
Detection and Characterisation of SARS-CoV-2 in Eastern Province of Zambia: A Retrospective Genomic Surveillance Study
 ,
,  , , , , , ,
, , , , , ,
Abstract
1. Introduction
2. Results
2.1. Demographic Summary
2.2. SARS-CoV-2 Genome Detection and Assembly
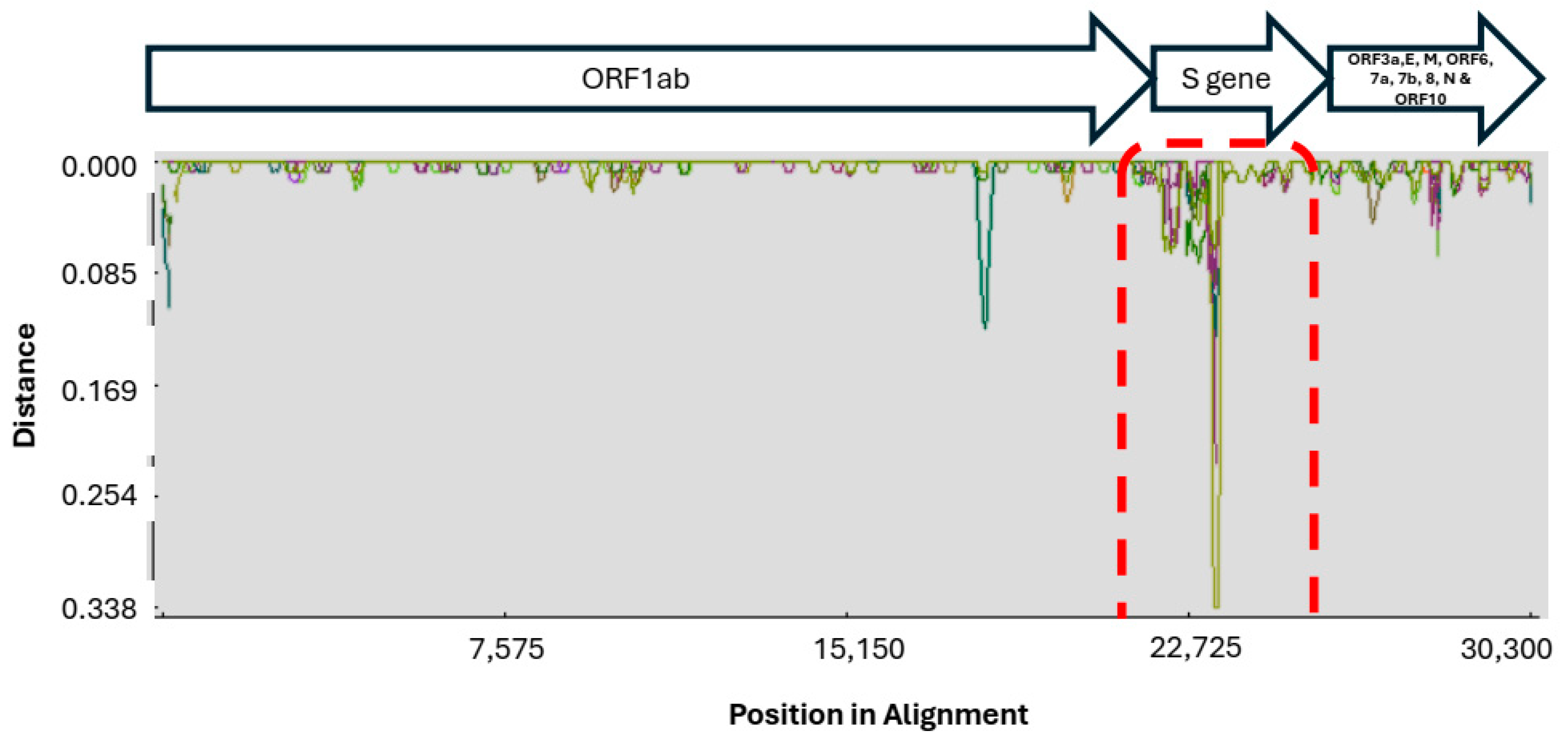
2.3. Genetic Diversity in SARS-CoV-2 Sequences
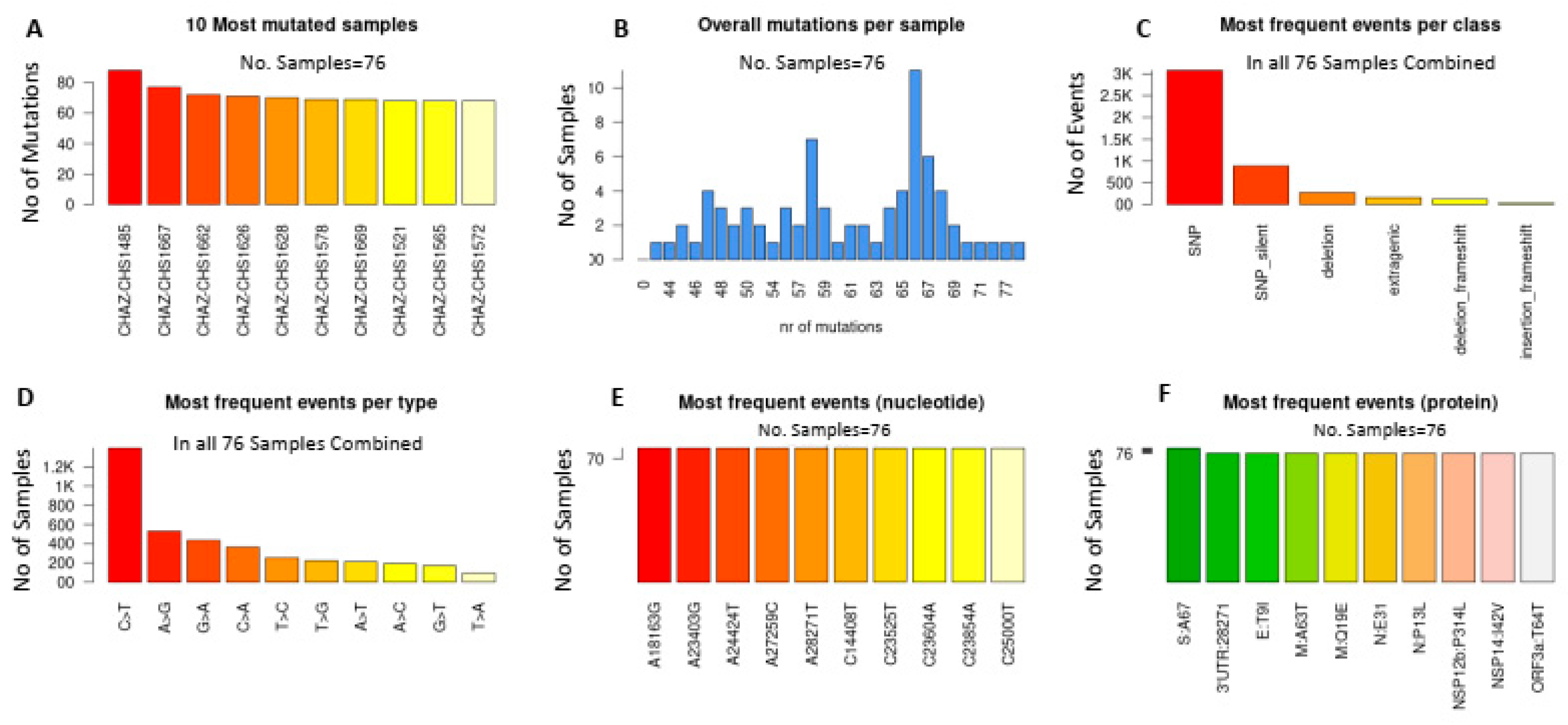
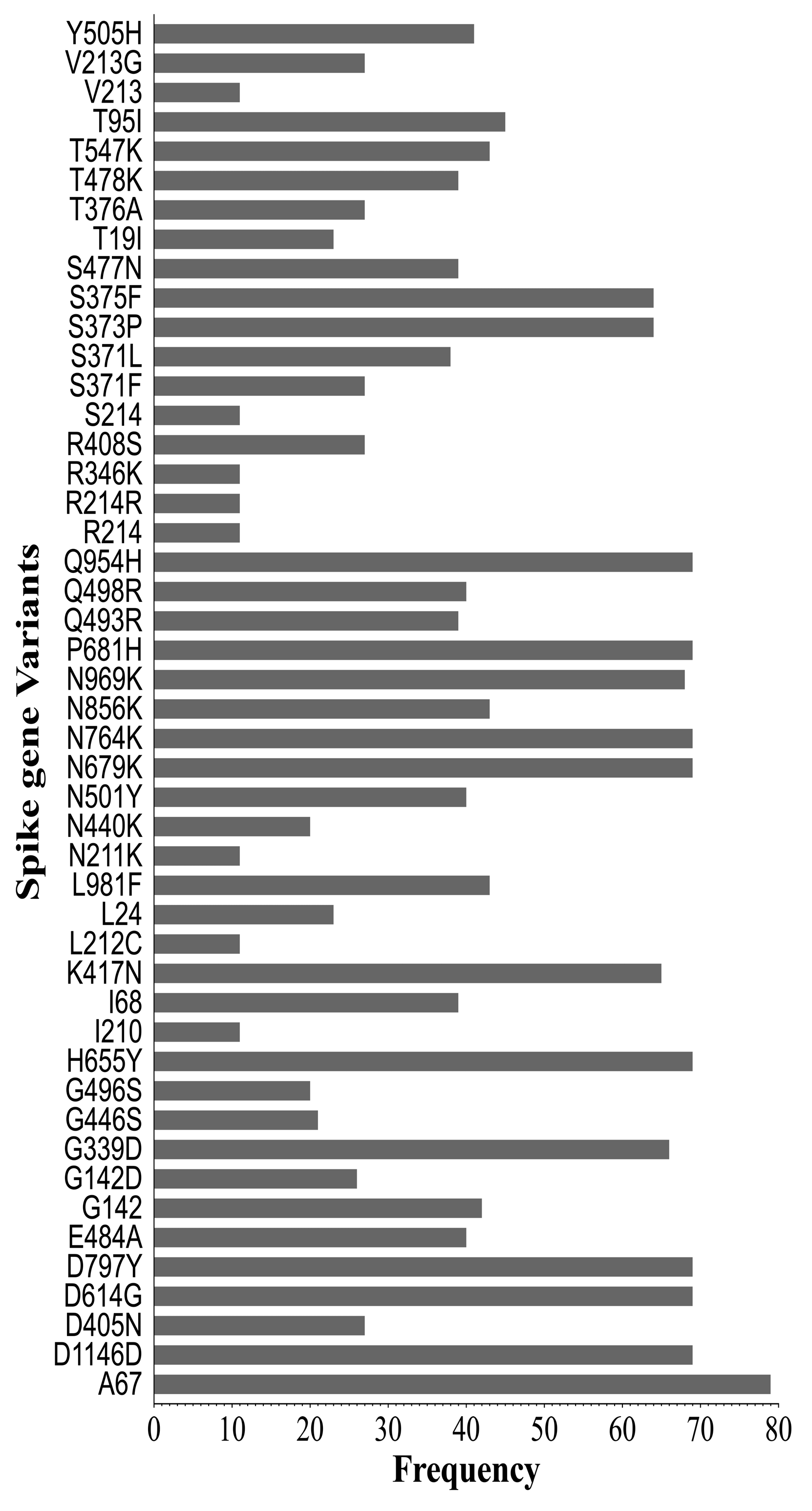
2.4. Synonymous and Non-Synonymous Mutations in SARS-CoV-2 Genomes
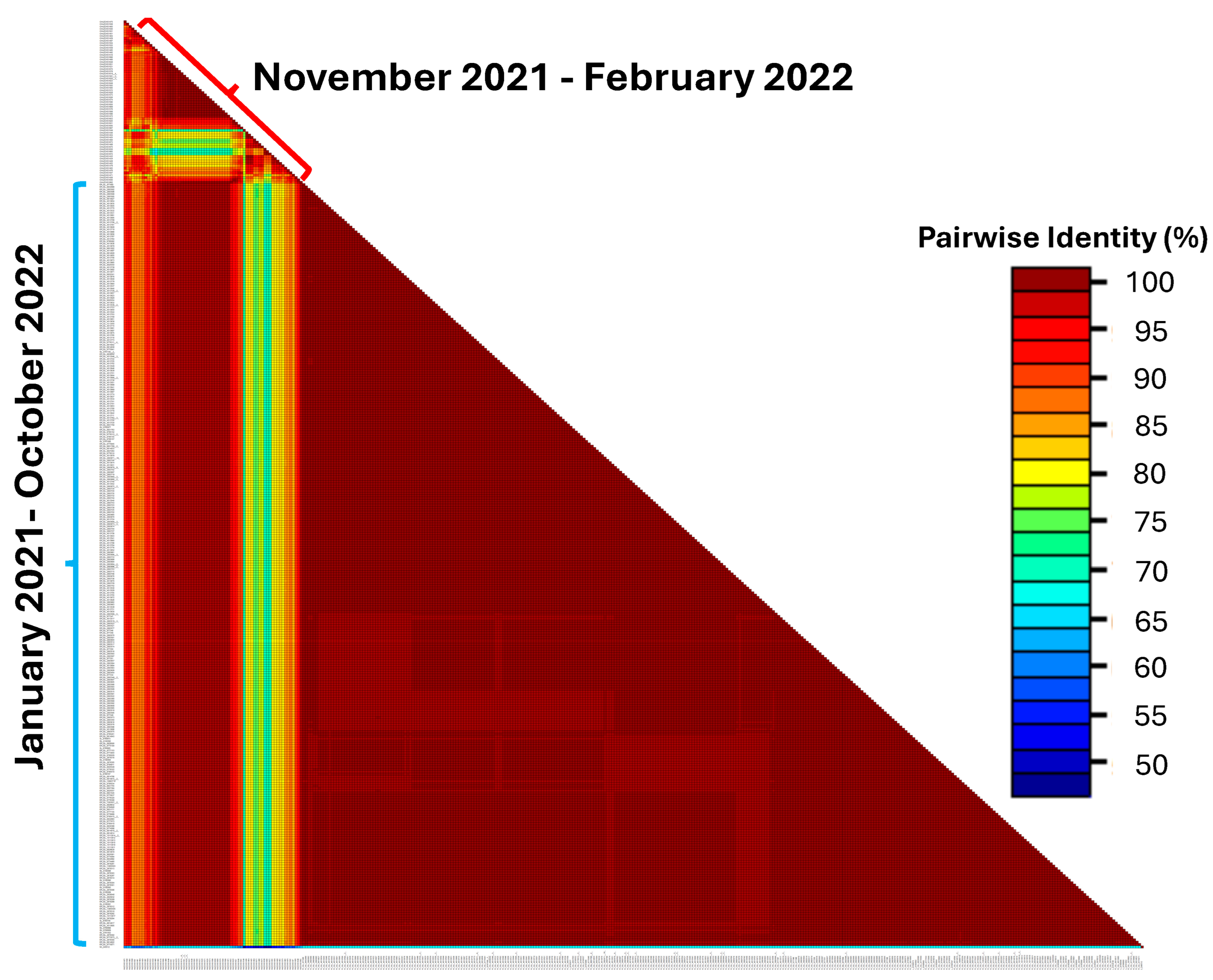
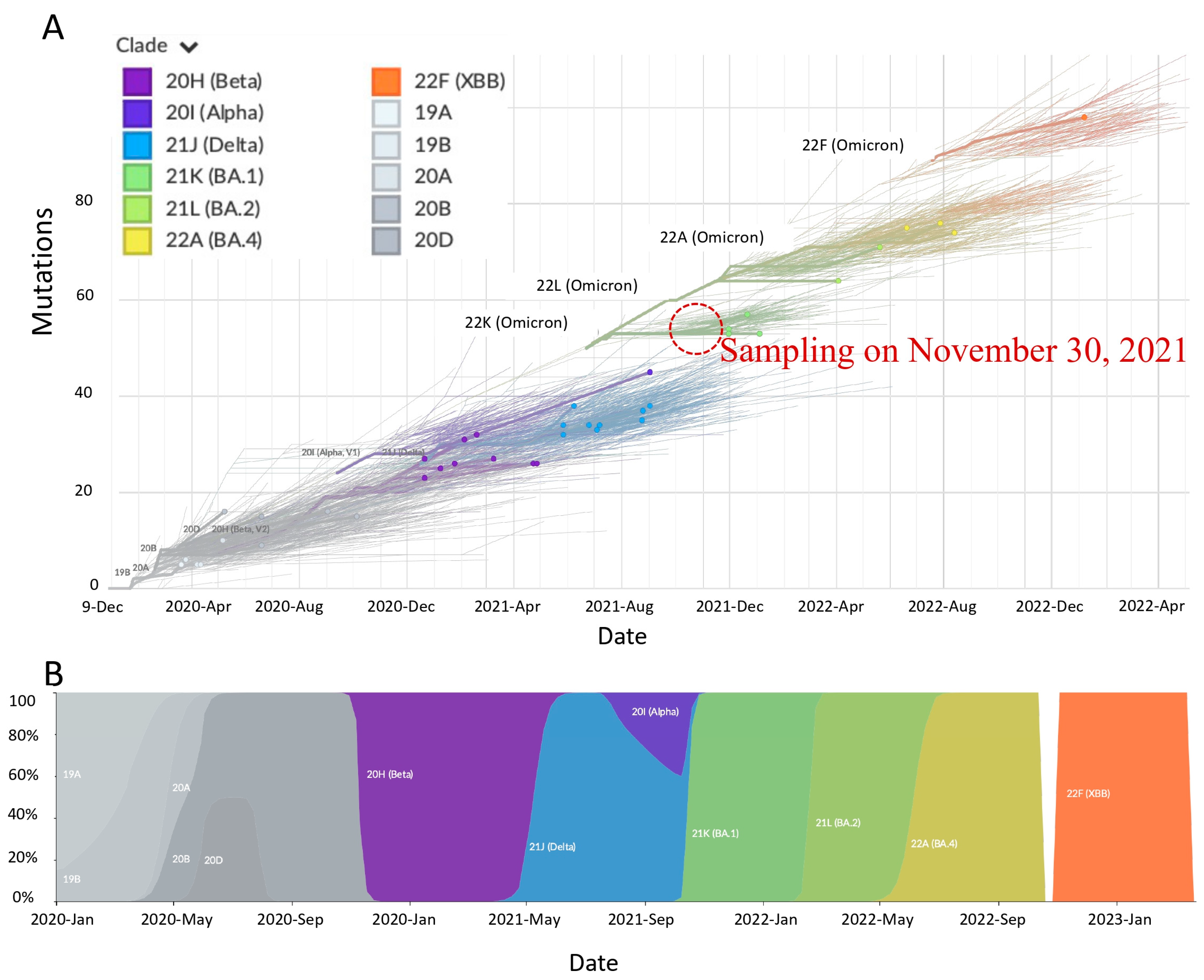
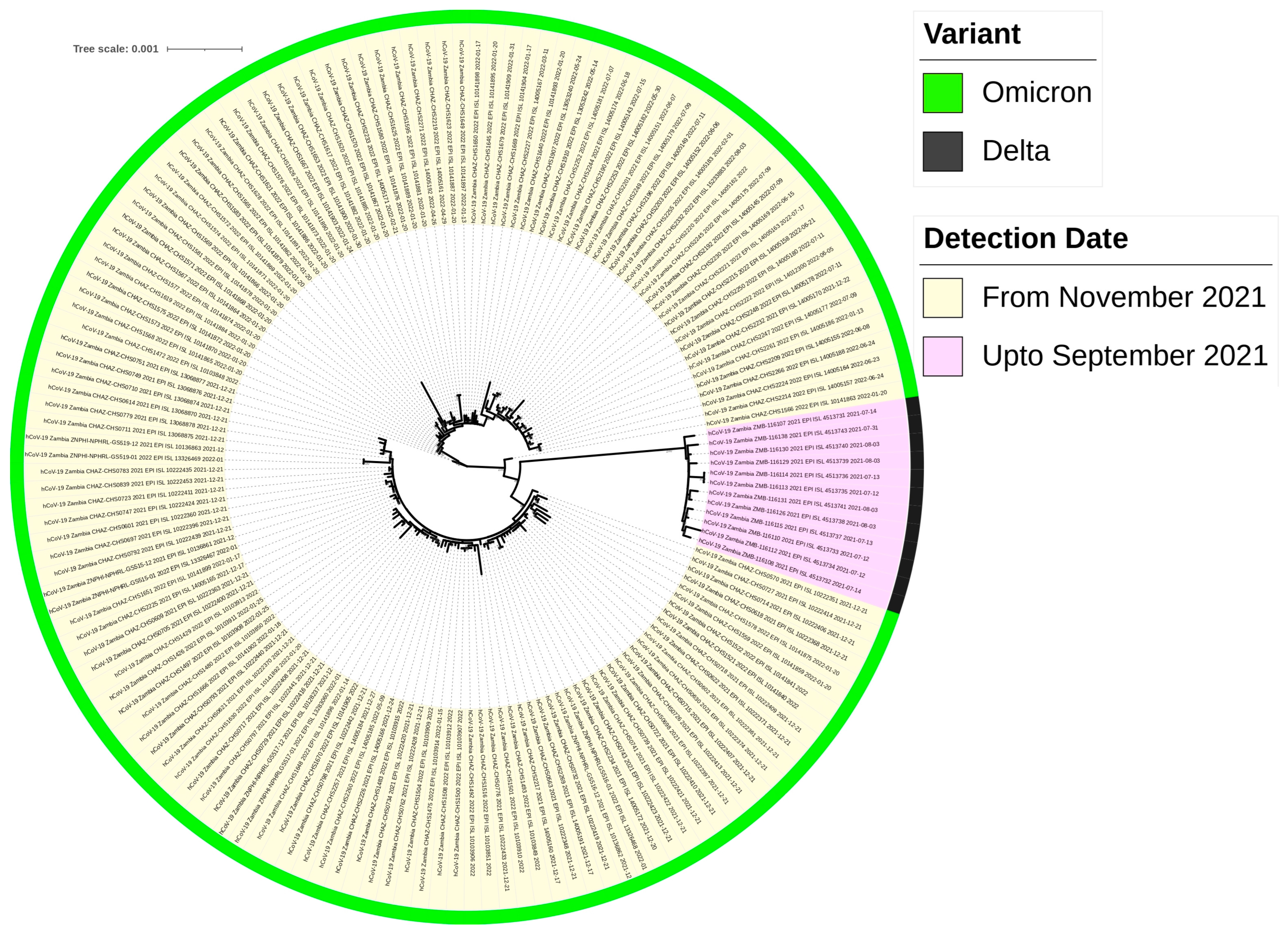
2.5. Highly Divergent Strain Rapidly Replaced Other Variants after 1 November 2021
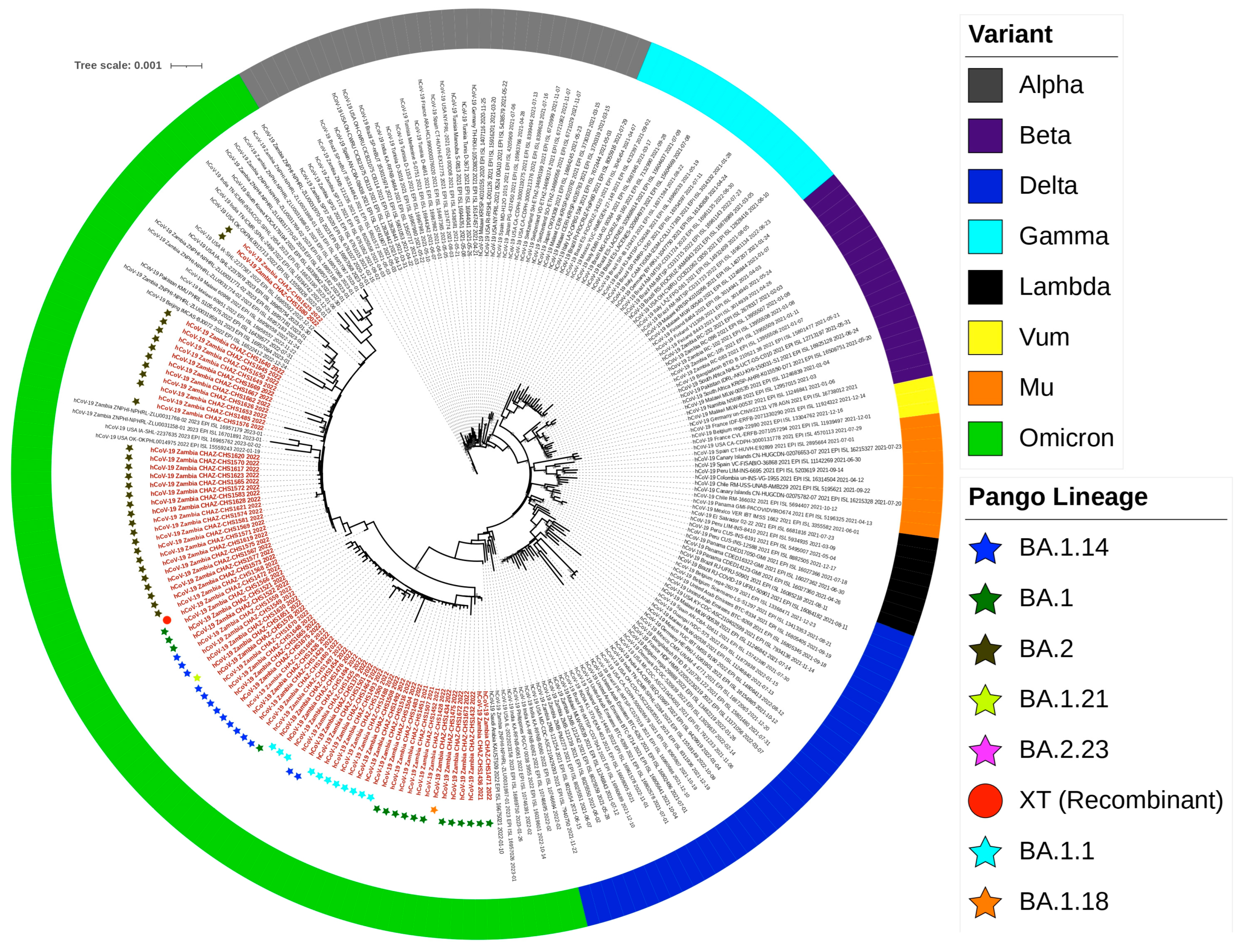
2.6. Lineage, Clade Assignment and Recombination Analysis
2.7. Phylogenetic Analysis
3. Discussion
4. Materials and Methods
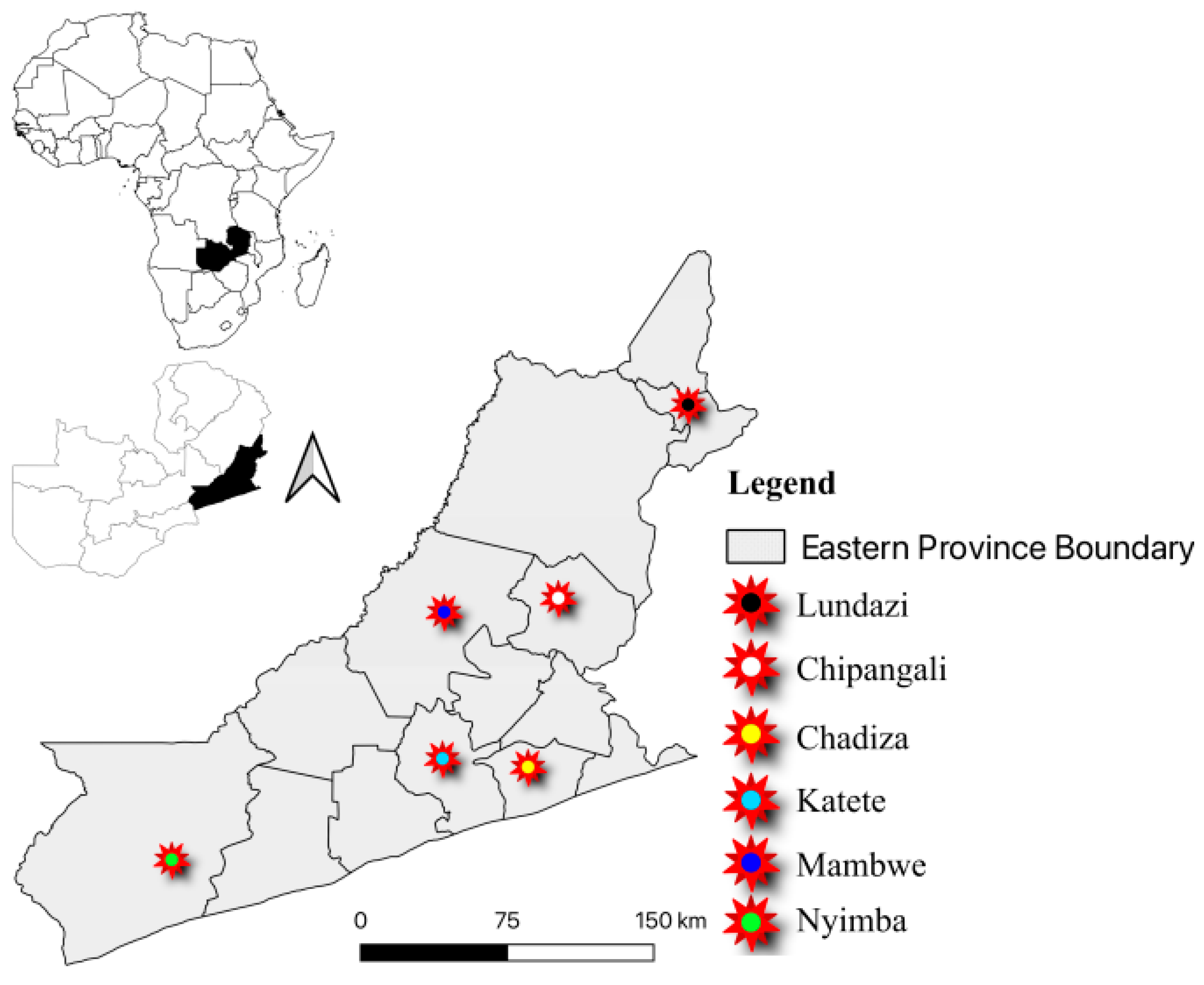
4.1. Study Area and Design
4.2. Sample Collection and RNA Extraction
4.3. SARS-CoV-2 Genome Detection by RT-qPCR
4.4. cDNA Synthesis and Amplification of SARS-CoV-2
4.5. Library Preparation, Illumina Sequencing and Genome Assembly
4.6. Genetic Diversity and Mutations of SARS-CoV-2 Genomes
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Swelum, A.A.; Shafi, M.E.; Albaqami, N.M.; El-Saadony, M.T.; Elsify, A.; Abdo, M.; Taha, A.E.; Abdel-Moneim, A.M.; Al-Gabri, N.A.; Almaiman, A.A.; et al. COVID-19 in Human, Animal, and Environment: A Review. Front. Vet. Sci. 2020, 7, 564334. [Google Scholar] [CrossRef]
- Santaniello, A.; Perruolo, G.; Cristiano, S.; Agognon, A.L.; Cabaro, S.; Amato, A.; Dipineto, L.; Borrelli, L.; Formisano, P.; Fioretti, A.; et al. SARS-CoV-2 Affects Both Humans and Animals: What Is the Potential Transmission Risk? A Literature Review. Microorganisms 2023, 11, 514. [Google Scholar] [CrossRef]
- Spernovasilis, N.; Tsiodras, S.; Poulakou, G. Emerging and Re-Emerging Infectious Diseases: Humankind’s Companions and Competitors. Microorganisms 2022, 10, 98. [Google Scholar] [CrossRef]
- Akhter, S.; Akhtar, S. Emerging coronavirus diseases and future perspectives. VirusDisease 2020, 31, 113. [Google Scholar] [CrossRef]
- da Costa, V.G.; Moreli, M.L.; Saivish, M.V. The emergence of SARS, MERS and novel SARS-2 coronaviruses in the 21st century. Arch. Virol. 2020, 165, 1517. [Google Scholar] [CrossRef]
- Pollard, C.A.; Morran, M.P.; Nestor-Kalinoski, A.L. The COVID-19 pandemic: A global health crisis. Physiol. Genom. 2020, 52, 549–557. [Google Scholar] [CrossRef]
- Jeanne, L.; Bourdin, S.; Nadou, F.; Noiret, G. Economic globalization and the COVID-19 pandemic: Global spread and inequalities. GeoJournal 2023, 88, 1181. [Google Scholar] [CrossRef]
- Telenti, A.; Arvin, A.; Corey, L.; Corti, D.; Diamond, M.S.; García-Sastre, A.; Garry, R.F.; Holmes, E.C.; Pang, P.S.; Virgin, H.W. After the pandemic: Perspectives on the future trajectory of COVID-19. Nature 2021, 596, 495–504. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Q.; Zhou, L.; Zhou, Y.; Yan, H.; Lan, K. Emerging SARS-CoV-2 variants: Why, how, and what’s next? Cell Insight 2022, 1, 100029. [Google Scholar] [CrossRef]
- Mahilkar, S.; Agrawal, S.; Chaudhary, S.; Parikh, S.; Sonkar, S.C.; Verma, D.K.; Chitalia, V.; Mehta, D.; Koner, B.C.; Vijay, N.; et al. SARS-CoV-2 variants: Impact on biological and clinical outcome. Front. Med. 2022, 9, 995960. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef]
- Callaway, E. Delta coronavirus variant: Scientists brace for impact. Nature 2021, 595, 17–18. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182, 1295–1310.e20. [Google Scholar] [CrossRef]
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, Á.; et al. Assessing transmissibility of SARS-CoV-2 lineage B.1.1.7 in England. Nature 2021, 593, 266–269. [Google Scholar] [CrossRef]
- Brown, K.A.; Gubbay, J.; Hopkins, J.; Patel, S.; Buchan, S.A.; Daneman, N.; Goneau, L.W. S-Gene Target Failure as a Marker of Variant B.1.1.7 among SARS-CoV-2 Isolates in the Greater Toronto Area, December 2020 to March 2021. JAMA 2021, 325, 2115–2116. [Google Scholar] [CrossRef]
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; Candido, D.D.; Mishra, S.; Crispim, M.A.; Sales, F.C.; Hawryluk, I.; McCrone, J.T.; et al. Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in Manaus, Brazil. Science 2021, 372, 815–821. [Google Scholar] [CrossRef]
- Wise, J. COVID-19: The E484K mutation and the risks it poses. BMJ 2021, 372, n359. [Google Scholar] [CrossRef]
- Ramanathan, M.; Ferguson, I.D.; Miao, W.; Khavari, P.A. SARS-CoV-2 B.1.1.7 and B.1.351 spike variants bind human ACE2 with increased affinity. Lancet Infect. Dis. 2021, 21, 1070. [Google Scholar] [CrossRef]
- Di Giacomo, S.; Mercatelli, D.; Rakhimov, A.; Giorgi, F.M. Preliminary report on severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) Spike mutation T478K. J. Med. Virol. 2021, 93, 5638–5643. [Google Scholar] [CrossRef]
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Shrestha, L.B.; Foster, C.; Rawlinson, W.; Tedla, N.; Bull, R.A. Evolution of the SARS-CoV-2 omicron variants BA.1 to BA.5: Implications for immune escape and transmission. Rev. Med. Virol. 2022, 32, e2381. [Google Scholar] [CrossRef]
- Maqsood, T.; Shahzad, K.; Naz, S.; Simsek, S.; Afzal, M.S.; Ali, S.; Ahmed, H.; Cao, J. A Cross-Sectional Study on the Association between Risk Factors of Toxoplasmosis and One Health Knowledge in Pakistan. Front. Vet. Sci. 2021, 8, 1390. [Google Scholar] [CrossRef]
- Ren, W.; Zhang, Y.; Rao, J.; Wang, Z.; Lan, J.; Liu, K.; Zhang, X.; Hu, X.; Yang, C.; Zhong, G.; et al. Evolution of Immune Evasion and Host Range Expansion by the SARS-CoV-2 B.1.1.529 (Omicron) Variant. MBio 2023, 14, e00416-23. [Google Scholar] [CrossRef]
- Karim, S.S.A.; Karim, Q.A. Omicron SARS-CoV-2 variant: A new chapter in the COVID-19 pandemic. Lancet 2021, 398, 2126–2128. [Google Scholar] [CrossRef]
- Park, A.K.; Kim, I.H.; Lee, C.Y.; Kim, J.A.; Lee, H.; Kim, H.M.; Lee, N.J.; Woo, S.; Lee, J.; Rhee, J.; et al. Rapid Emergence of the Omicron Variant of Severe Acute Respiratory Syndrome Coronavirus 2 in Korea. Ann. Lab. Med. 2023, 43, 211. [Google Scholar] [CrossRef]
- Dhama, K.; Nainu, F.; Frediansyah, A.; Yatoo, M.I.; Mohapatra, R.K.; Chakraborty, S.; Zhou, H.; Islam, M.R.; Mamada, S.S.; Kusuma, H.I.; et al. Global emerging Omicron variant of SARS-CoV-2: Impacts, challenges and strategies. J. Infect. Public Health 2023, 16, 4–14. [Google Scholar] [CrossRef]
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J. SARS-CoV-2 variant biology: Immune escape, transmission and fitness. Nat. Rev. Microbiol. 2023, 21, 162–177. [Google Scholar]
- Cameroni, E.; Bowen, J.E.; Rosen, L.E.; Saliba, C.; Zepeda, S.K.; Culap, K.; Pinto, D.; VanBlargan, L.A.; De Marco, A.; di Iulio, J.; et al. Broadly neutralizing antibodies overcome SARS-CoV-2 Omicron antigenic shift. Nature 2021, 602, 664–670. [Google Scholar] [CrossRef]
- Mwenda, M. Detection of B.1.351 SARS-CoV-2 Variant Strain—Zambia, December 2020. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 280–282. [Google Scholar] [CrossRef]
- Tosta, S.; Moreno, K.; Schuab, G.; Fonseca, V.; Segovia, F.M.; Kashima, S.; Elias, M.C.; Sampaio, S.C.; Ciccozzi, M.; Alcantara, L.C.; et al. Global SARS-CoV-2 genomic surveillance: What we have learned (so far). Infect. Genet. Evol. 2023, 108, 105405. [Google Scholar] [CrossRef]
- AlKalamouni, H.; Abou Hassan, F.F.; Bou Hamdan, M.; Page, A.J.; Lott, M.; Matthews, M.; Ghosn, N.; Rady, A.; Mahfouz, R.; Araj, G.F.; et al. Genomic surveillance of SARS-CoV-2 in COVID-19 vaccinated healthcare workers in Lebanon. BMC Med. Genom. 2023, 16, 14. [Google Scholar] [CrossRef]
- Simulundu, E.; Mupeta, F.; Chanda-Kapata, P.; Saasa, N.; Changula, K.; Muleya, W.; Chitanga, S.; Mwanza, M.; Simusika, P.; Chambaro, H.; et al. First COVID-19 case in Zambia—Comparative phylogenomic analyses of SARS-CoV-2 detected in African countries. Int. J. Infect. Dis. 2021, 102, 455–459. [Google Scholar] [CrossRef]
- Katowa, B.; Kalonda, A.; Mubemba, B.; Matoba, J.; Shempela, D.M.; Sikalima, J.; Kabungo, B.; Changula, K.; Chitanga, S.; Kasonde, M.; et al. Genomic Surveillance of SARS-CoV-2 in the Southern Province of Zambia: Detection and Characterization of Alpha, Beta, Delta, and Omicron Variants of Concern. Viruses 2022, 14, 1865. [Google Scholar] [CrossRef]
- Akande, O.W.; Carter, L.L.; Abubakar, A.; Achilla, R.; Barakat, A.; Gumede, N.; Guseinova, A.; Inbanathan, F.Y.; Kato, M.; Koua, E.; et al. Strengthening pathogen genomic surveillance for health emergencies: Insights from the World Health Organization’s regional initiatives. Front. Public Health 2023, 11, 1146730. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Khoosal, A.; Muhire, B. Detecting and Analyzing Genetic Recombination Using RDP4. Methods Mol. Biol. 2017, 1525, 433–460. [Google Scholar]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A Virus Classification Tool Based on Pairwise Sequence Alignment and Identity Calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef]
- Mercatelli, D.; Triboli, L.; Fornasari, E.; Ray, F.; Giorgi, F.M. Coronapp: A web application to annotate and monitor SARS-CoV-2 mutations. J. Med. Virol. 2021, 93, 3238–3245. [Google Scholar] [CrossRef]
- Martin, D.P.; Varsani, A.; Roumagnac, P.; Botha, G.; Maslamoney, S.; Schwab, T.; Kelz, Z.; Kumar, V.; Murrell, B. RDP5: A computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evol. 2021, 7, 87. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Graudenzi, A.; Maspero, D.; Angaroni, F.; Piazza, R.; Ramazzotti, D. Mutational signatures and heterogeneous host response revealed via large-scale characterization of SARS-CoV-2 genomic diversity. iScience 2021, 24, 102116. [Google Scholar] [CrossRef]
- Van Dorp, L.; Acman, M.; Richard, D.; Shaw, L.P.; Ford, C.E.; Ormond, L.; Owen, C.J.; Pang, J.; Tan, C.C.; Boshier, F.A.; et al. Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infect. Genet. Evol. 2020, 83, 104351. [Google Scholar] [CrossRef]
- Wang, L.; Berger, N.A.; Kaelber, D.C.; Davis, P.B.; Volkow, N.D.; Xu, R. COVID infection rates, clinical outcomes, and racial/ethnic and gender disparities before and after Omicron emerged in the US. medRxiv 2022. [Google Scholar] [CrossRef]
- Mseka, U.L.; Mandolo, J.; Nyoni, K.; Divala, O.; Kambalame, D.; Mapemba, D.; Kamzati, M.; Chibwe, I.; Henrion, M.Y.; Manda, K.; et al. Omicron B.1.1.529 variant infections associated with severe disease are uncommon in a COVID-19 under-vaccinated, high SARS-CoV-2 seroprevalence population in Malawi. eClinicalMedicine 2023, 56, 101800. [Google Scholar] [CrossRef]
- Taboada, B.; Zárate, S.; Iša, P.; Boukadida, C.; Vazquez-Perez, J.A.; Muñoz-Medina, J.E.; Ramírez-González, J.E.; Comas-García, A.; Grajales-Muñiz, C.; Rincón-Rubio, A.; et al. Genetic Analysis of SARS-CoV-2 Variants in Mexico during the First Year of the COVID-19 Pandemic. Viruses 2021, 13, 2161. [Google Scholar] [CrossRef]
- Bwire, G.M. Coronavirus: Why Men are More Vulnerable to COVID-19 Than Women? Sn Compr. Clin. Med. 2020, 2, 874. [Google Scholar] [CrossRef]
- Miao, Y.; Ren, Y.; Ren, T. Clinical Characteristics Profile of COVID-19 Patients with Omicron Variant Admitted in a Tertiary Hospital, Central China. Int. J. Gen. Med. 2023, 16, 2365. [Google Scholar] [CrossRef]
- Statsenko, Y.; Al Zahmi, F.; Habuza, T.; Almansoori, T.M.; Smetanina, D.; Simiyu, G.L.; Neidl-Van Gorkom, K.; Ljubisavljevic, M.; Awawdeh, R.; Elshekhali, H.; et al. Impact of Age and Sex on COVID-19 Severity Assessed from Radiologic and Clinical Findings. Front. Cell Infect. Microbiol. 2022, 11, 777070. [Google Scholar] [CrossRef]
- Monod, M.; Blenkinsop, A.; Xi, X.; Hebert, D.; Bershan, S.; Tietze, S.; Baguelin, M.; Bradley, V.C.; Chen, Y.; Coupland, H.; et al. Age groups that sustain resurging COVID-19 epidemics in the United States. Science 2021, 371, eabe8372. [Google Scholar] [CrossRef]
- Rahimi, A.; Mirzazadeh, A.; Tavakolpour, S. Genetics and genomics of SARS-CoV-2: A review of the literature with the special focus on genetic diversity and SARS-CoV-2 genome detection. Genomics 2021, 113, 1221–1232. [Google Scholar] [CrossRef]
- Rehman, S.U.; Shafique, L.; Ihsan, A.; Liu, Q. Evolutionary trajectory for the emergence of novel coronavirus SARS-CoV-2. Pathogens 2020, 9, 240. [Google Scholar] [CrossRef]
- da Silva, B.W.; Felix, P.T. Analysis of the genetic diversity of SARS-CoV-2 genomes carrying the Omicron B.1.1.529 mutation. medRxiv 2022. [Google Scholar] [CrossRef]
- Soratto, T.A.; Darban, H.; Bjerkner, A.; Coorens, M.; Albert, J.; Allander, T.; Andersson, B. Four SARS-CoV-2 Genome Sequences from Late April in Stockholm, Sweden, Reveal a Rare Mutation in the Spike Protein. Microbiol. Resour. Announc. 2020, 9, e00934-20. [Google Scholar] [CrossRef]
- Yin, C. Genotyping coronavirus SARS-CoV-2: Methods and implications. Genomics 2020, 112, 3588–3596. [Google Scholar] [CrossRef]
- Pachetti, M.; Marini, B.; Benedetti, F.; Giudici, F.; Mauro, E.; Storici, P.; Masciovecchio, C.; Angeletti, S.; Ciccozzi, M.; Gallo, R.C.; et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J. Transl. Med. 2020, 18, 179. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, W.; Liu, H.; Li, Y.; Yan, G.; Franzo, G.; Dai, J.; He, W.T. Mutation and codon bias analysis of the spike protein of Omicron, the recent variant of SARS-CoV-2. Int. J. Biol. Macromol. 2023, 250, 126080. [Google Scholar] [CrossRef]
- Ou, J.; Lan, W.; Wu, X.; Zhao, T.; Duan, B.; Yang, P.; Ren, Y.; Quan, L.; Zhao, W.; Seto, D.; et al. Tracking SARS-CoV-2 Omicron diverse spike gene mutations identifies multiple inter-variant recombination events. Signal Transduct. Target. Ther. 2022, 7, 138. [Google Scholar] [CrossRef]
- Saldivar-Espinoza, B.; Garcia-Segura, P.; Novau-Ferré, N.; Macip, G.; Martínez, R.; Puigbò, P.; Cereto-Massagué, A.; Pujadas, G.; Garcia-Vallve, S. The Mutational Landscape of SARS-CoV-2. Int. J. Mol. Sci. 2023, 24, 9072. [Google Scholar] [CrossRef]
- Yu, Q.; Tong, X.; Zuo, L.; Tao, X.; Xu, Z.; Li, X.; Liu, H.; Guan, W.; Liu, D.; Liu, H.; et al. Genomic Surveillance of SARS-CoV-2 Variants That Emerged in South and Southeast Asia during Early 2022. Viruses 2023, 15, 1355. [Google Scholar] [CrossRef]
- Pavlova, N.I.; Bochurov, A.A.; Alekseev, V.A.; Diakonova, A.T.; Dodokhov, V.V.; Kurtanov, K.A. Frequency of the Risk A Allele of rs17713054 Localized in the 3p21.31 COVID-19 Risk Locus in the Yakut Population. Int. J. Biomed. 2022, 12, 155–159. [Google Scholar] [CrossRef]
- Suzuki, T.; Aizawa, K.; Shibuya, K.; Yamanaka, S.; Anzai, Y.; Kurokawa, K.; Nagai, R. Prevalence of Asymptomatic SARS-CoV-2 Infection in Japan. JAMA Netw. Open 2022, 5, e2247704. [Google Scholar] [CrossRef]
- Lundberg, A.L.; Lorenzo-Redondo, R.; Ozer, E.A.; Hawkins, C.A.; Hultquist, J.F.; Welch, S.B.; Prasad, P.V.; Oehmke, J.F.; Achenbach, C.J.; Murphy, R.L.; et al. Has Omicron Changed the Evolution of the Pandemic? JMIR Public Health Surveill. 2022, 8, e35763. [Google Scholar] [CrossRef]
- Ke, H.; Chang, M.R.; Marasco, W.A. Immune Evasion of SARS-CoV-2 Omicron Subvariants. Vaccines 2022, 10, 1545. [Google Scholar] [CrossRef]
- Planas, D.; Bruel, T.; Staropoli, I.; Guivel-Benhassine, F.; Porrot, F.; Maes, P.; Grzelak, L.; Prot, M.; Mougari, S.; Planchais, C.; et al. Resistance of Omicron subvariants BA.2.75.2, BA.4.6, and BQ.1.1 to neutralizing antibodies. Nat. Commun. 2023, 14, 824. [Google Scholar] [CrossRef]
- da Costa, C.H.S.; de Freitas, C.A.B.; Alves, C.N.; Lameira, J. Assessment of mutations on RBD in the Spike protein of SARS-CoV-2 Alpha, Delta and Omicron variants. Sci. Rep. 2022, 12, 8540. [Google Scholar] [CrossRef]
- Adhikari, P.; Jawad, B.; Podgornik, R.; Ching, W.Y. Mutations of Omicron Variant at the Interface of the Receptor Domain Motif and Human Angiotensin-Converting Enzyme-2. Int. J. Mol. Sci. 2022, 23, 2870. [Google Scholar] [CrossRef]
- Peka, M.; Balatsky, V. The impact of mutation sets in receptor-binding domain of SARS-CoV-2 variants on stability of the RBD-ACE2 complex. Future Virol. 2023, 18, 225–242. [Google Scholar] [CrossRef]
- Chakraborty, C.; Bhattacharya, M.; Sharma, A.R.; Mallik, B. Omicron (B.1.1.529)—A new heavily mutated variant: Mapped location and probable properties of its mutations with an emphasis on S-glycoprotein. Int. J. Biol. Macromol. 2022, 219, 980–997. [Google Scholar] [CrossRef]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef]
- Alenquer, M.; Ferreira, F.; Lousa, D.; Valério, M.; Medina-Lopes, M.; Bergman, M.L.; Gonçalves, J.; Demengeot, J.; Leite, R.B.; Lilue, J.; et al. Signatures in SARS-CoV-2 spike protein conferring escape to neutralizing antibodies. PLoS Pathog. 2021, 17, e1009772. [Google Scholar] [CrossRef]
- Ching, W.Y.; Adhikari, P.; Jawad, B.; Podgornik, R. Effect of Delta and Omicron Mutations on the RBD-SD1 Domain of the Spike Protein in SARS-CoV-2 and the Omicron Mutations on RBD-ACE2 Interface Complex. Int. J. Mol. Sci. 2022, 23, 10091. [Google Scholar] [CrossRef]
- Patil, S.; Alzahrani, K.J.; Banjer, H.J.; Halawani, I.F.; Alzahrani, H.; Altayar, M.A.; Albogami, S.; Angeles, R.F.; Hassan, A.A.; Bhandi, S.; et al. Receptor binding domain of SARS-CoV-2 from Wuhan strain to Omicron B.1.1.529 attributes increased affinity to variable structures of human ACE2. J. Infect. Public Health 2022, 15, 781–787. [Google Scholar] [CrossRef]
- Shah, M.; Woo, H.G. Omicron: A Heavily Mutated SARS-CoV-2 Variant Exhibits Stronger Binding to ACE2 and Potently Escapes Approved COVID-19 Therapeutic Antibodies. Front. Immunol. 2022, 12, 830527. [Google Scholar] [CrossRef]
- Mannar, D.; Saville, J.W.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Tuttle, K.S.; Marquez, A.C.; Sekirov, I.; Subramaniam, S. SARS-CoV-2 Omicron variant: Antibody evasion and cryo-EM structure of spike protein–ACE2 complex. Science 2022, 375, 760–764. [Google Scholar] [CrossRef]
- Tong, C.; Shi, W.; Zhang, A.; Shi, Z. Tracking and controlling the spatiotemporal spread of SARS-CoV-2 Omicron variant in South Africa. Travel. Med. Infect. Dis. 2022, 46, 102252. [Google Scholar] [CrossRef]
- Eisen, J.A. Phylogenomics: Improving functional predictions for uncharacterized genes by evolutionary analysis. Genome Res. 1998, 8, 163–167. [Google Scholar] [CrossRef]
- Santiago, G.A.; Volkman, H.R.; Flores, B.; González, G.L.; Charriez, K.N.; Huertas, L.C.; Van Belleghem, S.M.; Rivera-Amill, V.; Major, C.; Colon, C.; et al. SARS-CoV-2 Omicron Replacement of Delta as Predominant Variant, Puerto Rico. Emerg. Infect. Dis. 2023, 29, 855–857. [Google Scholar] [CrossRef]
- Robles-Escajeda, E.; Mohl, J.E.; Contreras, L.; Betancourt, A.P.; Mancera, B.M.; Kirken, R.A.; Rodriguez, G. Rapid Shift from SARS-CoV-2 Delta to Omicron Sub-Variants within a Dynamic Southern U.S. Borderplex. Viruses 2023, 15, 658. [Google Scholar] [CrossRef]
- Vauhkonen, H.; Nguyen, P.T.; Kant, R.; Plyusnin, I.; Erdin, M.; Kurkela, S.; Liimatainen, H.; Ikonen, N.; Blomqvist, S.; Liitsola, K.; et al. Introduction and Rapid Spread of SARS-CoV-2 Omicron Variant and Dynamics of BA.1 and BA.1.1 Sublineages, Finland, December 2021. Emerg. Infect. Dis. 2022, 28, 1229–1232. [Google Scholar] [CrossRef]
- Sharma, D.; Notarte, K.I.; Fernandez, R.A.; Lippi, G.; Gromiha, M.M.; Henry, B.M. In silico evaluation of the impact of Omicron variant of concern sublineage BA.4 and BA.5 on the sensitivity of RT-qPCR assays for SARS-CoV-2 detection using whole genome sequencing. J. Med. Virol. 2023, 95, e28241. [Google Scholar] [CrossRef]
- Chibwana, M.G.; Thole, H.W.; Anscombe, C.; Ashton, P.M.; Green, E.; Barnes, K.G.; Cornick, J.; Turner, A.; Witte, D.; Nthala, S.; et al. Different clinical features in Malawian outpatients presenting with COVID-19 prior to and during Omicron variant dominance: A prospective observational study. PLOS Glob. Public Health 2023, 3, e0001575. [Google Scholar] [CrossRef]
- Simwanza, J.; Hines, J.Z.; Sinyange, D.; Sinyange, N.; Mulenga, C.; Hanyinza, S.; Sakubita, P.; Langa, N.; Nowa, H.; Gardner, P.; et al. COVID-19 Vaccine Effectiveness during a Prison Outbreak when Omicron was the Dominant Circulating Variant-Zambia, December 2021. Am. J. Trop. Med. Hyg. 2022, 107, 1055–1059. [Google Scholar] [CrossRef]
- Zambia Cenral Statistical Office; Government of the Republic of Zambia. Census of Population and Housing–2022. 2022. Available online: https://www.zamstats.gov.zm/population-by-province/ (accessed on 1 February 2024).
- Tcherepanov, V.; Ehlers, A.; Upton, C. Genome annotation transfer utility (GATU): Rapid annotation of viral genomes using a closely related reference genome. BMC Genom. 2006, 7, 150. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description | N | % | |
|---|---|---|---|
| Gender (n = 112) | Male | 45 | 40.2 |
| Female | 67 | 59.8 | |
| Unknown | 3 | 2.6 | |
| Age (n = 115) | 0–9 years | 3 | 3.4 |
| 10–19 years | 38 | 43.7 | |
| 20–29 years | 18 | 20.7 | |
| 30–39 years | 12 | 13.8 | |
| 40–49 years | 8 | 9.2 | |
| 50+ | 8 | 9.2 | |
| Unknown | 28 | 24.3 | |
| District (n = 115) | Chadiza | 9 | 7.8 |
| Chipangali | 16 | 13.9 | |
| Katete | 14 | 12.2 | |
| Lundazi | 12 | 10.4 | |
| Mambwe | 21 | 18.3 | |
| Nyimba | 43 | 37.4 |
| Target | |||
|---|---|---|---|
| S Gene | ORF1ab | N Gene | Number of Samples |
| − | + | + | 80 |
| + | + | + | 35 |
| Total | 115 | ||
| Genomic Region | Mutation Count | Annotation |
|---|---|---|
| 5′UTR | 41 | 5′ Untranslated region |
| NSP1 | 27 | RNA-dependent RNA polymerase |
| NSP2 | 8 | |
| NSP3 | 378 | |
| NSP4 | 156 | |
| NSP5 | 134 | |
| NSP6 | 122 | |
| NSP9 | 39 | |
| NSP10 | 43 | |
| NSP12b | 146 | |
| NSP13 | 33 | |
| NSP14 | 90 | |
| NSP15 | 68 | |
| NSP16 | 10 | |
| S | 1925 | Spike |
| ORF3a | 105 | Open reading frame 3a protein |
| E | 70 | Envelope protein |
| M | 198 | Membrane protein |
| ORF6 | 96 | Open reading frame 6 protein |
| ORF7a | 5 | Open reading frame 7a protein |
| ORF7b | 59 | Open reading frame 7b protein |
| ORF8 | 13 | Open reading frame 8 protein |
| N | 235 | Nucleocapsid protein |
| ORF10 | 2 | Open reading frame 10 protein |
| 3′UTR | 94 | 3′ Untranslated region |
| Total | 4097 |
| Receptor Binding Domain | ||
|---|---|---|
| Variant Class | Count | % |
| G339D | 66 | 1.6 |
| K417N | 65 | 1.6 |
| S375F | 64 | 1.6 |
| S373P | 64 | 1.6 |
| Y505H | 41 | 1.0 |
| E484A | 40 | 1.0 |
| Q498R | 40 | 1.0 |
| N501Y | 40 | 1.0 |
| S477N | 39 | 1.0 |
| T478K | 39 | 1.0 |
| Q493R | 39 | 1.0 |
| S371L | 38 | 0.9 |
| D405N | 27 | 0.7 |
| T376A | 27 | 0.7 |
| S371F | 27 | 0.7 |
| R408S | 27 | 0.7 |
| G446S | 21 | 0.5 |
| G496S | 20 | 0.5 |
| N440K | 20 | 0.5 |
| R346K | 11 | 0.3 |
| T547K | 6 | 0.1 |
| A372 | 1 | 0.0 |
| L368I | 1 | 0.0 |
| N370S | 1 | 0.0 |
| T430A | 1 | 0.0 |
| A397A | 1 | 0.0 |
| T376 | 1 | 0.0 |
| I410V | 1 | 0.0 |
| T385A | 1 | 0.0 |
| I434M | 1 | 0.0 |
| R403G | 1 | 0.0 |
| S399S | 1 | 0.0 |
| V367I | 1 | 0.0 |
| A475A | 1 | 0.0 |
| R454R | 1 | 0.0 |
| T333A | 1 | 0.0 |
| Total | 776 | 18.9 |
| Receptor Binding Motif | ||
| Y505H | 41 | 1.0 |
| Q498R | 40 | 1.0 |
| E484A | 40 | 1.0 |
| N501Y | 40 | 1.0 |
| T478K | 39 | 1.0 |
| S477N | 39 | 1.0 |
| Q493R | 39 | 1.0 |
| G446S | 21 | 0.5 |
| G496S | 20 | 0.5 |
| N440K | 2 | 0.0 |
| R454R | 1 | 0.0 |
| A475A | 1 | 0.0 |
| Total | 323 | 7.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shempela, D.M.; Chambaro, H.M.; Sikalima, J.; Cham, F.; Njuguna, M.; Morrison, L.; Mudenda, S.; Chanda, D.; Kasanga, M.; Daka, V.; et al. Detection and Characterisation of SARS-CoV-2 in Eastern Province of Zambia: A Retrospective Genomic Surveillance Study. Int. J. Mol. Sci. 2024, 25, 6338. https://doi.org/10.3390/ijms25126338
Shempela DM, Chambaro HM, Sikalima J, Cham F, Njuguna M, Morrison L, Mudenda S, Chanda D, Kasanga M, Daka V, et al. Detection and Characterisation of SARS-CoV-2 in Eastern Province of Zambia: A Retrospective Genomic Surveillance Study. International Journal of Molecular Sciences. 2024; 25(12):6338. https://doi.org/10.3390/ijms25126338
Chicago/Turabian StyleShempela, Doreen Mainza, Herman M. Chambaro, Jay Sikalima, Fatim Cham, Michael Njuguna, Linden Morrison, Steward Mudenda, Duncan Chanda, Maisa Kasanga, Victor Daka, and et al. 2024. "Detection and Characterisation of SARS-CoV-2 in Eastern Province of Zambia: A Retrospective Genomic Surveillance Study" International Journal of Molecular Sciences 25, no. 12: 6338. https://doi.org/10.3390/ijms25126338
APA StyleShempela, D. M., Chambaro, H. M., Sikalima, J., Cham, F., Njuguna, M., Morrison, L., Mudenda, S., Chanda, D., Kasanga, M., Daka, V., Kwenda, G., Musonda, K., Munsaka, S., Chilengi, R., Sichinga, K., & Simulundu, E. (2024). Detection and Characterisation of SARS-CoV-2 in Eastern Province of Zambia: A Retrospective Genomic Surveillance Study. International Journal of Molecular Sciences, 25(12), 6338. https://doi.org/10.3390/ijms25126338