Louis, MO, USA), penicillin–streptomycin solution 1% (GibcoTM, 15140122, Waltham, MA, USA), and Glutamax 2 mM (Fisher 35050038, Waltham, MA, USA). Both cell lines were cultured in a 175 m2 flask. The day before transfection, cells were detached from the flask by trypsinization (11590626, Fisher, Waltham, MA, USA), and subsequently, they were seeded into 96-well plates at a density of 1 × 104 cells/well. For transfection with a commercial cationic lipid, culture media was replaced with 90 μL of fresh media. Subsequently, a mixture of each mRNA (100 ng/well) and Lipofectamine MessengerMAXTM (Invitrogen 15397974; 0.2 μL/well, Waltham, MA, USA) was pre-incubated in OptiMEM media (31985062, Fisher, Waltham, MA, USA). The mRNA–lipofectamine mixture was added to the corresponding well in triplicate, directly resulting in a final mRNA concentration of 100 ng/well. Alternatively, the mRNA–lipofectamine mixture was diluted to half or a quarter of its concentration and then added to the cell culture, achieving final mRNA concentrations of 50 ng/well and 25 ng/well, respectively. For transfection with mRNA-LNPs, serial one-half dilutions in culture media were initially prepared. Then, 25 μL/well of the corresponding mRNA-LNP was added in triplicates to 100 μL of cells culture, resulting in final mRNA concentrations of 100 ng/well, 50 ng/well, or 25 ng/well. The cells, along with the mRNA-LNPs, were incubated for 24 h at 37 °C in a 5% CO2 atmosphere.
3.9. Firefly Luciferase Activity Quantification In Vitro
Cells were lysed 24 h post transfection by adding 100 μL of PBS-Triton 0.1%. Then, 98 μL of cell lysate was transferred to an opaque 96-well white plate. Buffered d-Luciferin (GoldBio LUCK−100 (St. Louis MO, USA) in 100 mM Tris-HCl pH 7.8, 5 mM MgCl2, 250 μM CoA, 150 μM ATP buffer) was added in 102 μL to each well, reaching a final concentration of 150 μg/mL. Cells that had not been incubated with any mRNA were employed as the negative control. Luminescence was measured after 5 min of incubation at room temperature in a FLUOstar Omega plate reader (BMG LABTECH, Ortenberg, Germany).
3.10. Statistical Analysis
Computer-based statistical analysis was carried out using the Prism® software (version 10.3.1, GraphPad Software, San Diego, CA, USA). All values are expressed as mean ± standard deviation (SD) of at least 3 experiments. Statistical significance was analyzed by using Student’s t-test. A p < 0.05 was considered statistically significant.
3.11. In Vivo Activity in Mice
Female BALB/c mice (Charles River Laboratories, Wilmington, MA, USA), 8–10-weeks old and weighting 18–23 g, were acclimatized to new conditions upon arrival at the experimental facilities for 3–7 days. Housing conditions were maintained at a room temperature of 20–24 °C, humidity of 50–70%, and light intensity of 60 lux, with a light–dark cycle of 12 h. For the measurement of firefly luciferase activity in mice, LNPs produced as described above, containing 1 μg of the indicated mRNA in a final volume of 50 μL, were injected intramuscularly. At 4 and 24 h post mRNA-LPN inoculation, mice were anesthetized by inhalation with 4% isoflurane using a vaporizer. The maintenance of anesthesia was sustained at 1.5% of isoflurane. Then, D-luciferin (12507, Quimigen, Madrid, Spain) was intraperitoneally injected at 150 mg/kg, typically 200 μL of the stock at 15 mg/mL in PBS for a 20 g mouse. Luciferase images were captured 10 min after luciferin inoculation using the IVIS Lumina XRMS Imaging System (PerkinElmer, Waltham, MA, USA) following the manufacturer’s instructions.
All procedures were carried out under Project Licence 59/21 approved by the Ethic Committee for Animal Experiments from the University of Zaragoza. The care and use of animals were performed accordingly with the Spanish Policy for Animal Protection RD53/2013, which meets the European Union Directive 2010/63 on the protection of animals used for experimental and other scientific purposes. Image acquisition was carried out by the “Imagen Médica y Fenotipado” service at Instituto Aragonés de Ciencias de la Salud.
4. Conclusions
In the present study, we carried out a comprehensive optimization of the freeze-drying process, from the selection of the best buffers and cryoprotectants to the fine-tuning of the lyophilization parameters, seeking to preserve the physicochemical properties and functionality of lyophilized LNPs. Our results revealed that Tris buffer was more effective than PBS in maintaining LNPs’ integrity and bioactivity, correlating with reduced RNA leakage and smaller size increments. On the other hand, the optimization of the freeze-drying recipe involved adjusting the temperatures of the primary and secondary drying steps and extending the primary drying phase to ensure complete water removal. These modifications led to improved mRNA encapsulation and particle size consistency, with sucrose and maltose used as lyoprotectants at 20% w/v yielding the best results. We next assessed the long-term stabilities of LNPs lyophilized using the optimized parameters and stored under various conditions over 12 months, including non-lyophilized formulations at 4 °C, −20 °C, and −80 °C and lyophilized LNPs stored at 4 °C, 25 °C, and 37 °C. The results demonstrated that lyophilized LNPs stored at 4 °C maintained their physicochemical properties, such as particle size, polydispersity index (PDI), zeta potential, and mRNA encapsulation efficiency, comparably to freshly prepared LNPs. Importantly, lyophilized LNPs stored at 4 °C retained in vivo functionality, achieving similar luciferase expression in mice to non-lyophilized LNPs stored at −80 °C. Conversely, LNPs stored at 25 °C and 37 °C showed decreased mRNA integrity and transfection efficiency over time, although, remarkably, some functionality persisted for up to 60 weeks at room temperature. In summary, herein we have described a detailed optimization process for obtaining lyophilized LNPs that are able to retain their physicochemical properties and in vivo activity for up to one year, offering significant advantages for the storage and distribution of mRNA vaccines by enabling long-term stability at 4 °C and potentially reducing cold chain logistics costs.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}