
BMPR2 Loss Activates AKT by Disrupting DLL4/NOTCH1 and PPARγ Signaling in Pulmonary Arterial Hypertension

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Silencing BMPR2 Activates AKT and Blocks Apoptosis
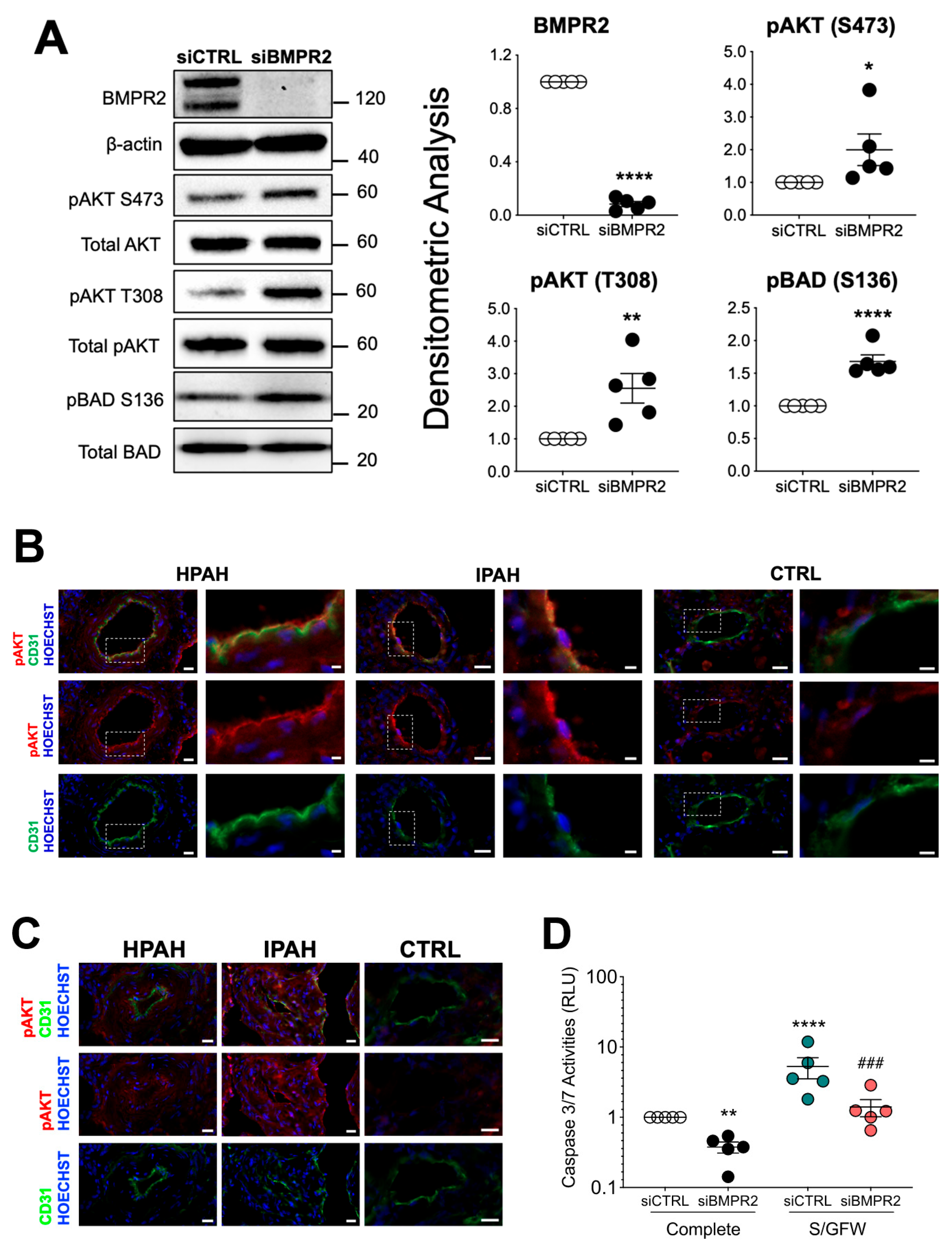
2.2. JNK1 Suppression Contributes to Apoptosis Resistance
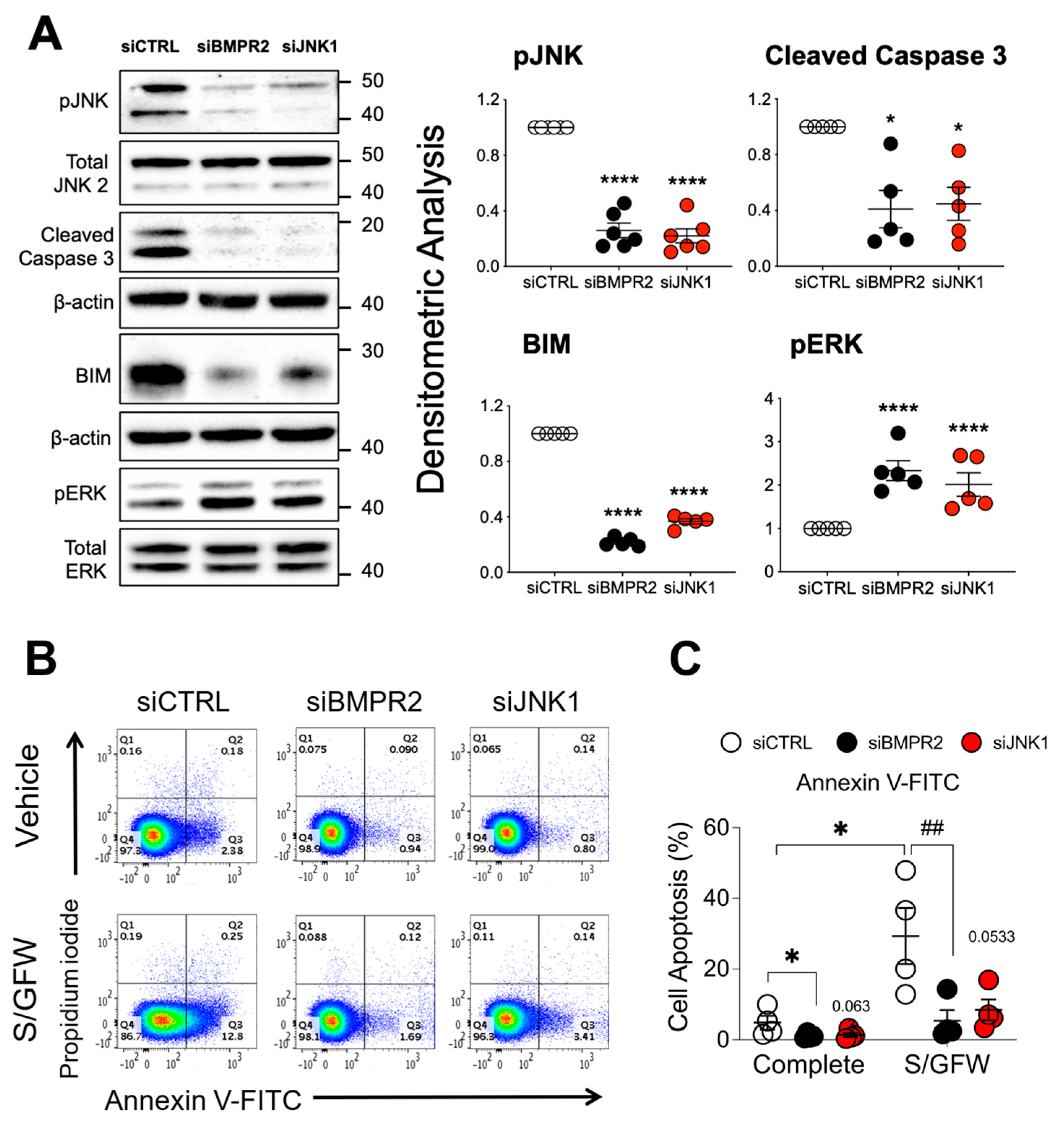
2.3. Leniolisib Attenuates AKT Activation, Inhibits Cell Proliferation and EndoMT, and Induces Apoptosis in BMPR2 and CAV1 Silenced PAECs
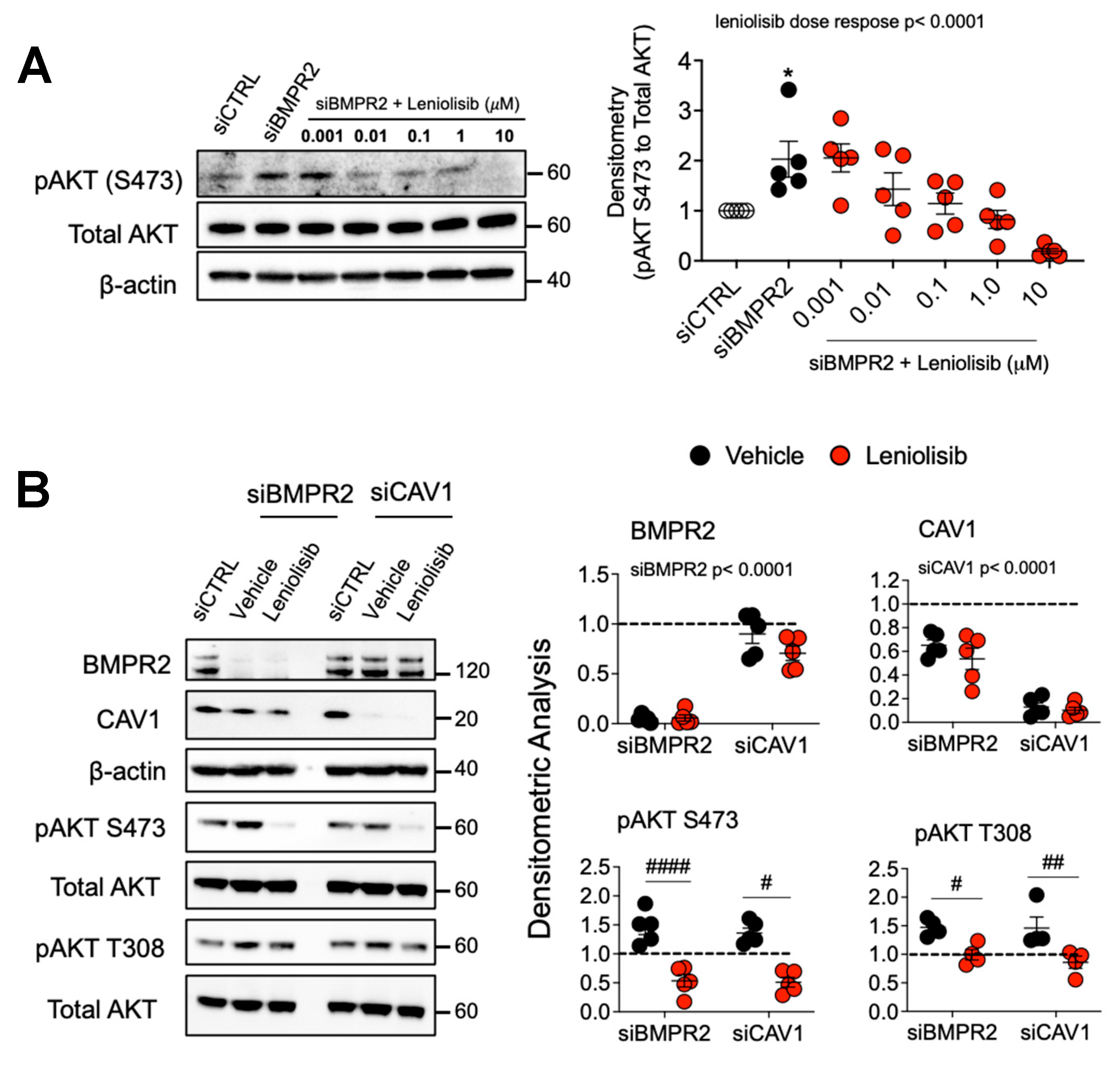
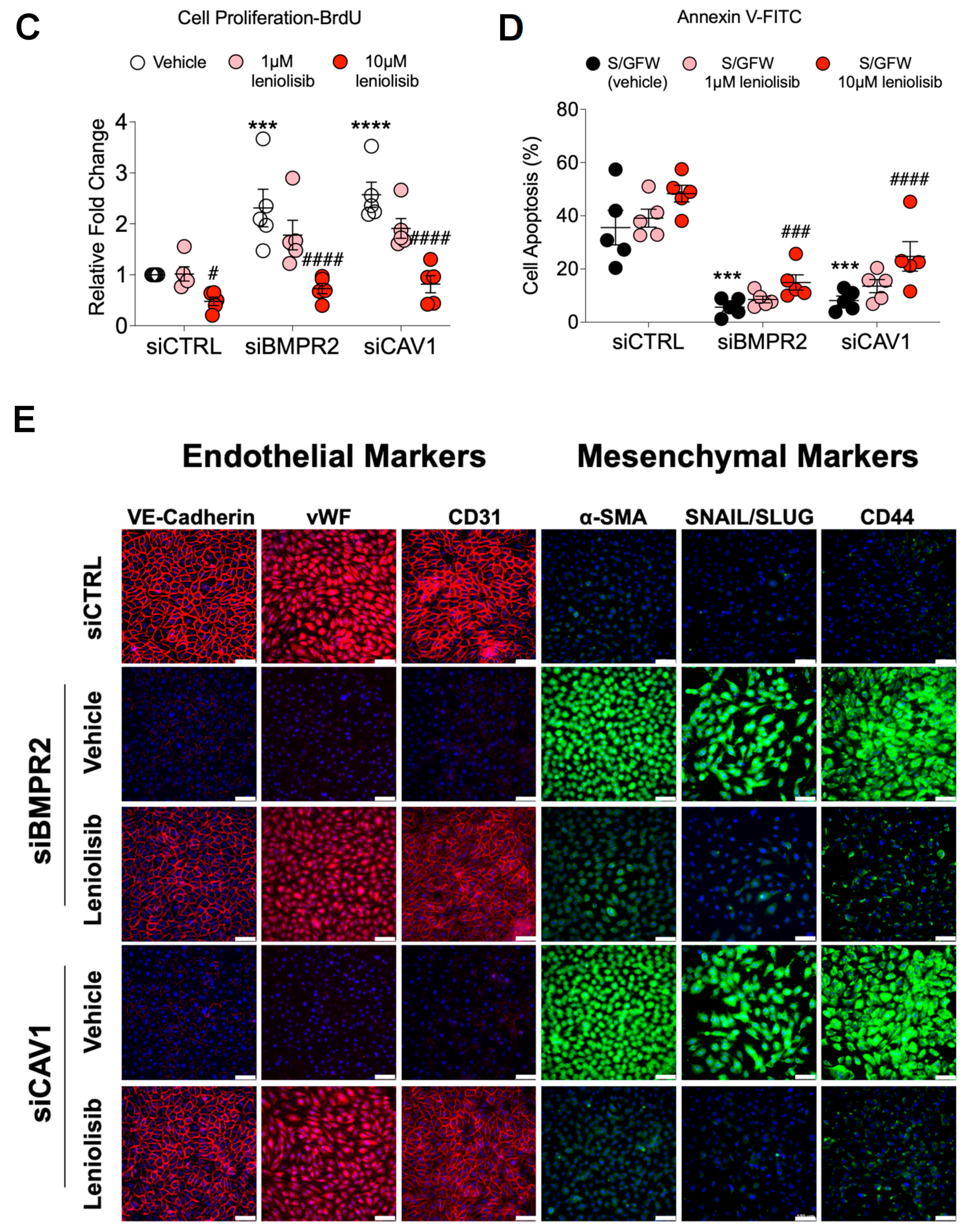
2.4. Loss of DLL4/NOTCH1 Signaling in BMPR2-Silenced PAECs and in PAH Lung and Its Reactivation Blocks AKT, Suppressing Cell Proliferation
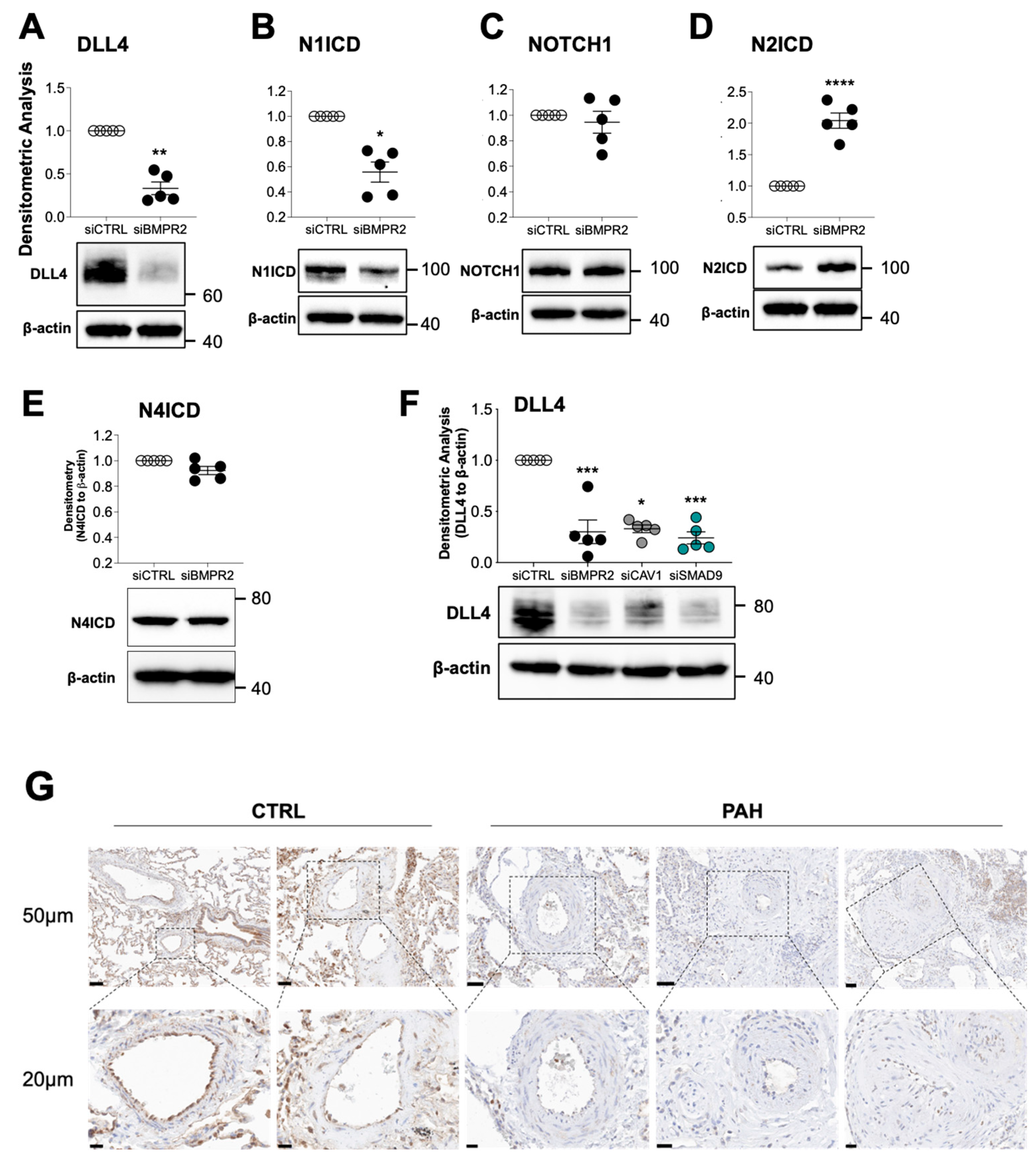

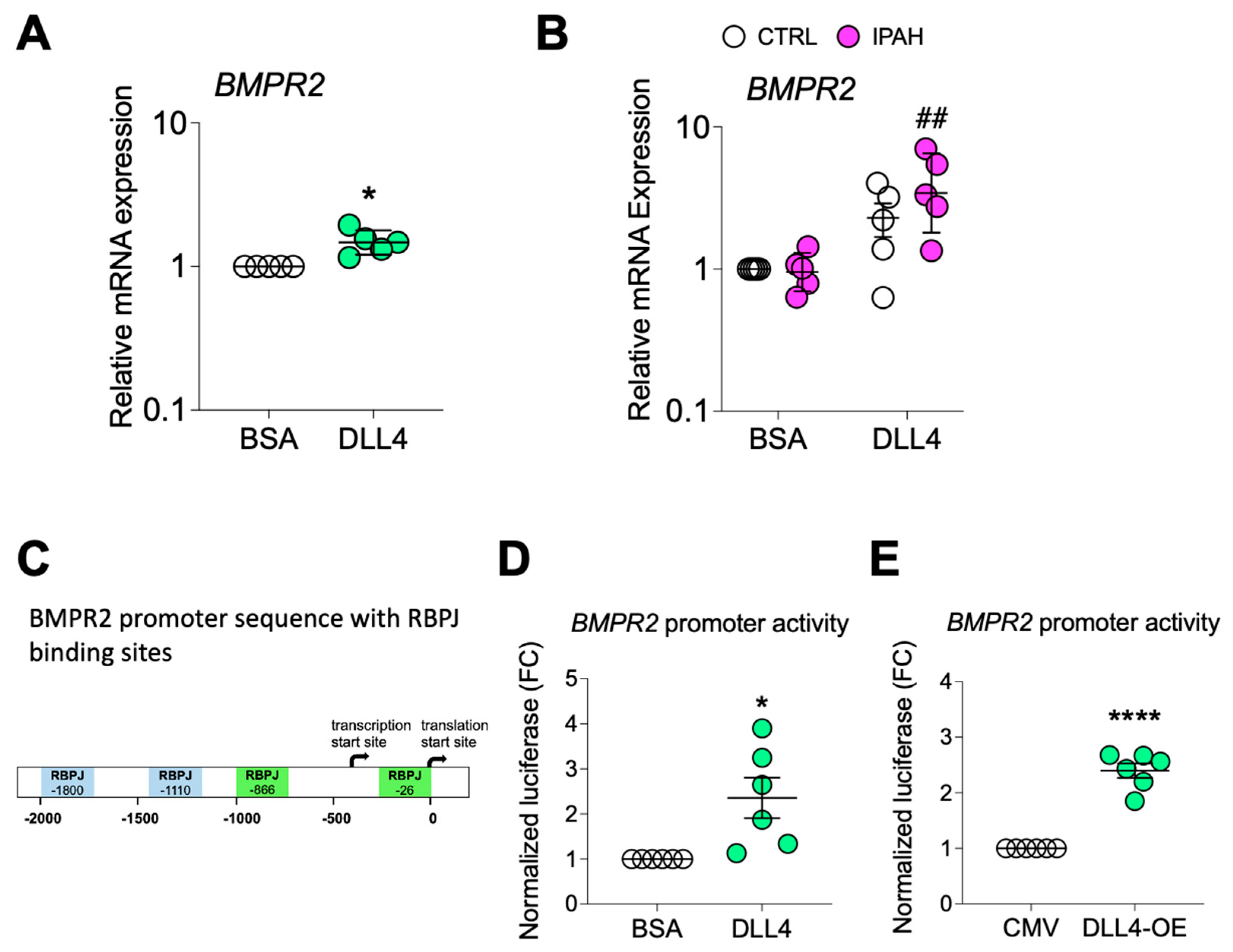
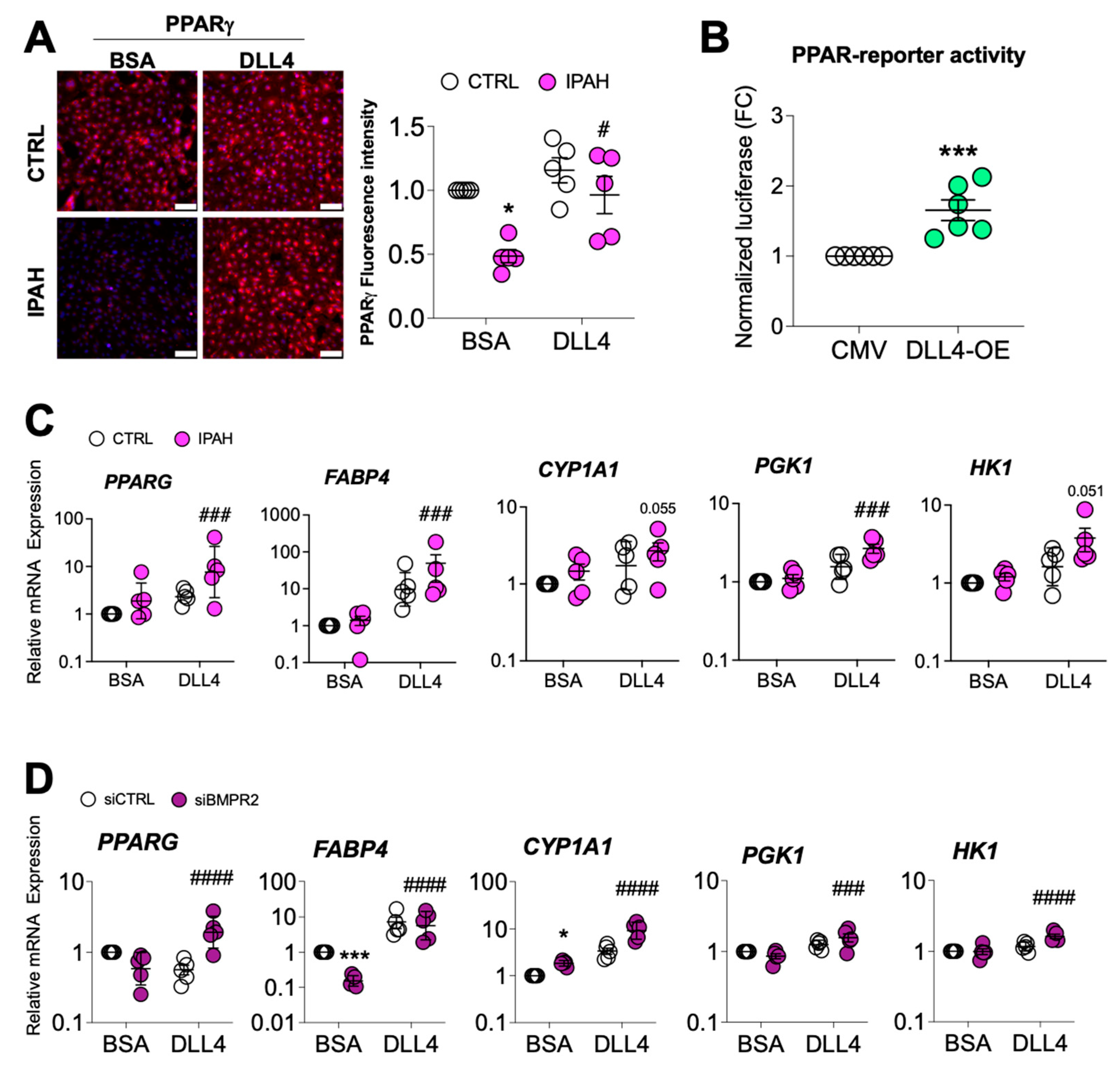
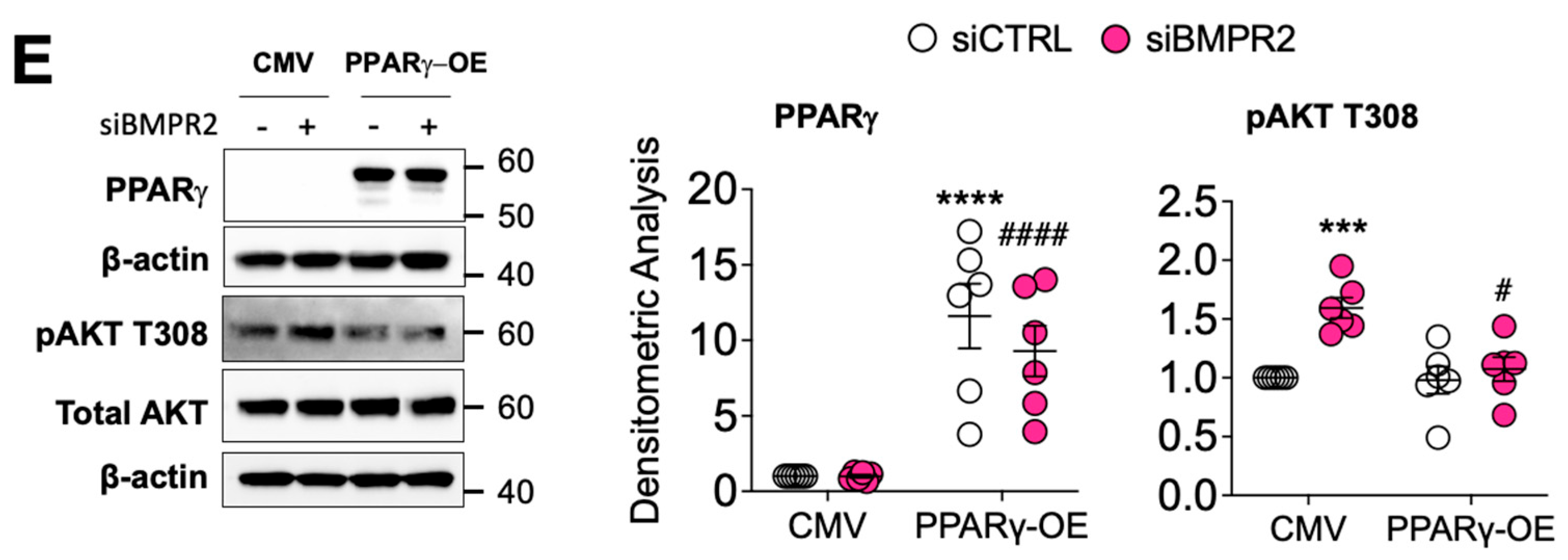
2.5. DLL4/N1ICD Upregulation of PPARγ Expression and Signaling Reduces AKT Activation
3. Discussion and Conclusions
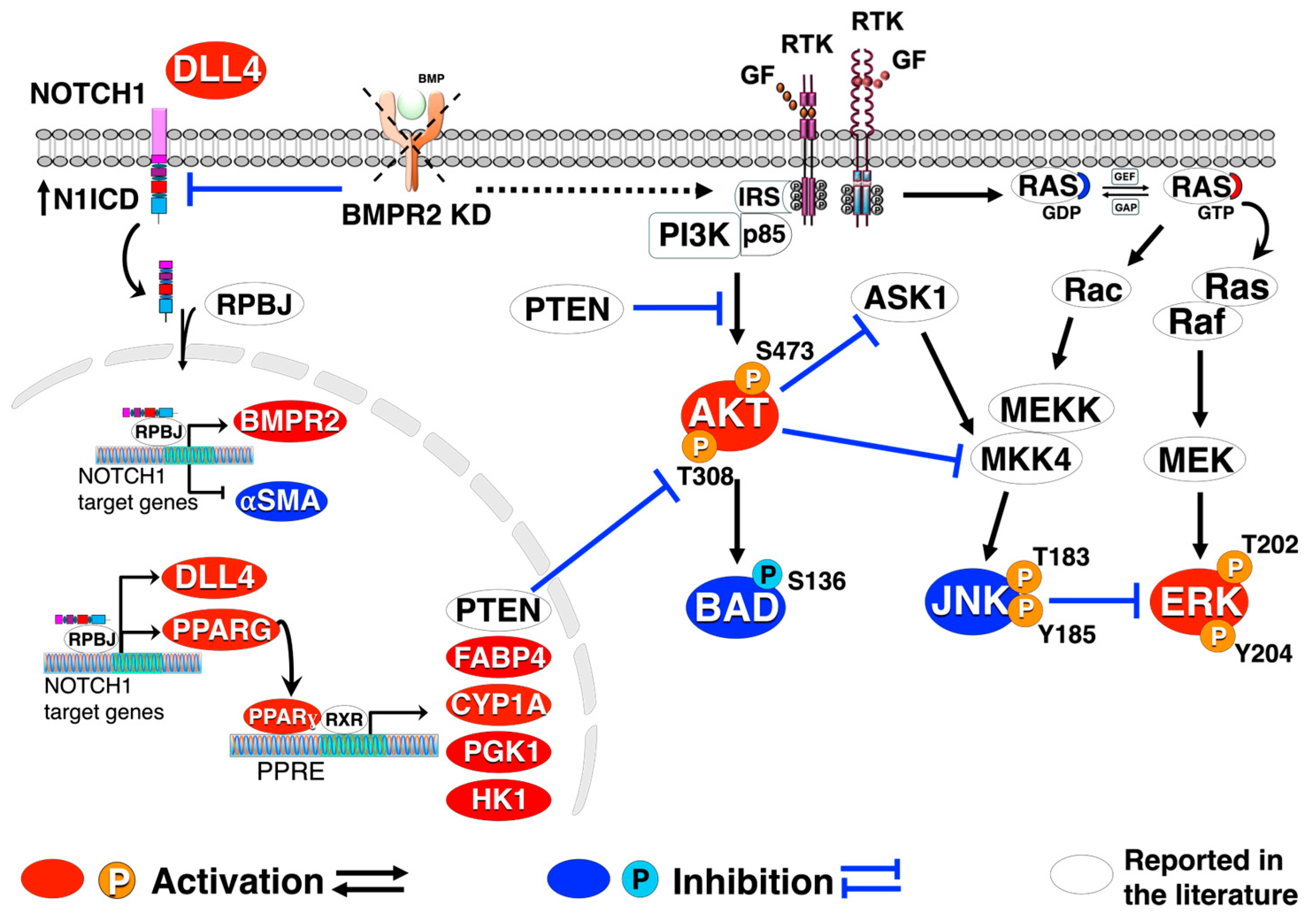
4. Materials and Methods
4.1. Cell Culturing
4.2. Gene Silencing
4.3. Immunofluorescence
4.4. Immunohistochemistry
4.5. Overexpression Studies
4.6. RNA Isolation, cDNA Synthesis and Quantitative Real-Time PCR
4.7. Western Blot
4.8. Cell Proliferation and Cell Apoptosis Assays
4.9. Statistics
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BMPR2 | Bone morphogenetic protein receptor 2 |
| PAH | Pulmonary arterial hypertension |
| PAEC | Pulmonary arterial endothelial cells |
| PI3K | Phosphoinositide 3-kinase |
| NOTCH1 | Neurogenic locus notch homolog protein 1 |
| DLL4 | Delta like canonical Notch ligand 4 |
| BSA | Bovine serum albumin |
| PPARɣ | Peroxisome proliferator activated receptor gamma |
| BrdU | Bromodeoxyuridine |
| S/GFW | Serum: growth factor withdrawal |
| LOF | Loss of function |
| ERK | Extracellular signal-regulated kinases |
| EndoMT | Endothelial to mesenchymal transition |
| CAV1 | Caveolin 1 |
| SMAD9 | Mothers against decapentaplegic homolog 9 |
| RTK | Receptor tyrosine kinases |
References
- Archer, S.; Rich, S. Primary pulmonary hypertension: A vascular biology and translational research “Work in progress”. Circulation 2000, 102, 2781–2791. [Google Scholar] [CrossRef]
- Bisserier, M.; Pradhan, N.; Hadri, L. Current and emerging therapeutic approaches to pulmonary hypertension. Rev. Cardiovasc. Med. 2020, 21, 163–179. [Google Scholar] [PubMed]
- Wilken, E.; Bennji, S.; Symons, G.; Williams, P.G.; Allwood, B. Treatment of pulmonary arterial hypertension: A review of drugs available for advanced therapy. Afr. J. Thorac. Crit. Care Med. 2019, 25, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Sakao, S.; Taraseviciene-Stewart, L.; Lee, J.D.; Wood, K.; Cool, C.D.; Voelkel, N.F. Initial apoptosis is followed by increased proliferation of apoptosis-resistant endothelial cells. FASEB J. 2005, 19, 1178–1180. [Google Scholar] [CrossRef]
- Tang, H.; Chen, J.; Fraidenburg, D.R.; Song, S.; Sysol, J.R.; Drennan, A.R.; Offermanns, S.; Ye, R.D.; Bonini, M.G.; Minshall, R.D.; et al. Deficiency of Akt1, but not Akt2, attenuates the development of pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L208–L220. [Google Scholar] [CrossRef]
- Garat, C.V.; Crossno, J.T., Jr.; Sullivan, T.M.; Reusch, J.E.; Klemm, D.J. Inhibition of phosphatidylinositol 3-kinase/Akt signaling attenuates hypoxia-induced pulmonary artery remodeling and suppresses CREB depletion in arterial smooth muscle cells. J. Cardiovasc. Pharmacol. 2013, 62, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Gu, Y.; Luo, J.; Ye, P.; Zheng, Y.; Yu, W.; Chen, S. Inhibition of Src activation reverses pulmonary vascular remodeling in experimental pulmonary arterial hypertension via Akt/mTOR/HIF-1<alpha> signaling pathway. Exp. Cell Res. 2019, 380, 36–46. [Google Scholar] [PubMed]
- Goncharov, D.A.; Kudryashova, T.V.; Ziai, H.; Ihida-Stansbury, K.; DeLisser, H.; Krymskaya, V.P.; Tuder, R.M.; Kawut, S.M.; Goncharova, E.A. Mammalian target of rapamycin complex 2 (mTORC2) coordinates pulmonary artery smooth muscle cell metabolism, proliferation, and survival in pulmonary arterial hypertension. Circulation 2014, 129, 864–874. [Google Scholar] [CrossRef]
- Budhiraja, R.; Tuder, R.M.; Hassoun, P.M. Endothelial dysfunction in pulmonary hypertension. Circulation. 2004, 109, 159–165. [Google Scholar] [CrossRef]
- Shiojima, I.; Walsh, K. Role of Akt signaling in vascular homeostasis and angiogenesis. Circ. Res. 2002, 90, 1243–1250. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef]
- Liu, Z.J.; Xiao, M.; Balint, K.; Soma, A.; Pinnix, C.C.; Capobianco, A.J.; Velazquez, O.C.; Herlyn, M. Inhibition of endothelial cell proliferation by Notch1 signaling is mediated by repressing MAPK and PI3K/Akt pathways and requires MAML1. FASEB J. 2006, 20, 1009–1011. [Google Scholar] [CrossRef]
- Bray, S.J. Notch signalling in context. Nat. Rev. Mol. Cell Biol. 2016, 17, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Noseda, M.; Chang, L.; McLean, G.; Grim, J.E.; Clurman, B.E.; Smith, L.L.; Karsan, A. Notch activation induces endothelial cell cycle arrest and participates in contact inhibition: Role of p21Cip1 repression. Mol. Cell Biol. 2004, 24, 8813–8822. [Google Scholar] [CrossRef] [PubMed]
- Briot, A.; Civelek, M.; Seki, A.; Hoi, K.; Mack, J.J.; Lee, S.D.; Kim, J.; Hong, C.; Yu, J.; Fishbein, G.A.; et al. Endothelial NOTCH1 is suppressed by circulating lipids and antagonizes inflammation during atherosclerosis. J. Exp. Med. 2015, 212, 2147–2163. [Google Scholar] [CrossRef]
- Mack, J.J.; Mosqueiro, T.S.; Archer, B.J.; Jones, W.M.; Sunshine, H.; Faas, G.C.; Briot, A.; Aragon, R.L.; Su, T.; Romay, M.C.; et al. NOTCH1 is a mechanosensor in adult arteries. Nat. Commun. 2017, 8, 1620. [Google Scholar] [CrossRef] [PubMed]
- Polacheck, W.J.; Kutys, M.L.; Yang, J.; Eyckmans, J.; Wu, Y.; Vasavada, H.; Hirschi, K.K.; Chen, C.S. A non-canonical Notch complex regulates adherens junctions and vascular barrier function. Nature 2017, 552, 258–262. [Google Scholar] [CrossRef]
- Shutter, J.R.; Scully, S.; Fan, W.; Richards, W.G.; Kitajewski, J.; Deblandre, G.A.; Kintner, C.R.; Stark, K.L. Dll4, a novel Notch ligand expressed in arterial endothelium. Genes Dev. 2000, 14, 1313–1318. [Google Scholar] [CrossRef]
- Sainson, R.C.; Johnston, D.A.; Chu, H.C.; Holderfield, M.T.; Nakatsu, M.N.; Crampton, S.P.; Davis, J.; Conn, E.; Hughes, C.C. TNF primes endothelial cells for angiogenic sprouting by inducing a tip cell phenotype. Blood. 2008, 111, 4997–5007. [Google Scholar] [CrossRef]
- Deng, J.; Liu, X.; Rong, L.; Ni, C.; Li, X.; Yang, W.; Lu, Y.; Yan, X.; Qin, C.; Zhang, L.; et al. IFNgamma-responsiveness of endothelial cells leads to efficient angiostasis in tumours involving down-regulation of Dll4. J. Pathol. 2014, 233, 170–182. [Google Scholar] [CrossRef]
- Soon, E.; Holmes, A.M.; Treacy, C.M.; Doughty, N.J.; Southgate, L.; Machado, R.D.; Trembath, R.C.; Jennings, S.; Barker, L.; Nicklin, P.; et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 2010, 122, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Elinoff, J.M.; Mazer, A.J.; Cai, R.; Lu, M.; Graninger, G.; Harper, B.; Ferreyra, G.A.; Sun, J.; Solomon, M.A.; Danner, R.L. Meta-analysis of blood genome-wide expression profiling studies in pulmonary arterial hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 318, L98–L111. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.C.; Eisenberg, P.D.; Manikhas, G.; Chugh, R.; Gubens, M.A.; Stagg, R.J.; Kapoun, A.M.; Xu, L.; Dupont, J.; Sikic, B. A phase I dose escalation and expansion study of the anticancer stem cell agent demcizumab (anti-DLL4) in patients with previously treated solid tumors. Clin. Cancer Res. 2014, 20, 6295–6303. [Google Scholar] [CrossRef] [PubMed]
- Chiorean, E.G.; LoRusso, P.; Strother, R.M.; Diamond, J.R.; Younger, A.; Messersmith, W.A.; Adriaens, L.; Liu, L.; Kao, R.J.; DiCioccio, A.T.; et al. A Phase I First-in-Human Study of Enoticumab (REGN421), a Fully Human Delta-like Ligand 4 (Dll4) Monoclonal Antibody in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2015, 21, 2695–2703. [Google Scholar] [CrossRef] [PubMed]
- McKeage, M.J.; Kotasek, D.; Markman, B.; Hidalgo, M.; Millward, M.J.; Jameson, M.B.; Harris, D.L.; Stagg, R.J.; Kapoun, A.M.; Xu, L.; et al. Phase IB Trial of the Anti-Cancer Stem Cell DLL4-Binding Agent Demcizumab with Pemetrexed and Carboplatin as First-Line Treatment of Metastatic Non-Squamous NSCLC. Target Oncol. 2018, 13, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, A.; Moore, K.N.; Gordon, M.; Chugh, R.; Diamond, J.R.; Aljumaily, R.; Mendelson, D.; Kapoun, A.M.; Xu, L.; Stagg, R.; et al. A first-in-human phase 1a study of the bispecific anti-DLL4/anti-VEGF antibody navicixizumab (OMP-305B83) in patients with previously treated solid tumors. Investig. New Drugs. 2019, 37, 461–472. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Wu, J.; Jiang, G.; Guo, J.; Wang, S.; Li, L.; Ge, J.; Zou, Y. Olmesartan attenuates cardiac remodeling through DLL4/Notch1 pathway activation in pressure overload mice. J. Cardiovasc. Pharmacol. 2013, 61, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Nasu, T.; Sonoda, H.; Ito, K.M.; Ikeda, M.; Ito, K. Evaluation of olmesartan medoxomil in the rat monocrotaline model of pulmonary hypertension. J. Cardiovasc. Pharmacol. 2008, 51, 18–23. [Google Scholar] [CrossRef]
- Miyagawa, K.; Shi, M.; Chen, P.I.; Hennigs, J.K.; Zhao, Z.; Wang, M.; Li, C.G.; Saito, T.; Taylor, S.; Sa, S.; et al. Smooth Muscle Contact Drives Endothelial Regeneration by BMPR2-Notch1-Mediated Metabolic and Epigenetic Changes. Circ. Res. 2019, 124, 211–224. [Google Scholar] [CrossRef]
- Wang, S.; Zhu, G.; Jiang, D.; Rhen, J.; Li, X.; Liu, H.; Lyu, Y.; Tsai, P.; Rose, Y.; Nguyen, T.; et al. Reduced Notch1 Cleavage Promotes the Development of Pulmonary Hypertension. Hypertension 2022, 79, 79–92. [Google Scholar] [CrossRef]
- Hiepen, C.; Jatzlau, J.; Hildebrandt, S.; Kampfrath, B.; Goktas, M.; Murgai, A.; Cuellar Camacho, J.L.; Haag, R.; Ruppert, C.; Sengle, G.; et al. BMPR2 acts as a gatekeeper to protect endothelial cells from increased TGFbeta responses and altered cell mechanics. PLoS Biol. 2019, 17, e3000557. [Google Scholar] [CrossRef] [PubMed]
- Kokeny, G.; Calvier, L.; Legchenko, E.; Chouvarine, P.; Mozes, M.M.; Hansmann, G. PPARgamma is a gatekeeper for extracellular matrix and vascular cell homeostasis: Beneficial role in pulmonary hypertension and renal/cardiac/pulmonary fibrosis. Curr. Opin. Nephrol. Hypertens. 2020, 29, 171–179. [Google Scholar] [CrossRef]
- Hansmann, G.; de Jesus Perez, V.A.; Alastalo, T.P.; Alvira, C.M.; Guignabert, C.; Bekker, J.M.; Schellong, S.; Urashima, T.; Wang, L.; Morrell, N.W.; et al. An antiproliferative BMP-2/PPARgamma/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J. Clin. Investig. 2008, 118, 1846–1857. [Google Scholar] [CrossRef] [PubMed]
- Ameshima, S.; Golpon, H.; Cool, C.D.; Chan, D.; Vandivier, R.W.; Gardai, S.J.; Wick, M.; Nemenoff, R.A.; Geraci, M.W.; Voelkel, N.F. Peroxisome proliferator-activated receptor gamma (PPARgamma) expression is decreased in pulmonary hypertension and affects endothelial cell growth. Circ. Res. 2003, 92, 1162–1169. [Google Scholar] [CrossRef] [PubMed]
- Aldred, M.A.; Vijayakrishnan, J.; James, V.; Soubrier, F.; Gomez-Sanchez, M.A.; Martensson, G.; Galie, N.; Manes, A.; Corris, P.; Simonneau, G.; et al. BMPR2 gene rearrangements account for a significant proportion of mutations in familial and idiopathic pulmonary arterial hypertension. Hum. Mutat. 2006, 27, 212–213. [Google Scholar] [CrossRef] [PubMed]
- Awad, K.S.; Elinoff, J.M.; Wang, S.; Gairhe, S.; Ferreyra, G.A.; Cai, R.; Sun, J.; Solomon, M.A.; Danner, R.L. Raf/ERK drives the proliferative and invasive phenotype of BMPR2-silenced pulmonary artery endothelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L187–L201. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.F.; Wang, J.; Tony To, S.S. The phosphatidylinositol 3-kinase/Akt and c-Jun N-terminal kinase signaling in cancer: Alliance or contradiction? (Review). Int. J. Oncol. 2015, 47, 429–436. [Google Scholar] [CrossRef]
- Rao, V.K.; Webster, S.; Dalm, V.; Sediva, A.; van Hagen, P.M.; Holland, S.; Rosenzweig, S.D.; Christ, A.D.; Sloth, B.; Cabanski, M.; et al. Effective "activated PI3Kdelta syndrome"-targeted therapy with the PI3Kdelta inhibitor leniolisib. Blood 2017, 130, 2307–2316. [Google Scholar] [CrossRef]
- Rao, V.K.; Webster, S.; Sediva, A.; Plebani, A.; Schuetz, C.; Shcherbina, A.; Conlon, N.; Coulter, T.; Dalm, V.A.; Trizzino, A.; et al. A randomized, placebo-controlled phase 3 trial of the PI3Kdelta inhibitor leniolisib for activated PI3Kdelta syndrome. Blood 2023, 141, 971–983. [Google Scholar] [CrossRef]
- Gairhe, S.; Awad, K.S.; Dougherty, E.J.; Ferreyra, G.A.; Wang, S.; Yu, Z.X.; Takeda, K.; Demirkale, C.Y.; Torabi-Parizi, P.; Austin, E.D.; et al. Type I interferon activation and endothelial dysfunction in caveolin-1 insufficiency-associated pulmonary arterial hypertension. Proc. Natl. Acad. Sci. USA 2021, 118, e2010206118. [Google Scholar] [CrossRef]
- Humbert, M.; Morrell, N.W.; Archer, S.L.; Stenmark, K.R.; MacLean, M.R.; Lang, I.M.; Christman, B.W.; Weir, E.K.; Eickelberg, O.; Voelkel, N.F.; et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2004, 43, 13S–24S. [Google Scholar] [CrossRef] [PubMed]
- Ranchoux, B.; Antigny, F.; Rucker-Martin, C.; Hautefort, A.; Pechoux, C.; Bogaard, H.J.; Dorfmuller, P.; Remy, S.; Lecerf, F.; Plante, S.; et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 2015, 131, 1006–1018. [Google Scholar] [CrossRef] [PubMed]
- Varnum-Finney, B.; Wu, L.; Yu, M.; Brashem-Stein, C.; Staats, S.; Flowers, D.; Griffin, J.D.; Bernstein, I.D. Immobilization of Notch ligand, Delta-1, is required for induction of notch signaling. J. Cell Sci. 2000, 113 Pt 23, 4313–4318. [Google Scholar] [CrossRef] [PubMed]
- Quillard, T.; Devalliere, J.; Chatelais, M.; Coulon, F.; Seveno, C.; Romagnoli, M.; Barille Nion, S.; Charreau, B. Notch2 signaling sensitizes endothelial cells to apoptosis by negatively regulating the key protective molecule survivin. PLoS ONE 2009, 4, e8244. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.; Li, Y.; de Jesus, D.; Sembrat, J.; Rojas, M.M.; Goncharova, E.; Cifuentes-Pagano, E.; Straub, A.C.; Pagano, P.J. Notch2 suppression mimicking changes in human pulmonary hypertension modulates Notch1 and promotes endothelial cell proliferation. Am. J. Physiol. Heart Circ. Physiol. 2021, 321, H542–H557. [Google Scholar] [CrossRef] [PubMed]
- Hamid, R.; Cogan, J.D.; Hedges, L.K.; Austin, E.; Phillips, J.A., 3rd; Newman, J.H.; Loyd, J.E. Penetrance of pulmonary arterial hypertension is modulated by the expression of normal BMPR2 allele. Hum. Mutat. 2009, 30, 649–654. [Google Scholar] [CrossRef] [PubMed]
- International PPH Consortium; Lane, K.B.; Machado, R.D.; Pauciulo, M.W.; Thomson, J.R.; Phillips, J.A., 3rd; Loyd, J.E.; Nichols, W.C.; Trembath, R.C. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat. Genet. 2000, 26, 81–84. [Google Scholar] [CrossRef]
- Atkinson, C.; Stewart, S.; Upton, P.D.; Machado, R.; Thomson, J.R.; Trembath, R.C.; Morrell, N.W. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation 2002, 105, 1672–1678. [Google Scholar] [CrossRef]
- Berghausen, E.M.; Janssen, W.; Vantler, M.; Gnatzy-Feik, L.L.; Krause, M.; Behringer, A.; Joseph, C.; Zierden, M.; Freyhaus, H.T.; Klinke, A.; et al. Disrupted PI3K subunit p110alpha signaling protects against pulmonary hypertension and reverses established disease in rodents. J. Clin. Investig. 2021, 131, e136939. [Google Scholar] [CrossRef]
- Zhabyeyev, P.; Chen, X.; Vanhaesebroeck, B.; Oudit, G.Y. PI3Kalpha in cardioprotection: Cytoskeleton, late Na(+) current, and mechanism of arrhythmias. Channels 2019, 13, 520–532. [Google Scholar] [CrossRef]
- Dougherty, E.J.; Chen, L.Y.; Awad, K.S.; Ferreyra, G.A.; Demirkale, C.Y.; Keshavarz, A.; Gairhe, S.; Johnston, K.A.; Hicks, M.E.; Sandler, A.B.; et al. Inflammation and DKK1-induced AKT activation contribute to endothelial dysfunction following NR2F2 loss. Am. J. Physiol. Lung Cell Mol. Physiol. 2023, 324, L783–L798. [Google Scholar] [CrossRef] [PubMed]
- Meester, J.A.; Southgate, L.; Stittrich, A.B.; Venselaar, H.; Beekmans, S.J.; den Hollander, N.; Bijlsma, E.K.; Helderman-van den Enden, A.; Verheij, J.B.; Glusman, G.; et al. Heterozygous Loss-of-Function Mutations in DLL4 Cause Adams-Oliver Syndrome. Am. J. Hum. Genet. 2015, 97, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Stittrich, A.B.; Lehman, A.; Bodian, D.L.; Ashworth, J.; Zong, Z.; Li, H.; Lam, P.; Khromykh, A.; Iyer, R.K.; Vockley, J.G.; et al. Mutations in NOTCH1 cause Adams-Oliver syndrome. Am. J. Hum. Genet. 2014, 95, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Welch, C.L.; Wang, J.; Allen, P.M.; Gonzaga-Jauregui, C.; Ma, L.; King, A.K.; Krishnan, U.; Rosenzweig, E.B.; Ivy, D.D.; et al. Rare variants in SOX17 are associated with pulmonary arterial hypertension with congenital heart disease. Genome Med. 2018, 10, 56. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.M.; Park, C.S.; Kim, S.H.; Noh, T.W.; Kim, J.H.; Park, S.; Lee, J.; Park, J.R.; Yoo, D.; Jung, H.H.; et al. Dll4 Suppresses Transcytosis for Arterial Blood-Retinal Barrier Homeostasis. Circ. Res. 2020, 126, 767–783. [Google Scholar] [CrossRef] [PubMed]
- Corada, M.; Orsenigo, F.; Morini, M.F.; Pitulescu, M.E.; Bhat, G.; Nyqvist, D.; Breviario, F.; Conti, V.; Briot, A.; Iruela-Arispe, M.L.; et al. Sox17 is indispensable for acquisition and maintenance of arterial identity. Nat. Commun. 2013, 4, 2609. [Google Scholar] [CrossRef] [PubMed]
- Gale, N.W.; Dominguez, M.G.; Noguera, I.; Pan, L.; Hughes, V.; Valenzuela, D.M.; Murphy, A.J.; Adams, N.C.; Lin, H.C.; Holash, J.; et al. Haploinsufficiency of delta-like 4 ligand results in embryonic lethality due to major defects in arterial and vascular development. Proc. Natl. Acad. Sci. USA 2004, 101, 15949–15954. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, K.; Satoh, M.; Ii, M.; Silver, M.; Limbourg, F.P.; Mukai, Y.; Rikitake, Y.; Radtke, F.; Gridley, T.; Losordo, D.W.; et al. Critical role of endothelial Notch1 signaling in postnatal angiogenesis. Circ. Res. 2007, 100, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Dabral, S.; Tian, X.; Kojonazarov, B.; Savai, R.; Ghofrani, H.A.; Weissmann, N.; Florio, M.; Sun, J.; Jonigk, D.; Maegel, L.; et al. Notch1 signalling regulates endothelial proliferation and apoptosis in pulmonary arterial hypertension. Eur. Respir. J. 2016, 48, 1137–1149. [Google Scholar] [CrossRef]
- Kopan, R.; Ilagan, M.X. The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef]
- Sangphech, N.; Keawvilai, P.; Palaga, T. Notch signaling increases PPARgamma protein stability and enhances lipid uptake through AKT in IL-4-stimulated THP-1 and primary human macrophages. FEBS Open Bio. 2020, 10, 1082–1095. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.Z.; Usher, M.G.; Mortensen, R.M. Peroxisome proliferator-activated receptor-gamma-mediated effects in the vasculature. Circ. Res. 2008, 102, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Guignabert, C.; Alvira, C.M.; Alastalo, T.P.; Sawada, H.; Hansmann, G.; Zhao, M.; Wang, L.; El-Bizri, N.; Rabinovitch, M. Tie2-mediated loss of peroxisome proliferator-activated receptor-gamma in mice causes PDGF receptor-beta-dependent pulmonary arterial muscularization. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L1082–L1090. [Google Scholar] [CrossRef]
- Legchenko, E.; Chouvarine, P.; Borchert, P.; Fernandez-Gonzalez, A.; Snay, E.; Meier, M.; Maegel, L.; Mitsialis, S.A.; Rog-Zielinska, E.A.; Kourembanas, S.; et al. PPARgamma agonist pioglitazone reverses pulmonary hypertension and prevents right heart failure via fatty acid oxidation. Sci. Transl. Med. 2018, 10, eaao0303. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Wang, G.; Zhang, D.; Zhang, Y.; Zhu, Y.; Li, F.; Li, S.; Li, M. Activation of peroxisome proliferator-activated receptor gamma ameliorates monocrotaline-induced pulmonary arterial hypertension in rats. Biomed. Rep. 2015, 3, 537–542. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Umesono, K.; Noonan, D.J.; Heyman, R.A.; Evans, R.M. Convergence of 9-cis retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature 1992, 358, 771–774. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Awad, K.S.; Wang, S.; Dougherty, E.J.; Keshavarz, A.; Demirkale, C.Y.; Yu, Z.X.; Miller, L.; Elinoff, J.M.; Danner, R.L. BMPR2 Loss Activates AKT by Disrupting DLL4/NOTCH1 and PPARγ Signaling in Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2024, 25, 5403. https://doi.org/10.3390/ijms25105403
Awad KS, Wang S, Dougherty EJ, Keshavarz A, Demirkale CY, Yu ZX, Miller L, Elinoff JM, Danner RL. BMPR2 Loss Activates AKT by Disrupting DLL4/NOTCH1 and PPARγ Signaling in Pulmonary Arterial Hypertension. International Journal of Molecular Sciences. 2024; 25(10):5403. https://doi.org/10.3390/ijms25105403
Chicago/Turabian StyleAwad, Keytam S., Shuibang Wang, Edward J. Dougherty, Ali Keshavarz, Cumhur Y. Demirkale, Zu Xi Yu, Latonia Miller, Jason M. Elinoff, and Robert L. Danner. 2024. "BMPR2 Loss Activates AKT by Disrupting DLL4/NOTCH1 and PPARγ Signaling in Pulmonary Arterial Hypertension" International Journal of Molecular Sciences 25, no. 10: 5403. https://doi.org/10.3390/ijms25105403
APA StyleAwad, K. S., Wang, S., Dougherty, E. J., Keshavarz, A., Demirkale, C. Y., Yu, Z. X., Miller, L., Elinoff, J. M., & Danner, R. L. (2024). BMPR2 Loss Activates AKT by Disrupting DLL4/NOTCH1 and PPARγ Signaling in Pulmonary Arterial Hypertension. International Journal of Molecular Sciences, 25(10), 5403. https://doi.org/10.3390/ijms25105403