


Tissue Kallikrein-1 Suppresses Type I Interferon Responses and Reduces Depressive-Like Behavior in the MRL/lpr Lupus-Prone Mouse Model


 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
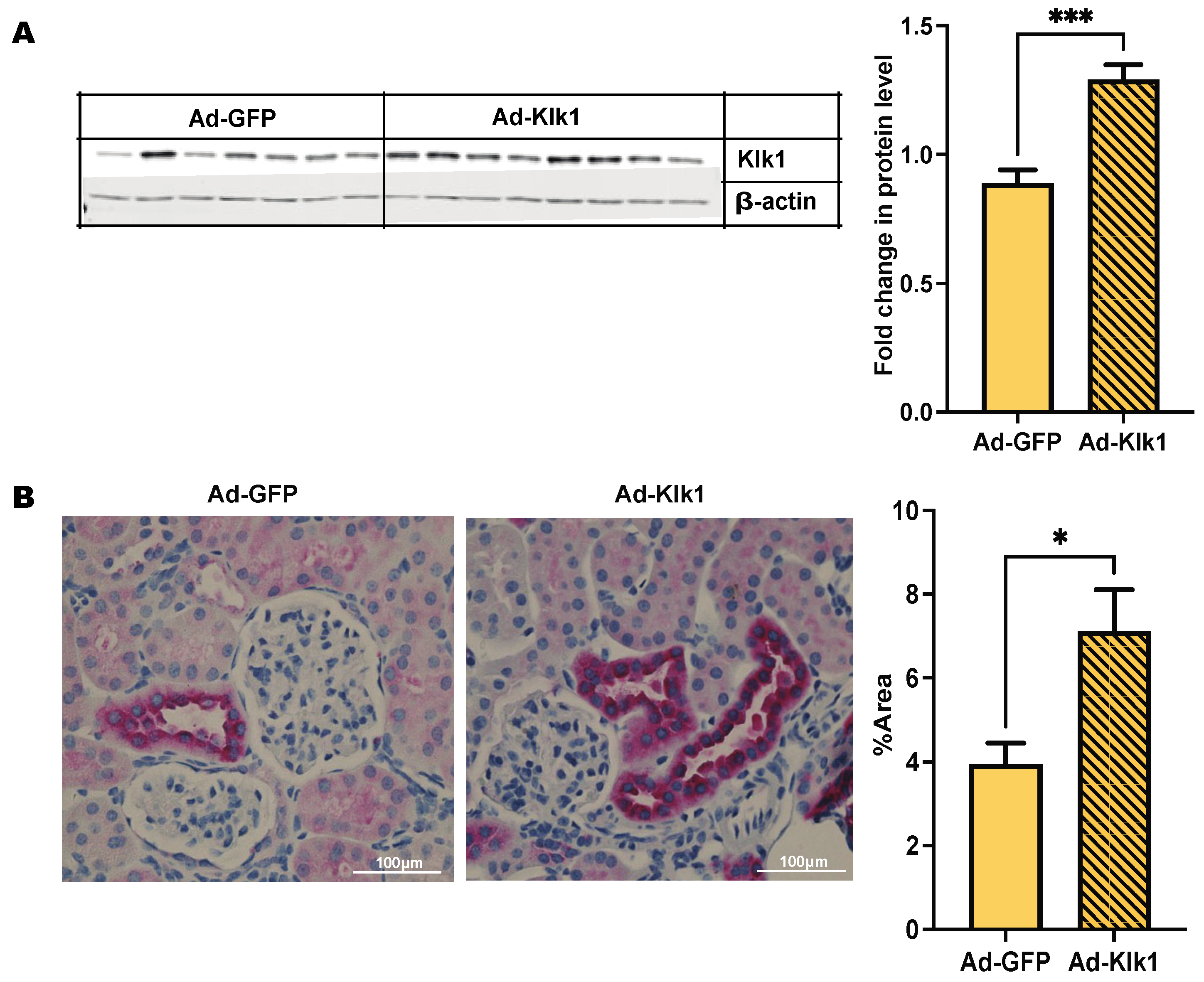
2.1. Adenoviral Vector Delivery of Mouse Klk1 Increases Tissue Protein Expression
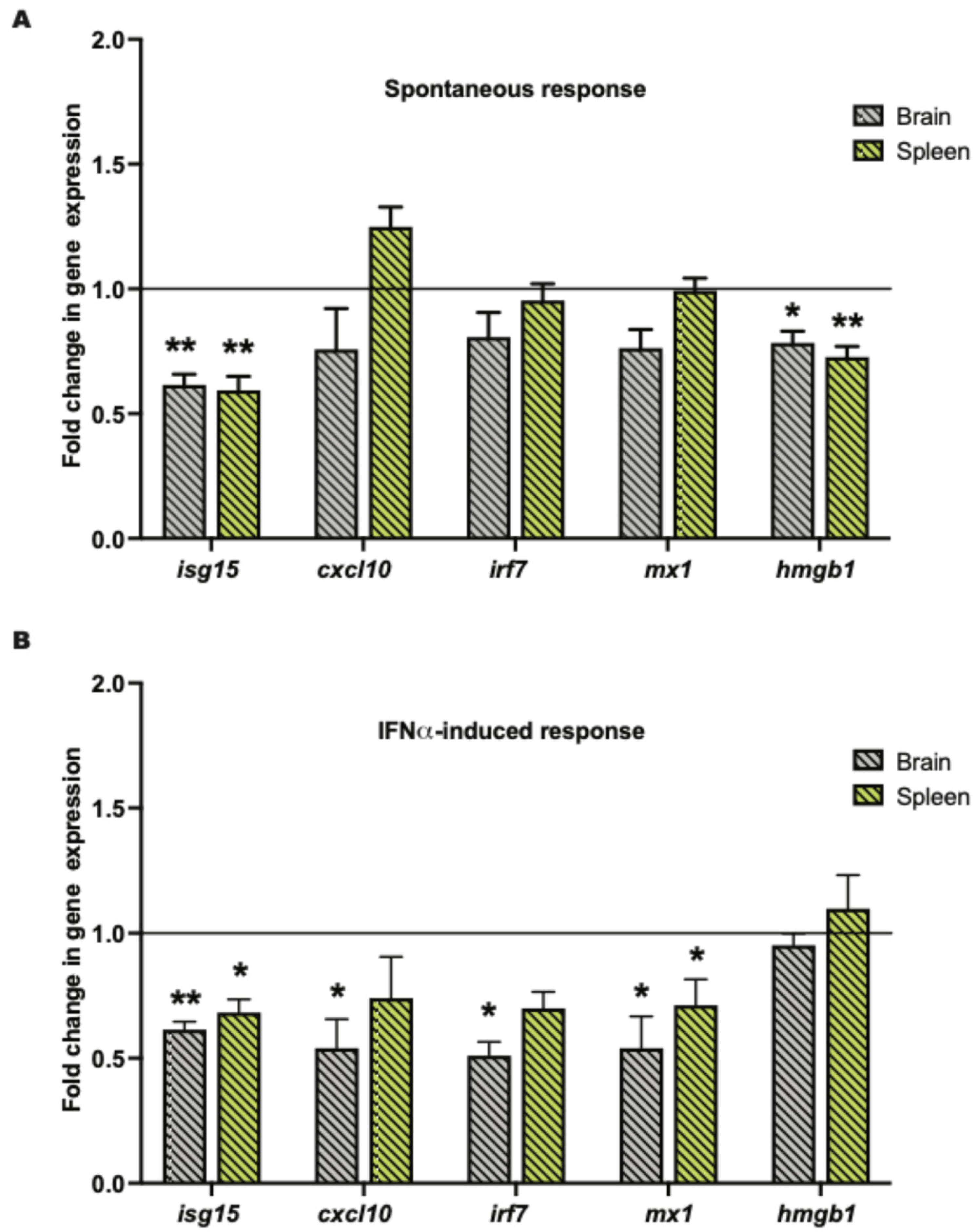
2.2. Tissue Klk1 Decreases IRG and hmgb1 Gene Expression in the Brain and Periphery
2.3. Tissue Klk1 Reduces Inflammatory Cytokine Levels in Plasma
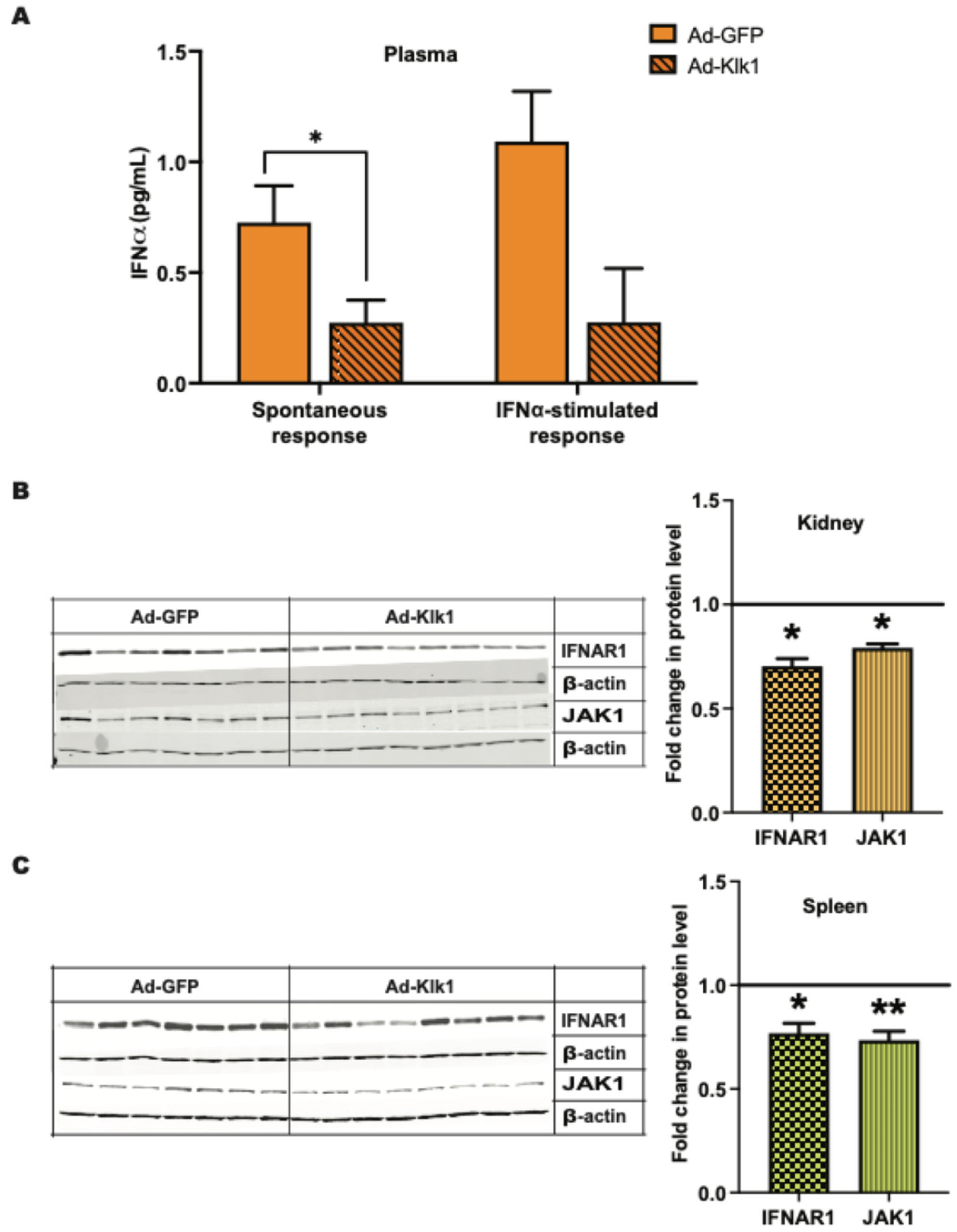
2.4. Klk1 Decreases Plasma IFNα Levels and Proteins in Type I IFN Signaling
2.5. Tissue Klk1 Reduces Depressive-Like Behavior in Lupus-Prone Mice
2.6. Klk1 Administration Reduces Microglial Inflammatory State in the Brain
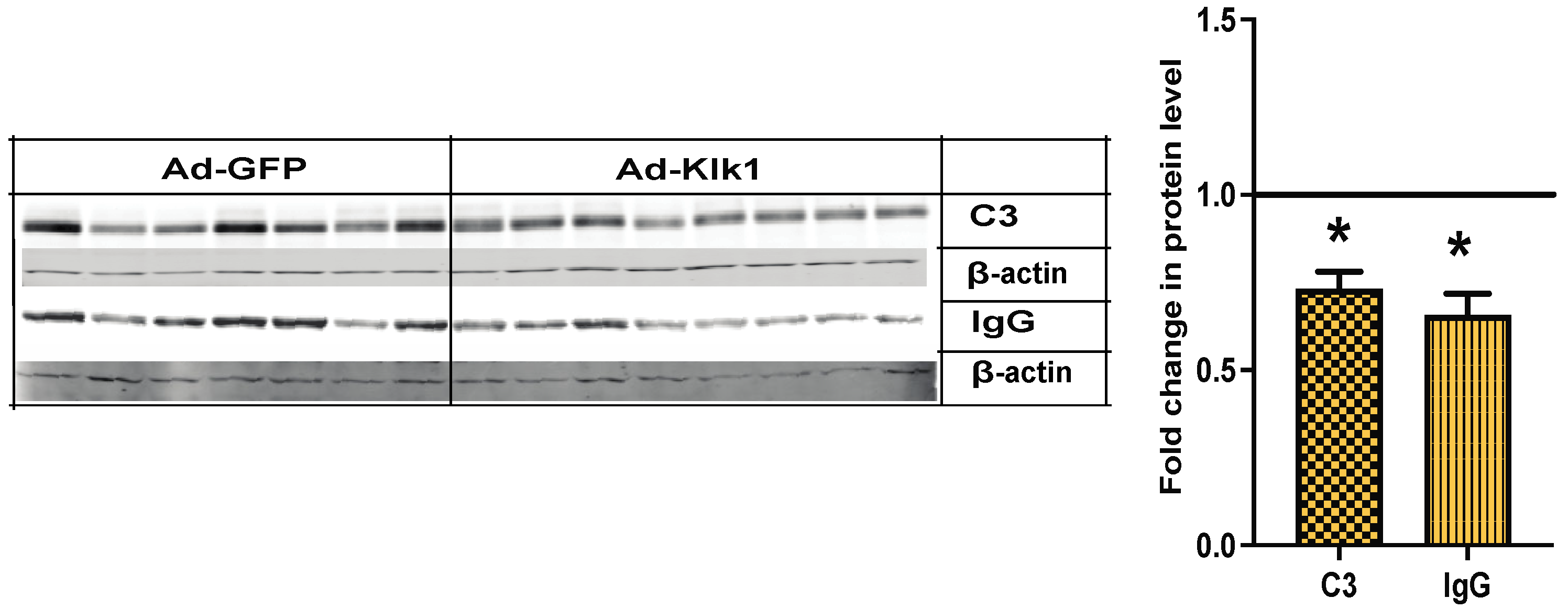
2.7. Klk1 Administration Reduces C3 and IgG Expression in the Kidney
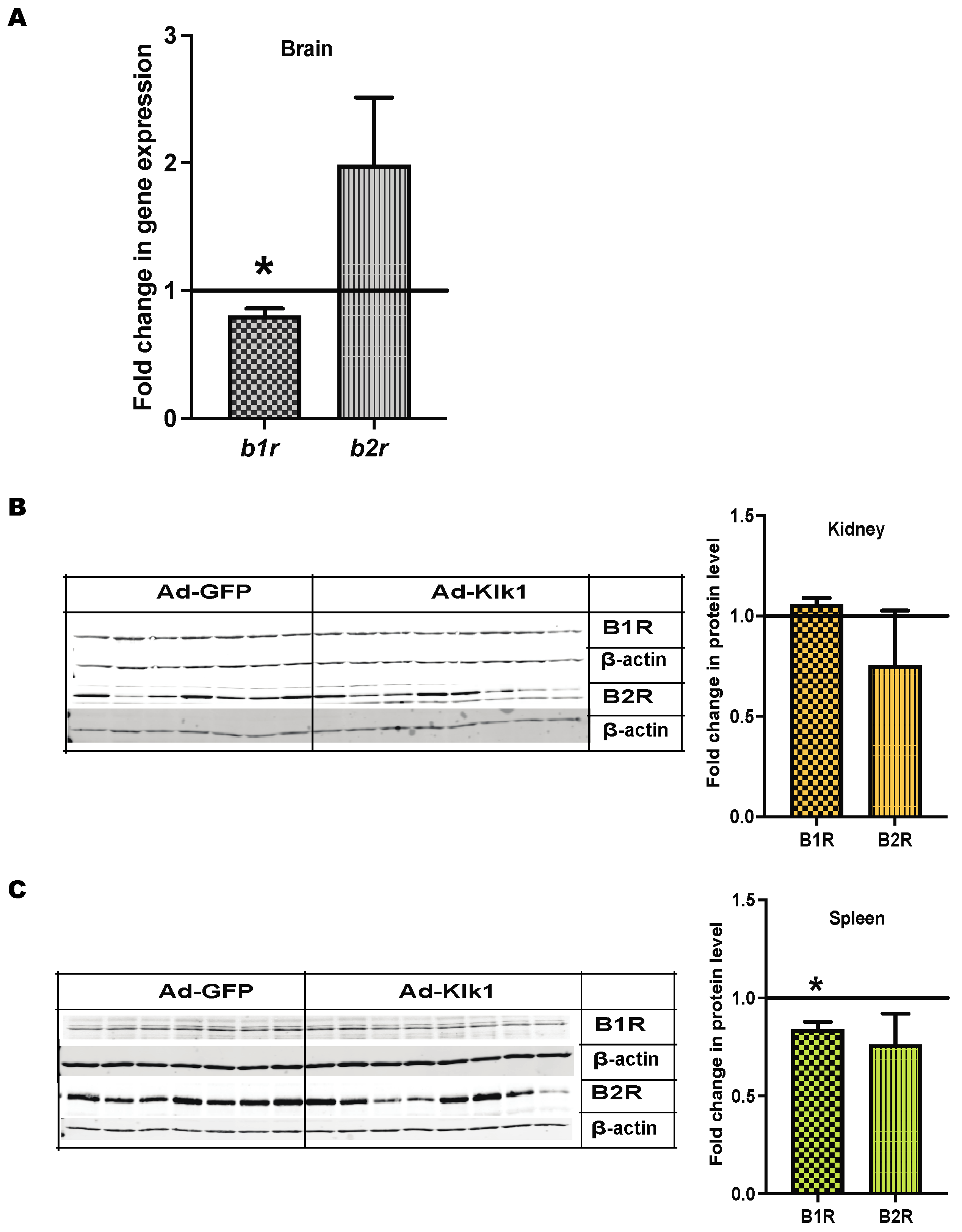
2.8. Klk1 Alters BK and PAR2 Receptor Expression
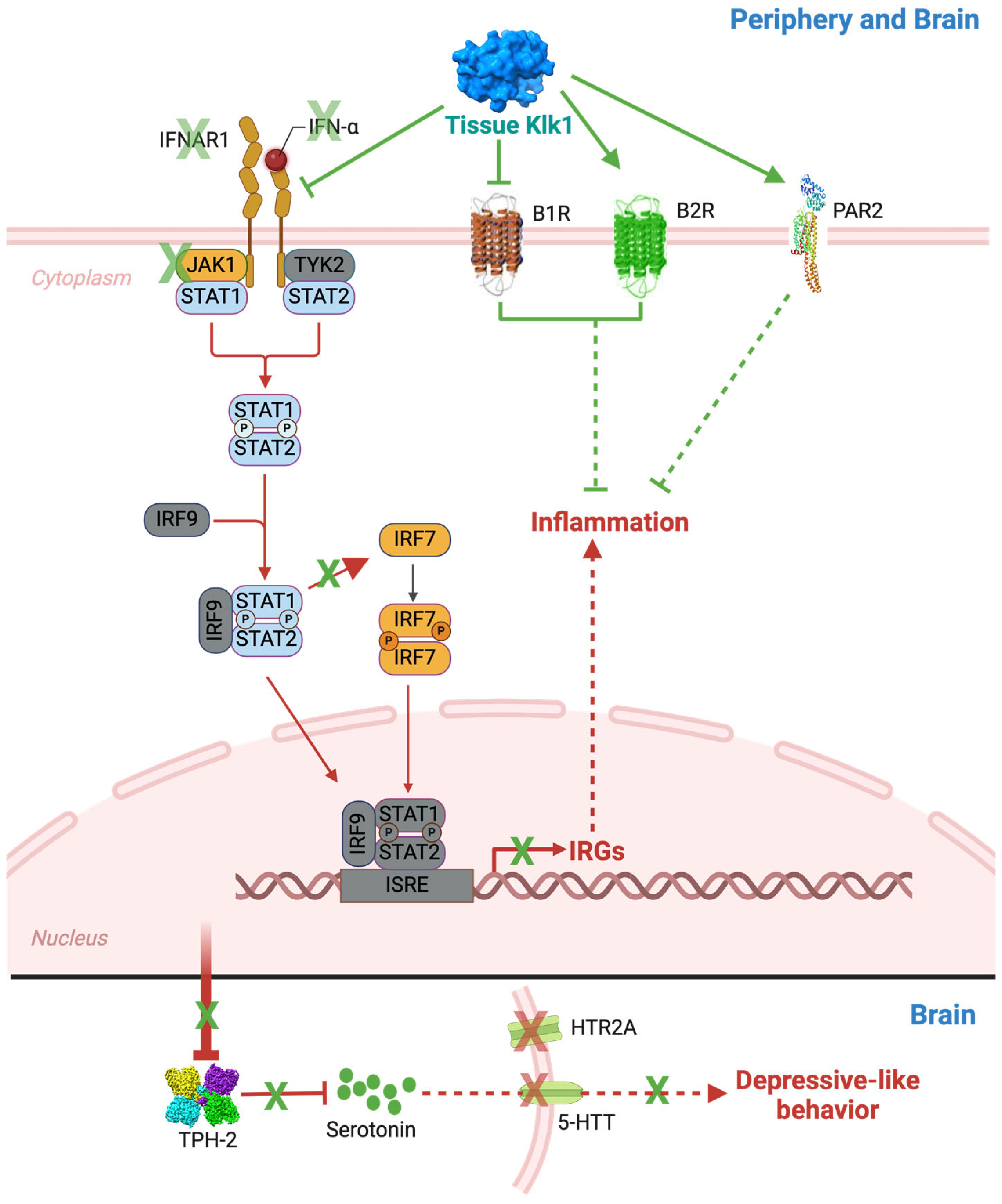
3. Discussion
4. Materials and Methods
4.1. Mice
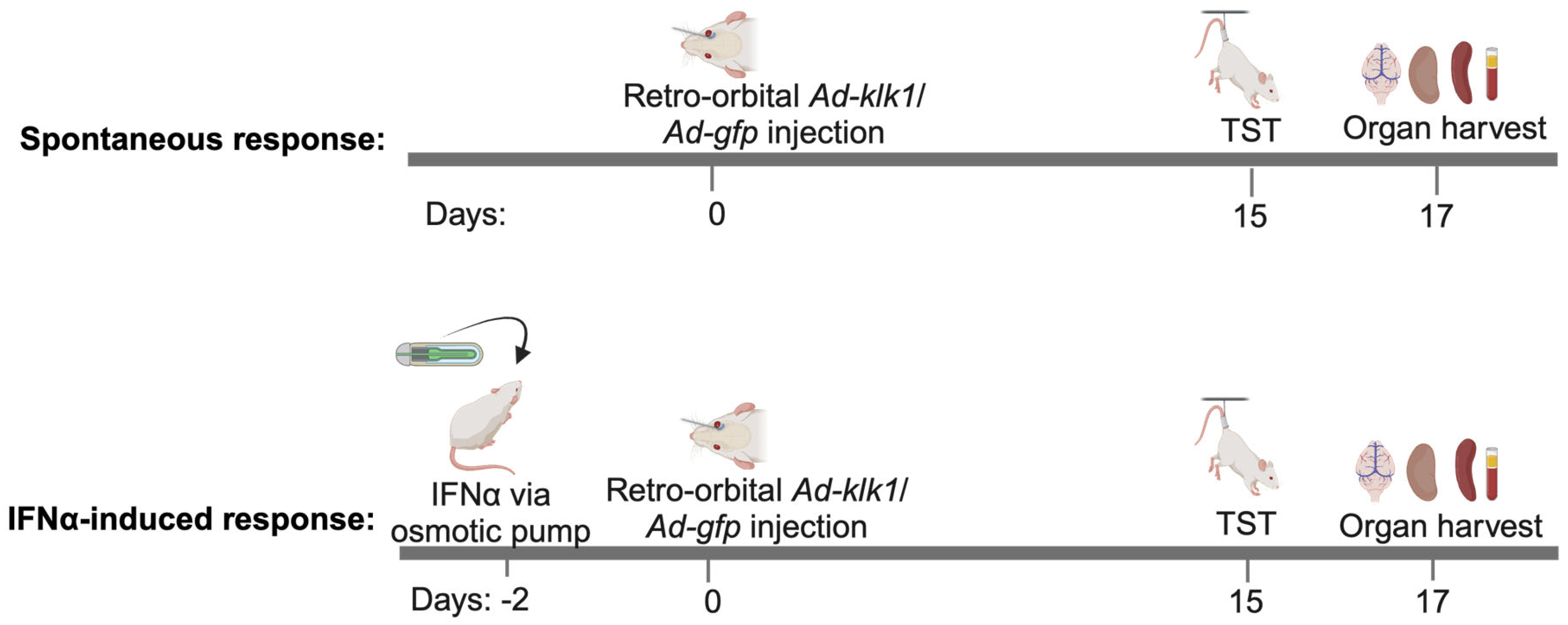
4.2. In Vivo Gene Delivery of Ad-klk1 and Administration of IFNα
4.3. Proteinuria Measurement
4.4. Blood Urea Nitrogen Analysis
4.5. RNA Extraction and qPCR
4.6. Western Blot
4.7. Proinflammatory Cytokine Analysis
4.8. IFNa ELISA
4.9. Autoantibody Detection
4.10. Immunohistochemistry
4.11. Plasma Serotonin Analysis
4.12. TST
4.13. Rotarod Test
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hawro, T.; Krupinska-Kun, M.; Rabe-Jablonska, J.; Sysa-Jedrzejowska, A.; Robak, E.; Bogaczewicz, J.; Wozniacka, A. Psychiatric disorders in patients with systemic lupus erythematosus: Association of anxiety disorder with shorter disease duration. Rheumatol. Int. 2011, 31, 1387–1391. [Google Scholar] [PubMed]
- Morrison, E.; Carpentier, S.; Shaw, E.; Doucette, S.; Hanly, J.G. Neuropsychiatric systemic lupus erythematosus: Association with global disease activity. Lupus 2014, 23, 370–377. [Google Scholar] [PubMed]
- Govoni, M.; Hanly, J.G. The management of neuropsychiatric lupus in the 21st century: Still so many unmet needs? Rheumatology 2020, 59 (Suppl. S5), v52–v62. [Google Scholar] [CrossRef]
- Palagini, L.; Mosca, M.; Tani, C.; Gemignani, A.; Mauri, M.; Bombardieri, S. Depression and systemic lupus erythematosus: A systematic review. Lupus 2013, 22, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Obermoser, G.; Pascual, V. The interferon-alpha signature of systemic lupus erythematosus. Lupus 2010, 19, 1012–1019. [Google Scholar] [CrossRef]
- Hagberg, N.; Ronnblom, L. Systemic Lupus Erythematosus--A Disease with A Dysregulated Type I Interferon System. Scand. J. Immunol. 2015, 82, 199–207. [Google Scholar] [CrossRef]
- Shiozawa, S.; Kuroki, Y.; Kim, M.; Hirohata, S.; Ogino, T. Interferon-alpha in lupus psychosis. Arthritis Rheum. 1992, 35, 417–422. [Google Scholar] [CrossRef]
- Bonaccorso, S.; Marino, V.; Biondi, M.; Grimaldi, F.; Ippoliti, F.; Maes, M. Depression induced by treatment with interferon-alpha in patients affected by hepatitis C virus. J. Affect. Disord. 2002, 72, 237–241. [Google Scholar]
- Pavol, M.A.; Meyers, C.A.; Rexer, J.L.; Valentine, A.D.; Mattis, P.J.; Talpaz, M. Pattern of neurobehavioral deficits associated with interferon alfa therapy for leukemia. Neurology 1995, 45, 947–950. [Google Scholar]
- Aw, E.; Zhang, Y.; Carroll, M. Microglial responses to peripheral type 1 interferon. J. Neuroinflamm. 2020, 17, 340. [Google Scholar] [CrossRef]
- Nocito, C.; Lubinsky, C.; Hand, M.; Khan, S.; Patel, T.; Seliga, A.; Winfield, M.; Zuluaga-Ramirez, V.; Fernandes, N.; Shi, X.; et al. Centrally Acting Angiotensin-Converting Enzyme Inhibitor Suppresses Type I Interferon Responses and Decreases Inflammation in the Periphery and the CNS in Lupus-Prone Mice. Front. Immunol. 2020, 11, 573677. [Google Scholar] [CrossRef] [PubMed]
- Seliga, A.; Lee, M.H.; Fernandes, N.C.; Zuluaga-Ramirez, V.; Didukh, M.; Persidsky, Y.; Potula, R.; Gallucci, S.; Sriram, U. Kallikrein–Kinin System Suppresses Type I Interferon Responses: A Novel Pathway of Interferon Regulation. Front. Immunol. 2018, 9, 156. [Google Scholar] [CrossRef]
- Kalinska, M.; Meyer-Hoffert, U.; Kantyka, T.; Potempa, J. Kallikreins—The melting pot of activity and function. Biochimie 2016, 122, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Blaber, S.I.; Ciric, B.; Christophi, G.P.; Bernett, M.J.; Blaber, M.; Rodriguez, M.; Scarisbrick, I.A. Targeting kallikrein 6 proteolysis attenuates CNS inflammatory disease. FASEB J. 2004, 18, 920–922. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Shen, B.; Gao, L.; Xia, C.F.; Bledsoe, G.; Chao, L. Tissue kallikrein in cardiovascular, cerebrovascular and renal diseases and skin wound healing. Biol. Chem. 2010, 391, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Moreau, M.E.; Garbacki, N.; Molinaro, G.; Brown, N.J.; Marceau, F.; Adam, A. The kallikrein-kinin system: Current and future pharmacological targets. J. Pharmacol. Sci. 2005, 99, 6–38. [Google Scholar]
- Herring, A.; Munster, Y.; Akkaya, T.; Moghaddam, S.; Deinsberger, K.; Meyer, J.; Zahel, J.; Sanchez-Mendoza, E.; Wang, Y.; Hermann, D.M.; et al. Kallikrein-8 inhibition attenuates Alzheimer’s disease pathology in mice. Alzheimers Dement. 2016, 12, 1273–1287. [Google Scholar] [CrossRef]
- Spencer, B.; Valera, E.; Rockenstein, E.; Trejo-Morales, M.; Adame, A.; Masliah, E. A brain-targeted, modified neurosin (kallikrein-6) reduces alpha-synuclein accumulation in a mouse model of multiple system atrophy. Mol. Neurodegener. 2015, 10, 48. [Google Scholar] [CrossRef]
- Yoon, H.; Scarisbrick, I.A. Kallikrein-related peptidase 6 exacerbates disease in an autoimmune model of multiple sclerosis. Biol. Chem. 2016, 397, 1277–1286. [Google Scholar] [CrossRef]
- Bhoola, K.D.; Figueroa, C.D.; Worthy, K. Bioregulation of kinins: Kallikreins, kininogens, and kininases. Pharmacol. Rev. 1992, 44, 1–80. [Google Scholar]
- Avgeris, M.; Mavridis, K.; Scorilas, A. Kallikrein-related peptidase genes as promising biomarkers for prognosis and monitoring of human malignancies. Biol. Chem. 2010, 391, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Chao, L. Kallikrein-kinin in stroke, cardiovascular and renal disease. Exp. Physiol. 2005, 90, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Regoli, D.; Plante, G.E.; Gobeil, F., Jr. Impact of kinins in the treatment of cardiovascular diseases. Pharmacol. Ther. 2012, 135, 94–111. [Google Scholar] [CrossRef] [PubMed]
- Heuberger, D.M.; Schuepbach, R.A. Protease-activated receptors (PARs): Mechanisms of action and potential therapeutic modulators in PAR-driven inflammatory diseases. Thromb. J. 2019, 17, 4. [Google Scholar] [CrossRef]
- Lewkowich, I.P.; Day, S.B.; Ledford, J.R.; Zhou, P.; Dienger, K.; Wills-Karp, M.; Page, K. Protease-activated receptor 2 activation of myeloid dendritic cells regulates allergic airway inflammation. Respir. Res. 2011, 12, 122. [Google Scholar] [CrossRef]
- Ossovskaya, V.S.; Bunnett, N.W. Protease-activated receptors: Contribution to physiology and disease. Physiol. Rev. 2004, 84, 579–621. [Google Scholar] [CrossRef]
- Rothmeier, A.S.; Ruf, W. Protease-activated receptor 2 signaling in inflammation. Semin. Immunopathol. 2012, 34, 133–149. [Google Scholar] [CrossRef]
- Yamaguchi, R.; Yamamoto, T.; Sakamoto, A.; Ishimaru, Y.; Narahara, S.; Sugiuchi, H.; Yamaguchi, Y. A protease-activated receptor 2 agonist (AC-264613) suppresses interferon regulatory factor 5 and decreases interleukin-12p40 production by lipopolysaccharide-stimulated macrophages: Role of p53. Cell Biol. Int. 2016, 40, 629–641. [Google Scholar] [CrossRef]
- Zhong, B.; Ma, S.; Wang, D.H. Protease-activated receptor 2 protects against myocardial ischemia-reperfusion injury through the lipoxygenase pathway and TRPV1 channels. Exp. Ther. Med. 2019, 18, 3636–3642. [Google Scholar] [CrossRef]
- Ponticelli, C.; Meroni, P.L. Kallikreins and lupus nephritis. J. Clin. Investig. 2009, 119, 768–771. [Google Scholar] [CrossRef]
- Liu, K.; Li, Q.Z.; Delgado-Vega, A.M.; Abelson, A.K.; Sanchez, E.; Kelly, J.A.; Li, L.; Liu, Y.; Zhou, J.; Yan, M.; et al. Kallikrein genes are associated with lupus and glomerular basement membrane-specific antibody-induced nephritis in mice and humans. J. Clin. Investig. 2009, 119, 911–923. [Google Scholar] [CrossRef]
- Li, Q.Z.; Zhou, J.; Yang, R.; Yan, M.; Ye, Q.; Liu, K.; Liu, S.; Shao, X.; Li, L.; Zhou, X.J.; et al. The lupus-susceptibility gene kallikrein downmodulates antibody-mediated glomerulonephritis. Genes. Immun. 2009, 10, 503–508. [Google Scholar] [CrossRef]
- Banchereau, J.; Pascual, V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity 2006, 25, 383–392. [Google Scholar] [CrossRef]
- Niewold, T.B.; Clark, D.N.; Salloum, R.; Poole, B.D. Interferon alpha in systemic lupus erythematosus. J. Biomed. Biotechnol. 2010, 2010, 948364. [Google Scholar] [CrossRef]
- Theofilopoulos, A.N.; Baccala, R.; Beutler, B.; Kono, D.H. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu. Rev. Immunol. 2005, 23, 307–336. [Google Scholar] [CrossRef]
- Figueiredo-Braga, M.; Cornaby, C.; Cortez, A.; Bernardes, M.; Terroso, G.; Figueiredo, M.; Mesquita, C.D.S.; Costa, L.; Poole, B.D. Depression and anxiety in systemic lupus erythematosus: The crosstalk between immunological, clinical, and psychosocial factors. Medicine 2018, 97, e11376. [Google Scholar] [CrossRef]
- Gao, H.X.; Campbell, S.R.; Cui, M.H.; Zong, P.; Hee-Hwang, J.; Gulinello, M.; Putterman, C. Depression is an early disease manifestation in lupus-prone MRL/lpr mice. J. Neuroimmunol. 2009, 207, 45–56. [Google Scholar] [CrossRef]
- Can, A.; Dao, D.T.; Terrillion, C.E.; Piantadosi, S.C.; Bhat, S.; Gould, T.D. The tail suspension test. J. Vis. Exp. 2012, 59, e3769. [Google Scholar] [CrossRef]
- Curzon, P.; Zhang, M.; Radek, R.J.; Fox, G.B. The Behavioral Assessment of Sensorimotor Processes in the Mouse: Acoustic Startle, Sensory Gating, Locomotor Activity, Rotarod, and Beam Walking. In Methods of Behavior Analysis in Neuroscience, 2nd ed.; Buccafusco, J.J., Ed.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2009. [Google Scholar]
- Catena-Dell’Osso, M.; Rotella, F.; Dell’Osso, A.; Fagiolini, A.; Marazziti, D. Inflammation, serotonin and major depression. Curr. Drug Targets 2013, 14, 571–577. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia states and nomenclature: A field at its crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef]
- Weissmann, G. Rheumatoid arthritis and systemic lupus erythematosus as immune complex diseases. Bull. NYU Hosp. Jt. Dis. 2009, 67, 251–253. [Google Scholar]
- Qin, L.; Du, Y.; Ding, H.; Haque, A.; Hicks, J.; Pedroza, C.; Mohan, C. Bradykinin 1 receptor blockade subdues systemic autoimmunity, renal inflammation, and blood pressure in murine lupus nephritis. Arthritis Res. Ther. 2019, 21, 12. [Google Scholar] [CrossRef]
- Shao, X.; Yang, R.; Yan, M.; Li, Y.; Du, Y.; Raman, I.; Zhang, B.; Wakeland, E.K.; Igarashi, P.; Mohan, C.; et al. Inducible expression of kallikrein in renal tubular cells protects mice against spontaneous lupus nephritis. Arthritis Rheum. 2013, 65, 780–791. [Google Scholar] [CrossRef]
- Loncharich, M.F.; Anderson, C.W. Interferon Inhibition for Lupus with Anifrolumab: Critical Appraisal of the Evidence Leading to FDA Approval. ACR Open Rheumatol. 2022, 4, 486–491. [Google Scholar] [CrossRef]
- Crow, M.K.; Olferiev, M.; Kirou, K.A. Targeting of type I interferon in systemic autoimmune diseases. Transl. Res. 2015, 165, 296–305. [Google Scholar] [CrossRef]
- Sforzini, L.; Cattaneo, A.; Ferrari, C.; Turner, L.; Mariani, N.; Enache, D.; Hastings, C.; Lombardo, G.; Nettis, M.A.; Nikkheslat, N.; et al. Higher immune-related gene expression in major depression is independent of CRP levels: Results from the BIODEP study. Transl. Psychiatry 2023, 13, 185. [Google Scholar] [CrossRef]
- Liu, T.; Son, M.; Diamond, B. HMGB1 in Systemic Lupus Erythematosus. Front. Immunol. 2020, 11, 1057. [Google Scholar] [CrossRef]
- Xu, S.Y.; Jia, J.Q.; Sun, M.; Bao, X.Y.; Xia, S.N.; Shu, S.; Liu, P.Y.; Ji, S.L.; Ye, L.; Cao, X.; et al. QHRD106 ameliorates ischemic stroke injury as a long-acting tissue kallikrein preparation. iScience 2023, 26, 107268. [Google Scholar] [CrossRef]
- Vital, E.M.; Merrill, J.T.; Morand, E.F.; Furie, R.A.; Bruce, I.N.; Tanaka, Y.; Manzi, S.; Kalunian, K.C.; Kalyani, R.N.; Streicher, K.; et al. Anifrolumab efficacy and safety by type I interferon gene signature and clinical subgroups in patients with SLE: Post hoc analysis of pooled data from two phase III trials. Ann. Rheum. Dis. 2022, 81, 951–961. [Google Scholar] [CrossRef]
- Bruce, I.N.; Furie, R.A.; Morand, E.F.; Manzi, S.; Tanaka, Y.; Kalunian, K.C.; Merrill, J.T.; Puzio, P.; Maho, E.; Kleoudis, C.; et al. Concordance and discordance in SLE clinical trial outcome measures: Analysis of three anifrolumab phase 2/3 trials. Ann. Rheum. Dis. 2022, 81, 962–969. [Google Scholar] [CrossRef]
- Fushida, N.; Horii, M.; Oishi, K.; Matsushita, T. Anifrolumab for systemic lupus erythematosus: A clinical study of Japanese patients in Kanazawa University Hospital. J. Dermatol. 2024, 51, 607–611. [Google Scholar] [CrossRef]
- Tanaka, Y.; Luo, Y.; O’Shea, J.J.; Nakayamada, S. Janus kinase-targeting therapies in rheumatology: A mechanisms-based approach. Nat. Rev. Rheumatol. 2022, 18, 133–145. [Google Scholar] [CrossRef]
- Gonzales, E.L.; Jeon, S.J.; Han, K.M.; Yang, S.J.; Kim, Y.; Remonde, C.G.; Ahn, T.J.; Ham, B.J.; Shin, C.Y. Correlation between immune-related genes and depression-like features in an animal model and in humans. Brain Behav. Immun. 2023, 113, 29–43. [Google Scholar] [CrossRef]
- Fischer, C.W.; Eskelund, A.; Budac, D.P.; Tillmann, S.; Liebenberg, N.; Elfving, B.; Wegener, G. Interferon-alpha treatment induces depression-like behaviour accompanied by elevated hippocampal quinolinic acid levels in rats. Behav. Brain Res. 2015, 293, 166–172. [Google Scholar] [CrossRef]
- Zeng, J.; Meng, X.; Zhou, P.; Yin, Z.; Xie, Q.; Zou, H.; Shen, N.; Ye, Z.; Tang, Y. Interferon-alpha exacerbates neuropsychiatric phenotypes in lupus-prone mice. Arthritis Res. Ther. 2019, 21, 205. [Google Scholar] [CrossRef]
- Lood, C.; Tyden, H.; Gullstrand, B.; Klint, C.; Wenglen, C.; Nielsen, C.T.; Heegaard, N.H.; Jonsen, A.; Kahn, R.; Bengtsson, A.A. Type I interferon-mediated skewing of the serotonin synthesis is associated with severe disease in systemic lupus erythematosus. PLoS ONE 2015, 10, e0125109. [Google Scholar] [CrossRef]
- Saldanha, D.; Kumar, N.; Ryali, V.; Srivastava, K.; Pawar, A.A. Serum Serotonin Abnormality in Depression. Med. J. Armed Forces India 2009, 65, 108–112. [Google Scholar] [CrossRef]
- Wang, N.; Tian, B. Brain-derived neurotrophic factor in autoimmune inflammatory diseases (Review). Exp. Ther. Med. 2021, 22, 1292. [Google Scholar] [CrossRef]
- Zheng, Q.; Xu, M.J.; Cheng, J.; Chen, J.M.; Zheng, L.; Li, Z.G. Serum levels of brain-derived neurotrophic factor are associated with depressive symptoms in patients with systemic lupus erythematosus. Psychoneuroendocrinology 2017, 78, 246–252. [Google Scholar] [CrossRef]
- Quilter, M.C.; Hiraki, L.T.; Korczak, D.J. Depressive and anxiety symptom prevalence in childhood-onset systemic lupus erythematosus: A systematic review. Lupus 2019, 28, 878–887. [Google Scholar] [CrossRef]
- Kollias, G.; Kontoyiannis, D.; Douni, E.; Kassiotis, G. The role of TNF/TNFR in organ-specific and systemic autoimmunity: Implications for the design of optimized ‘anti-TNF’ therapies. Curr. Dir. Autoimmun. 2002, 5, 30–50. [Google Scholar] [PubMed]
- Godsell, J.; Rudloff, I.; Kandane-Rathnayake, R.; Hoi, A.; Nold, M.F.; Morand, E.F.; Harris, J. Clinical associations of IL-10 and IL-37 in systemic lupus erythematosus. Sci. Rep. 2016, 6, 34604. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Gallo, P.M.; Hooper, K.M.; Corradetti, C.; Ganea, D.; Caricchio, R.; Gallucci, S. The cytokine network type I IFN-IL-27-IL-10 is augmented in murine and human lupus. J. Leukoc. Biol. 2019, 106, 967–975. [Google Scholar] [CrossRef]
- Iikuni, N.; Okamoto, H.; Yoshio, T.; Sato, E.; Kamitsuji, S.; Iwamoto, T.; Momohara, S.; Taniguchi, A.; Yamanaka, H.; Minota, S.; et al. Raised monocyte chemotactic protein-1 (MCP-1)/CCL2 in cerebrospinal fluid of patients with neuropsychiatric lupus. Ann. Rheum. Dis. 2006, 65, 253–256. [Google Scholar] [CrossRef]
- Lee, Y.H.; Song, G.G. Urinary MCP-1 as a biomarker for lupus nephritis: A meta-analysis. Z. Rheumatol. 2017, 76, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H.; Iikuni, N.; Kamitsuji, S.; Yoshio, T.; Minota, S.; Kamatani, N. IP-10/MCP-1 ratio in CSF is an useful diagnostic marker of neuropsychiatric lupus patients. Rheumatology 2006, 45, 232–234. [Google Scholar] [CrossRef] [PubMed]
- Sarrand, J.; Soyfoo, M. Involvement of IL-33 in the Pathophysiology of Systemic Lupus Erythematosus: Review. Int. J. Mol. Sci. 2022, 23, 3138. [Google Scholar] [CrossRef]
- Sun, Y.; Wen, Y.; Wang, L.; Wen, L.; You, W.; Wei, S.; Mao, L.; Wang, H.; Chen, Z.; Yang, X. Therapeutic Opportunities of Interleukin-33 in the Central Nervous System. Front. Immunol. 2021, 12, 654626. [Google Scholar] [CrossRef]
- Georgakis, S.; Gkirtzimanaki, K.; Papadaki, G.; Gakiopoulou, H.; Drakos, E.; Eloranta, M.L.; Makridakis, M.; Kontostathi, G.; Zoidakis, J.; Baira, E.; et al. NETs decorated with bioactive IL-33 infiltrate inflamed tissues and induce IFN-alpha production in patients with SLE. JCI Insight 2021, 6, e147671. [Google Scholar] [CrossRef]
- Hrycek, E.; Franek, A.; Blaszczak, E.; Dworak, J.; Hrycek, A. Serum levels of selected chemokines in systemic lupus erythematosus patients. Rheumatol. Int. 2013, 33, 2423–2427. [Google Scholar] [CrossRef]
- Carneiro, J.R.; Fuzii, H.T.; Kayser, C.; Alberto, F.L.; Soares, F.A.; Sato, E.I.; Andrade, L.E. IL-2, IL-5, TNF-alpha and IFN-gamma mRNA expression in epidermal keratinocytes of systemic lupus erythematosus skin lesions. Clinics 2011, 66, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Boulougoura, A.; Endo, Y.; Tsokos, G.C. Abnormalities of T cells in systemic lupus erythematosus: New insights in pathogenesis and therapeutic strategies. J. Autoimmun. 2022, 132, 102870. [Google Scholar] [CrossRef] [PubMed]
- Nestor, J.; Arinuma, Y.; Huerta, T.S.; Kowal, C.; Nasiri, E.; Kello, N.; Fujieda, Y.; Bialas, A.; Hammond, T.; Sriram, U.; et al. Lupus antibodies induce behavioral changes mediated by microglia and blocked by ACE inhibitors. J. Exp. Med. 2018, 215, 2554–2566. [Google Scholar] [CrossRef] [PubMed]
- Makinde, H.M.; Winter, D.R.; Procissi, D.; Mike, E.V.; Stock, A.D.; Kando, M.J.; Gadhvi, G.T.; Droho, S.; Bloomfield, C.L.; Dominguez, S.T.; et al. A Novel Microglia-Specific Transcriptional Signature Correlates with Behavioral Deficits in Neuropsychiatric Lupus. Front. Immunol. 2020, 11, 230. [Google Scholar] [CrossRef] [PubMed]
- Vanarsa, K.; Henderson, J.; Soomro, S.; Qin, L.; Zhang, T.; Jordan, N.; Putterman, C.; Blanco, I.; Saxena, R.; Mohan, C. Upregulation of Proinflammatory Bradykinin Peptides in Systemic Lupus Erythematosus and Rheumatoid Arthritis. J. Immunol. 2020, 205, 369–376. [Google Scholar] [CrossRef]
- Gobel, K.; Pankratz, S.; Schneider-Hohendorf, T.; Bittner, S.; Schuhmann, M.K.; Langer, H.F.; Stoll, G.; Wiendl, H.; Kleinschnitz, C.; Meuth, S.G. Blockade of the kinin receptor B1 protects from autoimmune CNS disease by reducing leukocyte trafficking. J. Autoimmun. 2011, 36, 106–114. [Google Scholar] [CrossRef]
- Manolis, A.J.; Marketou, M.E.; Gavras, I.; Gavras, H. Cardioprotective properties of bradykinin: Role of the B(2) receptor. Hypertens. Res. 2010, 33, 772–777. [Google Scholar] [CrossRef]
- Itto, R.; Oe, Y.; Imaruoka, K.; Sato, E.; Sekimoto, A.; Yamakage, S.; Kumakura, S.; Sato, H.; Ito, S.; Takahashi, N. Glomerular Injury Is Exacerbated in Lupus-Prone MRL/lpr Mice Treated with a Protease-Activated Receptor 2 Antagonist. Tohoku J. Exp. Med. 2019, 249, 127–133. [Google Scholar] [CrossRef]
- Seo, Y.; Mun, C.H.; Park, S.H.; Jeon, D.; Kim, S.J.; Yoon, T.; Ko, E.; Jo, S.; Park, Y.B.; Namkung, W.; et al. Punicalagin Ameliorates Lupus Nephritis via Inhibition of PAR2. Int. J. Mol. Sci. 2020, 21, 4975. [Google Scholar] [CrossRef]
- Iablokov, V.; Hirota, C.L.; Peplowski, M.A.; Ramachandran, R.; Mihara, K.; Hollenberg, M.D.; MacNaughton, W.K. Proteinase-activated receptor 2 (PAR2) decreases apoptosis in colonic epithelial cells. J. Biol. Chem. 2014, 289, 34366–34377. [Google Scholar] [CrossRef]
- Sriram, U.; Varghese, L.; Bennett, H.L.; Jog, N.R.; Shivers, D.K.; Ning, Y.; Behrens, E.M.; Caricchio, R.; Gallucci, S. Myeloid dendritic cells from B6.NZM Sle1/Sle2/Sle3 lupus-prone mice express an IFN signature that precedes disease onset. J. Immunol. 2012, 189, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Rom, S.; Heldt, N.A.; Gajghate, S.; Seliga, A.; Reichenbach, N.L.; Persidsky, Y. Hyperglycemia and advanced glycation end products disrupt BBB and promote occludin and claudin-5 protein secretion on extracellular microvesicles. Sci. Rep. 2020, 10, 7274. [Google Scholar] [CrossRef]
- Marcinko, K.; Parsons, T.; Lerch, J.P.; Sled, J.G.; Sakic, B. Effects of prolonged treatment with memantine in the MRL model of CNS lupus. Clin. Exp. Neuroimmunol. 2012, 3, 116–128. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhoj, P.S.; Nocito, C.; Togre, N.S.; Winfield, M.; Lubinsky, C.; Khan, S.; Mogadala, N.; Seliga, A.; Unterwald, E.M.; Persidsky, Y.; et al. Tissue Kallikrein-1 Suppresses Type I Interferon Responses and Reduces Depressive-Like Behavior in the MRL/lpr Lupus-Prone Mouse Model. Int. J. Mol. Sci. 2024, 25, 10080. https://doi.org/10.3390/ijms251810080
Bhoj PS, Nocito C, Togre NS, Winfield M, Lubinsky C, Khan S, Mogadala N, Seliga A, Unterwald EM, Persidsky Y, et al. Tissue Kallikrein-1 Suppresses Type I Interferon Responses and Reduces Depressive-Like Behavior in the MRL/lpr Lupus-Prone Mouse Model. International Journal of Molecular Sciences. 2024; 25(18):10080. https://doi.org/10.3390/ijms251810080
Chicago/Turabian StyleBhoj, Priyanka S., Cassandra Nocito, Namdev S. Togre, Malika Winfield, Cody Lubinsky, Sabeeya Khan, Nikhita Mogadala, Alecia Seliga, Ellen M. Unterwald, Yuri Persidsky, and et al. 2024. "Tissue Kallikrein-1 Suppresses Type I Interferon Responses and Reduces Depressive-Like Behavior in the MRL/lpr Lupus-Prone Mouse Model" International Journal of Molecular Sciences 25, no. 18: 10080. https://doi.org/10.3390/ijms251810080
APA StyleBhoj, P. S., Nocito, C., Togre, N. S., Winfield, M., Lubinsky, C., Khan, S., Mogadala, N., Seliga, A., Unterwald, E. M., Persidsky, Y., & Sriram, U. (2024). Tissue Kallikrein-1 Suppresses Type I Interferon Responses and Reduces Depressive-Like Behavior in the MRL/lpr Lupus-Prone Mouse Model. International Journal of Molecular Sciences, 25(18), 10080. https://doi.org/10.3390/ijms251810080