

The Long Non-Coding RNA MALAT1 Modulates NR4A1 Expression through a Downstream Regulatory Element in Specific Cancer Cell Types

 ,
,  , , and
, , and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
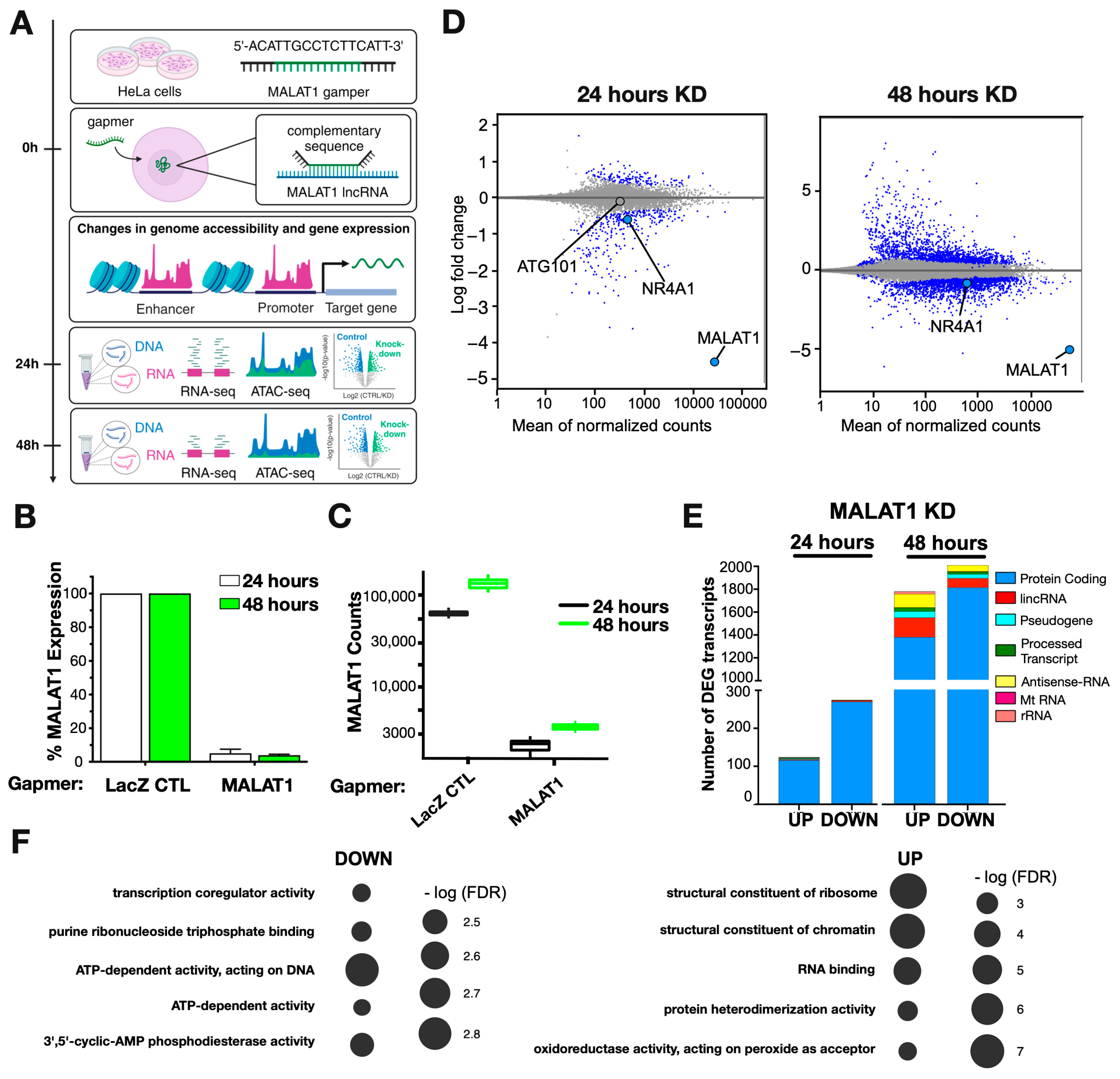
2.1. MALAT1 Primarily Regulates Protein-Coding Genes
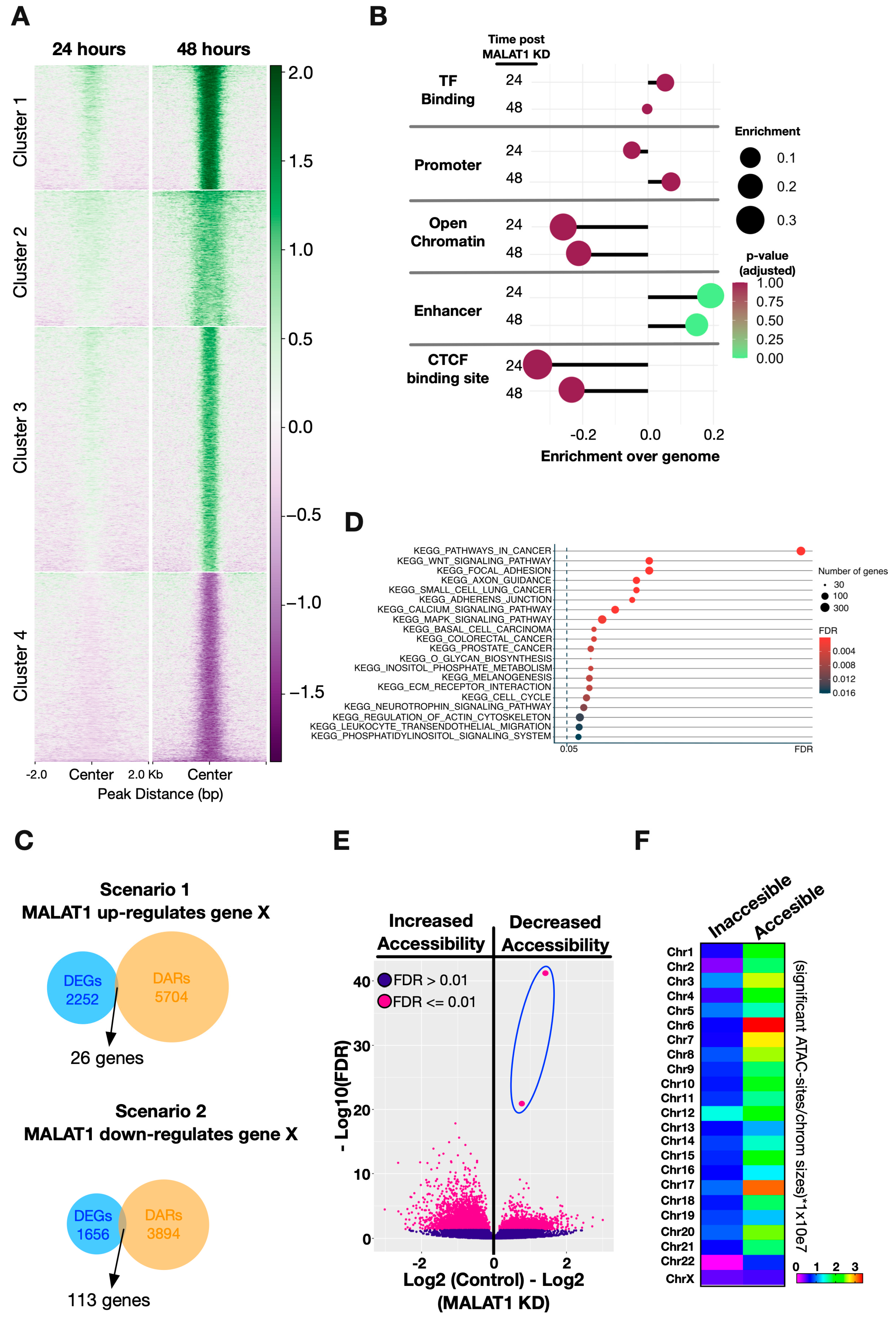
2.2. Chromatin Accessibility Changes after MALAT1 Downregulation
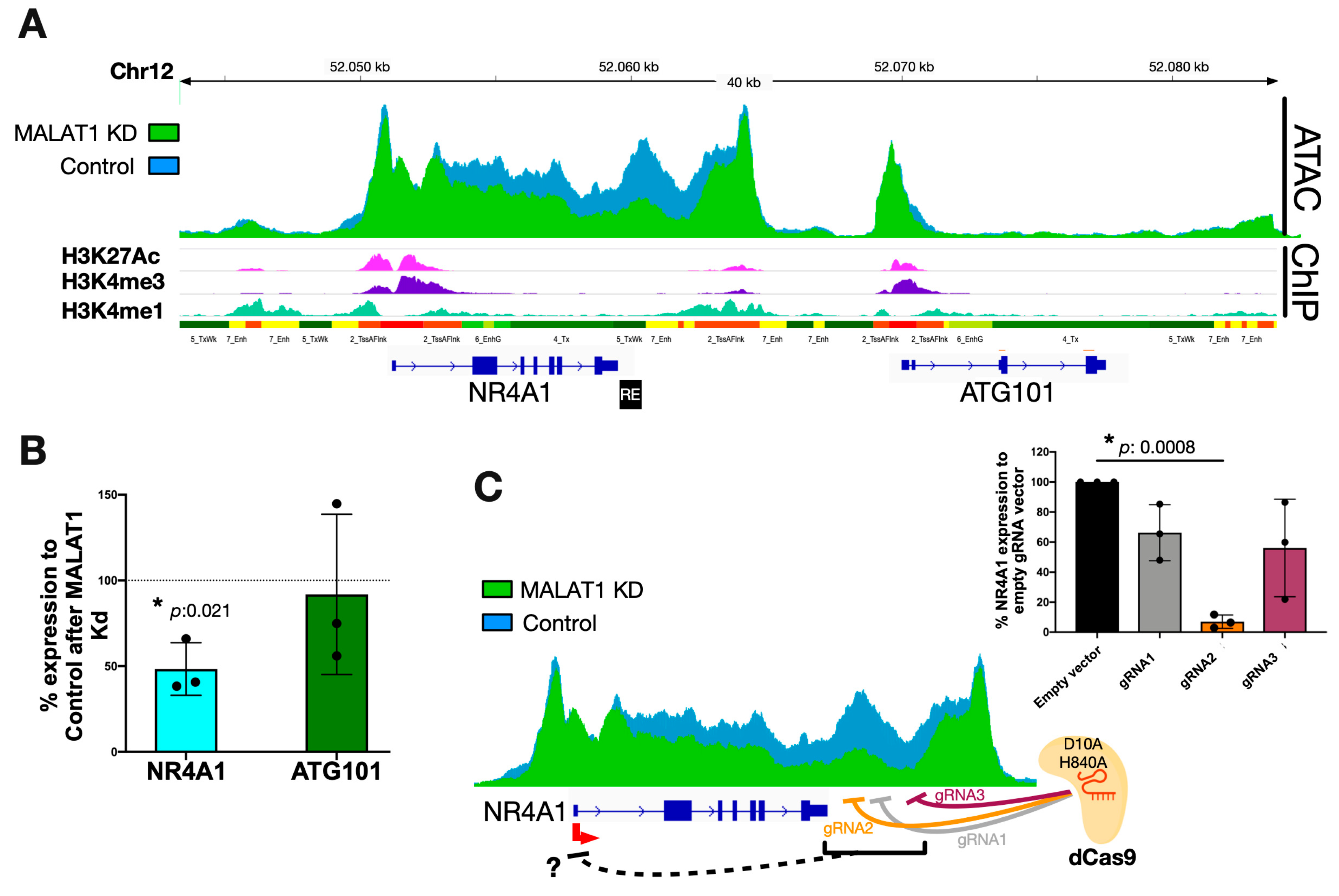
2.3. MALAT1 Regulates NR4A1 Expression by Modulating the Chromatin Accessibility of a Downstream RE
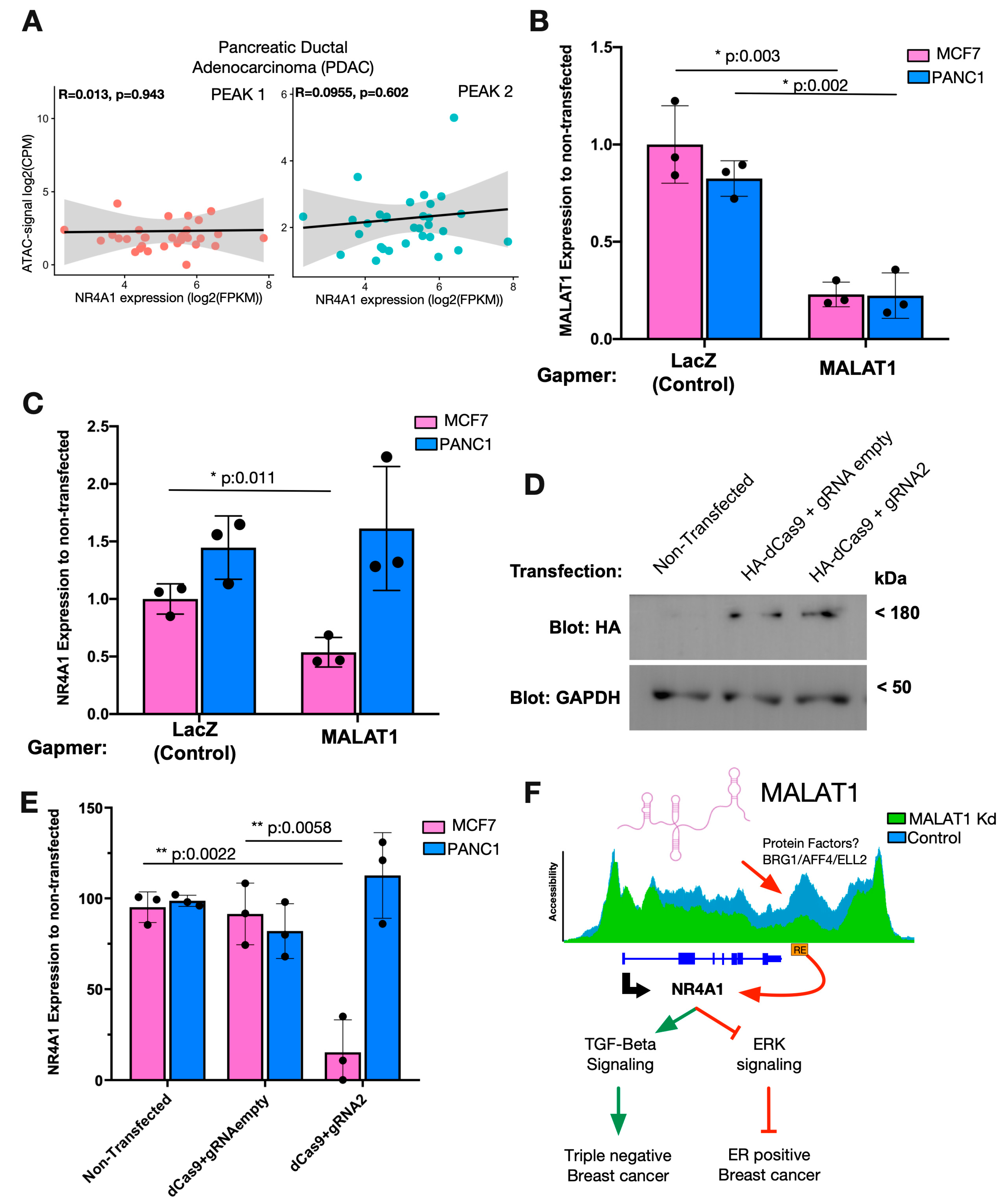
2.4. The Accessibility of the NR4A1-Downstream RE Correlates with NR4A1 Expression in Specific Cancer Types
2.5. The MALAT1/NR4A1 Axis Is Functional in Breast Cancer Cells but Not in Pancreatic Cancer Cells
3. Discussion
3.1. MALAT1-Dependent Changes in Chromatin Accessibility
3.2. The Impact of the MALAT1/NR4A1 Axis on Breast Cancer Development
4. Materials and Methods
4.1. Cell Culture
4.2. Western Blot
4.3. Real-Time PCR
4.4. Cloning of Guide RNAs
4.5. Transfections
4.6. Custom Genome
4.7. Annotation
4.8. RNA Purification
4.9. RNA Sequencing and Transcriptome Analysis
4.10. ATAC-seq Assays, Sequencing, and Analysis
4.11. Correlations with TCGA and Pancreatic Cancer Data
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- Marchese, F.P.; Raimondi, I.; Huarte, M. The multidimensional mechanisms of long noncoding RNA function. Genome Biol. 2017, 18, 206. [Google Scholar] [CrossRef] [PubMed]
- Statello, L.; Guo, J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fu, X.D. Chromatin-associated RNAs as facilitators of functional genomic interactions. Nat. Rev. Genet. 2019, 20, 503–519. [Google Scholar] [CrossRef] [PubMed]
- Volders, P.J.; Anckaert, J.; Verheggen, K.; Nuytens, J.; Martens, L.; Mestdagh, P.; Vandesompele, J. Lncipedia 5: Towards a reference set of human long non-coding rnas. Nucleic Acids Res. 2019, 47, D135–D139. [Google Scholar] [CrossRef]
- Flynn, R.A.; Chang, H.Y. Long noncoding RNAs in cell-fate programming and reprogramming. Cell Stem Cell 2014, 14, 752–761. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, S. Long noncoding RNAs in cell differentiation and pluripotency. Cell Tissue Res. 2016, 366, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Much, C.; Lasda, E.L.; Lewandowski, J.P.; Smallegan, M.J.; Rinn, J.L. The temporal dynamics of lncRNA Firre-mediated epigenetic and transcriptional regulation. bioRxiv 2022. [Google Scholar] [CrossRef]
- Arab, K.; Park, Y.J.; Lindroth, A.M.; Schäfer, A.; Oakes, C.; Weichenhan, D.; Lukanova, A.; Lundin, E.; Risch, A.; Meister, M.; et al. Long noncoding RNA TARID directs demethylation and activation of the tumor suppressor TCF21 via GADD45A. Mol. Cell 2014, 55, 604–614. [Google Scholar] [CrossRef]
- Nandwani, A.; Rathore, S.; Datta, M. LncRNAs in cancer: Regulatory and therapeutic implications. Cancer Lett. 2021, 501, 162–171. [Google Scholar] [CrossRef]
- Lin, W.; Zhou, Q.; Wang, C.Q.; Zhu, L.; Bi, C.; Zhang, S.; Wang, X.; Jin, H. LncRNAs regulate metabolism in cancer. Int. J. Biol. Sci. 2020, 16, 1194–1206. [Google Scholar] [CrossRef]
- Peng, W.X.; Koirala, P.; Mo, Y.Y. LncRNA-mediated regulation of cell signaling in cancer. Oncogene 2017, 36, 5661–5667. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.N.; Ensminger, A.W.; Clemson, C.M.; Lynch, C.R.; Lawrence, J.B.; Chess, A. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genom. 2007, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Ji, P.; Diederichs, S.; Wang, W.; Böing, S.; Metzger, R.; Schneider, P.M.; Tidow, N.; Brandt, B.; Buerger, H.; Bulk, E.; et al. MALAT-1, a novel noncoding RNA, and thymosin β4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 2003, 22, 8031–8041. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E.; Freier, S.M.; Spector, D.L. 3′ End Processing of a Long Nuclear-Retained Noncoding RNA Yields a tRNA-like Cytoplasmic RNA. Cell 2008, 135, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E.; JnBaptiste, C.K.; Lu, L.Y.; Kuhn, C.-D.; Joshua-Tor, L.; Sharp, P.A. A triple helix stabilizes the 3′ ends of long noncoding RNAs that lack poly(A) tails. Genes Dev. 2012, 26, 2392–2407. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Valenstein, M.L.; Yario, T.A.; Tycowski, K.T.; Steitz, J.A. Formation of triple-helical structures by the 3′-end sequences of MALAT1 and MENβ noncoding RNAs. Proc. Natl. Acad. Sci. USA 2012, 109, 19202–19207. [Google Scholar] [CrossRef]
- Engreitz, J.M.; Sirokman, K.; McDonel, P.; Shishkin, A.A.; Surka, C.; Russell, P.; Grossman, S.R.; Chow, A.Y.; Guttman, M.; Lander, E.S. RNA-RNA interactions enable specific targeting of noncoding RNAs to nascent pre-mRNAs and chromatin sites. Cell 2014, 159, 188–199. [Google Scholar] [CrossRef]
- West, J.A.; Davis, C.P.; Sunwoo, H.; Simon, M.D.; Sadreyev, R.I.; Wang, P.I.; Tolstorukov, M.Y.; Kingston, R.E. The Long Noncoding RNAs NEAT1 and MALAT1 Bind Active Chromatin Sites. Mol. Cell 2014, 55, 791–802. [Google Scholar] [CrossRef]
- Goyal, B.; Yadav, S.R.M.; Awasthee, N.; Gupta, S.; Kunnumakkara, A.B.; Gupta, S.C. Diagnostic, prognostic, and therapeutic significance of long non-coding RNA MALAT1 in cancer. Biochim. Biophys. Acta-Rev. Cancer 2021, 1875, 188502. [Google Scholar] [CrossRef]
- Sun, Y.; Ma, L. New insights into long non-coding rna malat1 in cancer and metastasis. Cancers 2019, 11, 216. [Google Scholar] [CrossRef]
- Hedrick, E.; Lee, S.O.; Safe, S. The nuclear orphan receptor NR4A1 regulates β1-integrin expression in pancreatic and colon cancer cells and can be targeted by NR4A1 antagonists. Mol. Carcinog. 2017, 56, 2066–2075. [Google Scholar] [CrossRef]
- Palumbo-Zerr, K.; Zerr, P.; Distler, A.; Fliehr, J.; Mancuso, R.; Huang, J.; Mielenz, D.; Tomcik, M.; Fürnrohr, B.G.; Scholtysek, C.; et al. Orphan nuclear receptor NR4A1 regulates transforming growth factor-β 2 signaling and fibrosis. Nat. Med. 2015, 21, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, Y.; Lu, H.; Li, J.; Yan, X.; Xiao, M.; Hao, J.; Alekseev, A.; Khong, H.; Chen, T.; et al. Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature 2019, 567, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; López-Moyado, I.F.; Seo, H.; Lio, C.-W.J.; Hempleman, L.J.; Sekiya, T.; Yoshimura, A.; Scott-Browne, J.P.; Rao, A. NR4A transcription factors limit CAR T cell function in solid tumours. Nature 2019, 567, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Drabsch, Y.; Dekker, T.J.A.; de Vinuesa, A.G.; Li, Y.; Hawinkels, L.J.A.C.; Sheppard, K.-A.; Goumans, M.-J.; Luwor, R.B.; de Vries, C.J.; et al. Nuclear receptor NR4A1 promotes breast cancer invasion and metastasis by activating TGF-β signalling. Nat. Commun. 2014, 5, 3388. [Google Scholar] [CrossRef]
- Kim, Y.C.; Kim, C.Y.; Oh, J.H.; Kim, M.H. Nr4a1 regulates tamoxifen resistance by suppressing erk signaling in er-positive breast cancer. Cells 2021, 10, 1633. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.D.; Campbell, M.J.; Kejariwal, A.; Mi, H.; Karlak, B.; Daverman, R.; Diemer, K.; Muruganujan, A.; Narechania, A. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef]
- Thomas, P.D.; Kejariwal, A.; Guo, N.; Mi, H.; Campbell, M.J.; Muruganujan, A.; Lazareva-Ulitsky, B. Applications for protein sequence-function evolution data: mRNA/protein expression analysis and coding SNP scoring tools. Nucleic Acids Res. 2006, 34, 645–650. [Google Scholar] [CrossRef]
- Corces, M.R.; Granja, J.M.; Shams, S.; Louie, B.H.; Seoane, J.A.; Zhou, W.; Silva, T.C.; Groeneveld, C.; Wong, C.K.; Cho, S.W.; et al. The chromatin accessibility landscape of primary human cancers. Science 2018, 362, eaav1898. [Google Scholar] [CrossRef]
- Lin, C.Y.; Loven, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef]
- Martini, M.; De Santis, M.C.; Braccini, L.; Gulluni, F.; Hirsch, E. PI3K/AKT signaling pathway and cancer: An updated review. Ann. Med. 2014, 46, 372–383. [Google Scholar] [CrossRef]
- Altomare, D.A.; Testa, J.R. Perturbations of the AKT signaling pathway in human cancer. Oncogene 2005, 24, 7455–7464. [Google Scholar] [CrossRef] [PubMed]
- Schubert, T.; Pusch, M.C.; Diermeier, S.; Benes, V.; Kremmer, E.; Imhof, A. Df31 Protein and snoRNAs Maintain Accessible Higher-Order Structures of Chromatin. Mol. Cell 2012, 48, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Fujita, R.; Yamamoto, T.; Arimura, Y.; Fujiwara, S.; Tachiwana, H.; Ichikawa, Y.; Sakata, Y.; Yang, L.; Maruyama, R.; Hamada, M.; et al. Nucleosome destabilization by nuclear non-coding RNAs. Commun. Biol. 2020, 3, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Yang, Y.W.; Liu, B.; Sanyal, A.; Corces-Zimmerman, R.; Chen, Y.; Lajoie, B.R.; Protacio, A.; Flynn, R.A.; Gupta, R.A.; et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 2011, 472, 120–126. [Google Scholar] [CrossRef]
- Engreitz, J.M.; Pandya-Jones, A.; McDonel, P.; Shishkin, A.; Sirokman, K.; Surka, C.; Kadri, S.; Xing, J.; Goren, A.; Lander, E.S.; et al. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science 2013, 341, 1237973. [Google Scholar] [CrossRef]
- van Ouwerkerk, A.F.; Bosada, F.M.; van Duijvenboden, K.; Hill, M.C.; Montefiori, L.E.; Scholman, K.T.; Liu, J.; de Vries, A.A.F.; Boukens, B.J.; Ellinor, P.T.; et al. Identification of atrial fibrillation associated genes and functional non-coding variants. Nat. Commun. 2019, 10, 4755. [Google Scholar] [CrossRef]
- Drissen, R.; Palstra, R.-J.; Gillemans, N.; Splinter, E.; Grosveld, F.; Philipsen, S.; de Laat, W. The active spatial organization of the β-globin locus requires the transcription factor EKLF. Genes Dev. 2004, 18, 2485–2490. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Wang, H.; Hu, X.; Cao, X. lncRNA MALAT1 binds chromatin remodeling subunit BRG1 to epigenetically promote inflammation-related hepatocellular carcinoma progression. Oncoimmunology 2019, 8, e1518628. [Google Scholar] [CrossRef]
- Lin, C.; Smith, E.R.; Takahashi, H.; Lai, K.C.; Martin-Brown, S.; Florens, L.; Washburn, M.P.; Conaway, J.W.; Conaway, R.C.; Shilatifard, A. AFF4, a Component of the ELL/P-TEFb Elongation Complex and a Shared Subunit of MLL Chimeras, Can Link Transcription Elongation to Leukemia. Mol. Cell 2010, 37, 429–437. [Google Scholar] [CrossRef]
- Xie, X.; Lin, J.; Liu, J.; Huang, M.; Zhong, Y.; Liang, B.; Song, X.; Gu, S.; Chang, X.; Huang, D.; et al. A novel lncRNA NR4A1AS up-regulates orphan nuclear receptor NR4A1 expression by blocking UPF1-mediated mRNA destabilization in colorectal cancer. Clin. Sci. 2019, 133, 1457–1473. [Google Scholar] [CrossRef]
- Leeman-Neill, R.J.; Song, D.; Bizarro, J.; Wacheul, L.; Rothschild, G.; Singh, S.; Yang, Y.; Sarode, A.Y.; Gollapalli, K.; Wu, L.; et al. Noncoding mutations cause super-enhancer retargeting resulting in protein synthesis dysregulation during B cell lymphoma progression. Nat. Genet. 2023, 55, 2160–2174. [Google Scholar] [CrossRef]
- Chen, Q.; Zhu, C.; Jin, Y. The Oncogenic and Tumor Suppressive Functions of the Long Noncoding RNA MALAT1: An Emerging Controversy. Front. Genet. 2020, 11, 505991. [Google Scholar] [CrossRef]
- Bahrami, S.; Drabløs, F. Gene regulation in the immediate-early response process. Adv. Biol. Regul. 2016, 62, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.G.; Vassiliou, L.V.F.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; Ditullio, R.A., Jr.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef]
- Guo, H.; Golczer, G.; Wittner, B.S.; Langenbucher, A.; Zachariah, M.; Dubash, T.D.; Hong, X.; Comaills, V.; Burr, R.; Ebright, R.Y.; et al. NR4A1 regulates expression of immediate early genes, suppressing replication stress in cancer. Mol. Cell 2021, 81, 4041–4058.e15. [Google Scholar] [CrossRef]
- Perez-Pinera, P.; Kocak, D.D.; Vockley, C.M.; Adler, A.F.; Kabadi, A.M.; Polstein, L.R.; Thakore, P.I.; Glass, K.A.; Ousterout, D.G.; Leong, K.W.; et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat. Methods 2013, 10, 973–976. [Google Scholar] [CrossRef] [PubMed]
- Mars, J.; Sabourin-Felix, M.; Tremblay, M.; Moss, T. A deconvolution protocol for ChIP-seq reveals analogous enhancer structures on the mouse and human ribosomal RNA genes. G3 Genes Genomes Genet. 2018, 8, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FASTQC: A Quality Control Tool for High Throughput Sequence Data. Nucleic Acid Res. 2010, 41, e108. [Google Scholar]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Bolger, A.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2015, 30, 2114. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapters from high-throughput sequencing reads. EMBnet.J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.; Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acid Res. 2013, 41, e108. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.; Ibrahim, J.G.; Love, M.I. Heavy-Tailed prior distributions for sequence count data: Removing the noise and preserving large differences. Bioinformatics 2019, 35, 2084–2092. [Google Scholar] [CrossRef] [PubMed]
- Corces, M.R.; Trevino, A.E.; Hamilton, E.G.; Greenside, P.G.; Sinnott-Armstrong, N.A.; Vesuna, S.; Satpathy, A.T.; Rubin, A.J.; Montine, K.S.; Wu, B.; et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 2017, 14, 959–962. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Ramírez, F.; Dündar, F.; Diehl, S.; Grüning, B.A.; Manke, T. DeepTools: A flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014, 42, 187–191. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]
- McLean, C.Y.; Bristor, D.; Hiller, M.; Clarke, S.L.; Schaar, B.T.; Lowe, C.B.; Wenger, A.M.; Bejerano, G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010, 28, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wernig-Zorc, S.; Schwartz, U.; Martínez-Rodríguez, P.; Inalef, J.; Pavicic, F.; Ehrenfeld, P.; Längst, G.; Maldonado, R. The Long Non-Coding RNA MALAT1 Modulates NR4A1 Expression through a Downstream Regulatory Element in Specific Cancer Cell Types. Int. J. Mol. Sci. 2024, 25, 5515. https://doi.org/10.3390/ijms25105515
Wernig-Zorc S, Schwartz U, Martínez-Rodríguez P, Inalef J, Pavicic F, Ehrenfeld P, Längst G, Maldonado R. The Long Non-Coding RNA MALAT1 Modulates NR4A1 Expression through a Downstream Regulatory Element in Specific Cancer Cell Types. International Journal of Molecular Sciences. 2024; 25(10):5515. https://doi.org/10.3390/ijms25105515
Chicago/Turabian StyleWernig-Zorc, Sara, Uwe Schwartz, Paulina Martínez-Rodríguez, Josefa Inalef, Francisca Pavicic, Pamela Ehrenfeld, Gernot Längst, and Rodrigo Maldonado. 2024. "The Long Non-Coding RNA MALAT1 Modulates NR4A1 Expression through a Downstream Regulatory Element in Specific Cancer Cell Types" International Journal of Molecular Sciences 25, no. 10: 5515. https://doi.org/10.3390/ijms25105515
APA StyleWernig-Zorc, S., Schwartz, U., Martínez-Rodríguez, P., Inalef, J., Pavicic, F., Ehrenfeld, P., Längst, G., & Maldonado, R. (2024). The Long Non-Coding RNA MALAT1 Modulates NR4A1 Expression through a Downstream Regulatory Element in Specific Cancer Cell Types. International Journal of Molecular Sciences, 25(10), 5515. https://doi.org/10.3390/ijms25105515