
Delineated 3-1-BenCarMethInYlPro-Phosphonic Acid’s Adroit Activity against Lung Cancer through Multitargeted Docking, MM\GBSA, QM-DFT and Multiscale Simulations
 , and
, and
Abstract
1. Introduction
2. Results
2.1. Protein Structural Analysis
2.2. Protein–Ligand Interaction Analysis
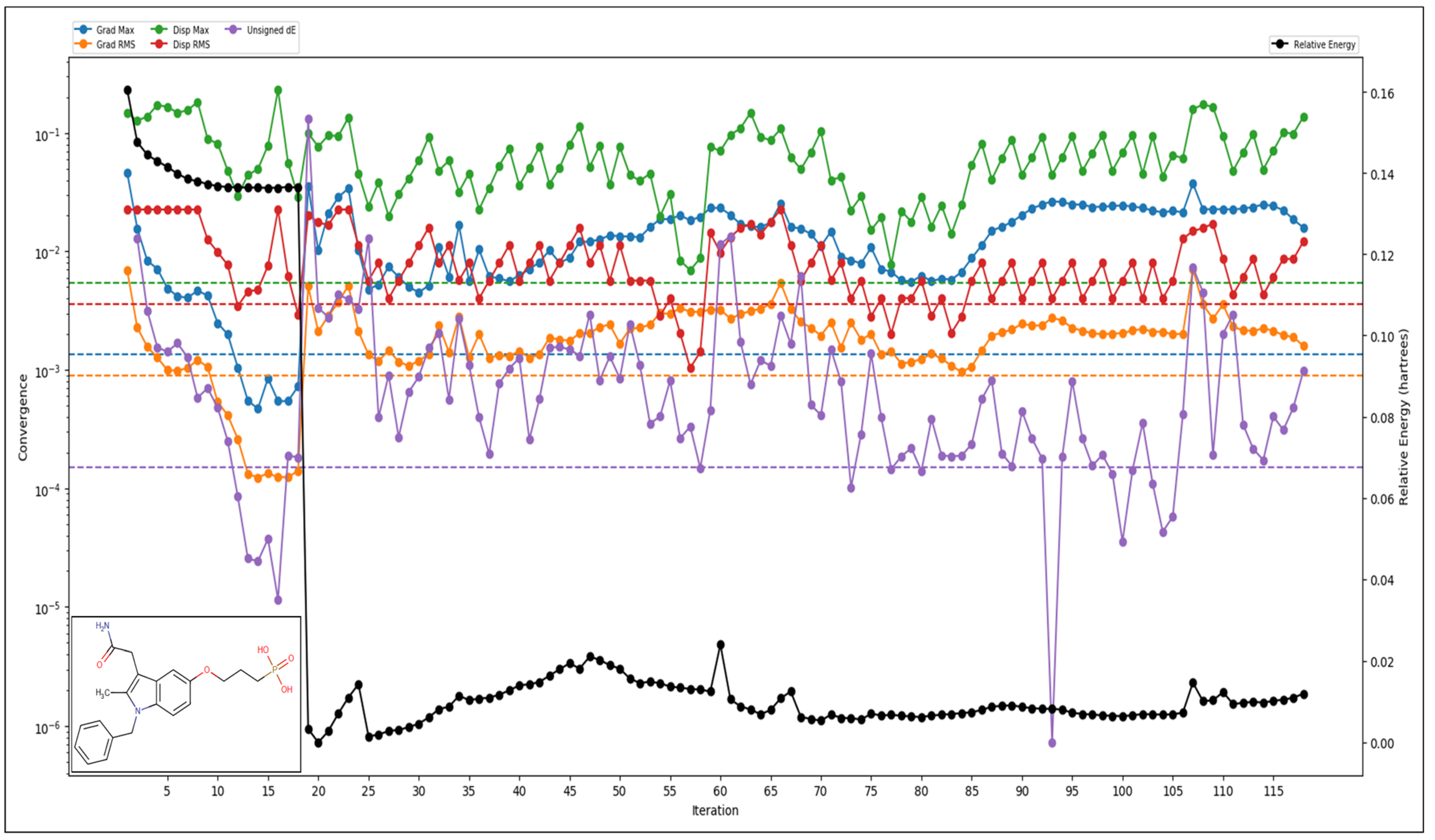
2.3. Pharmacokinetics and QM-Based DFT Studies
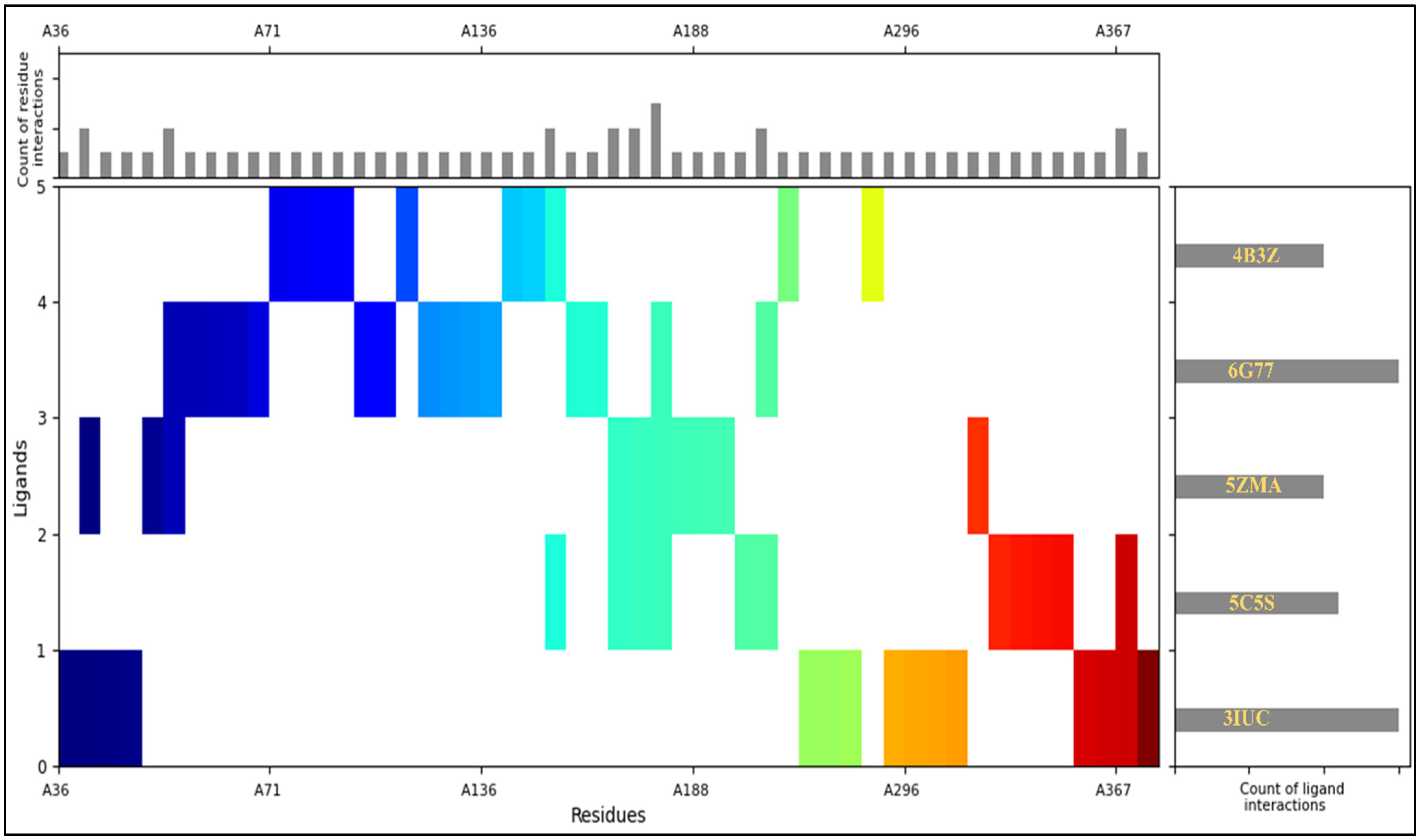
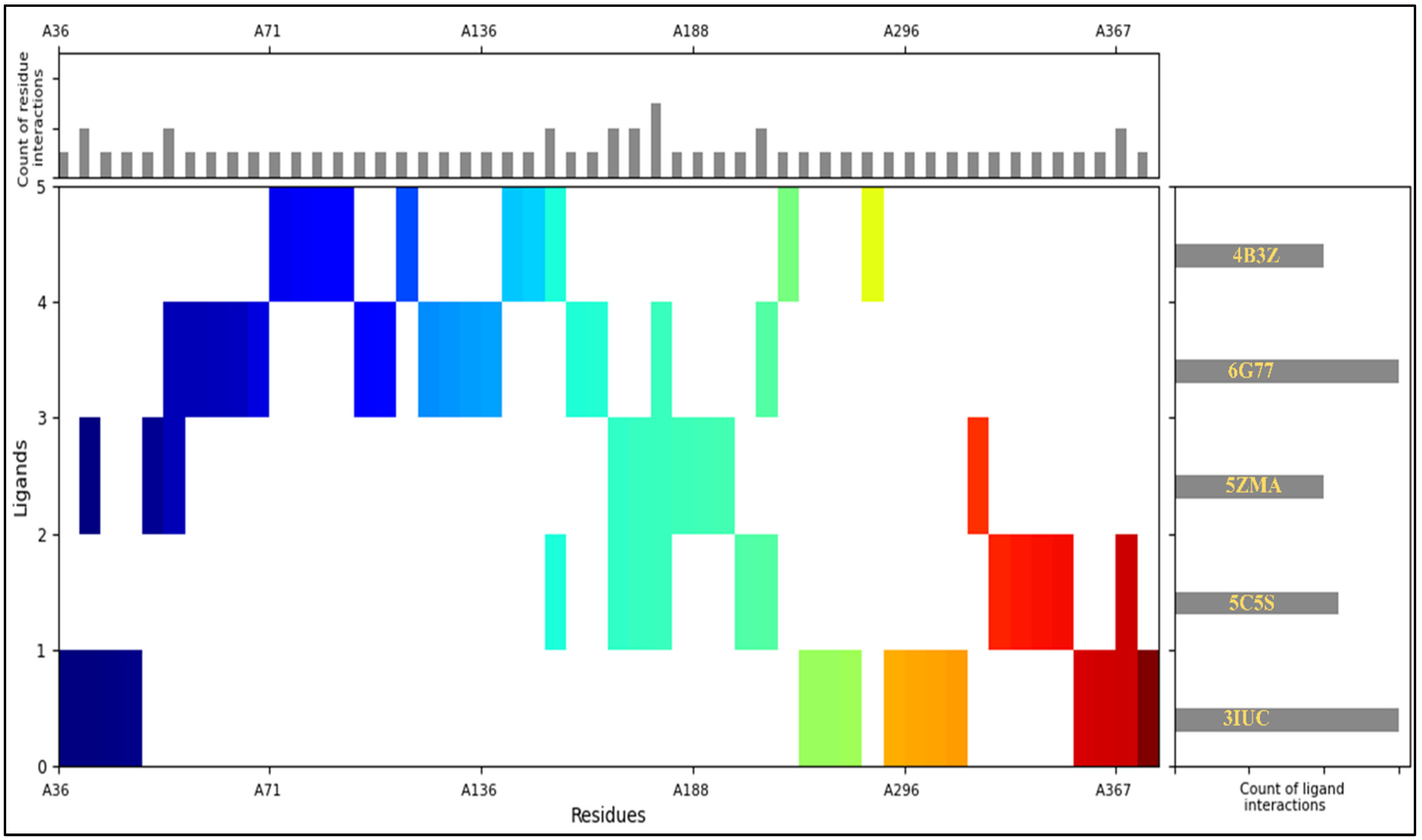
2.4. Interaction Fingerprints
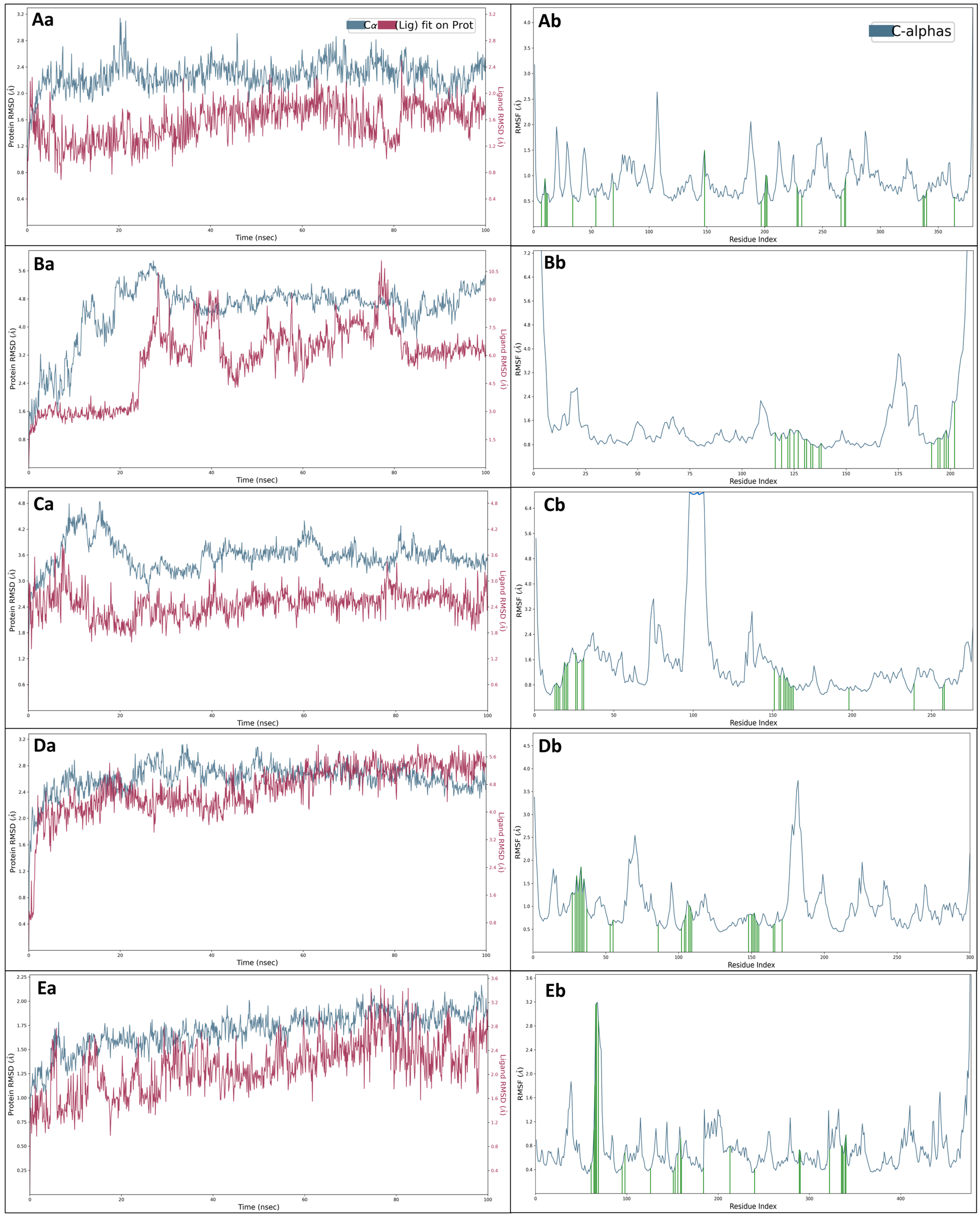
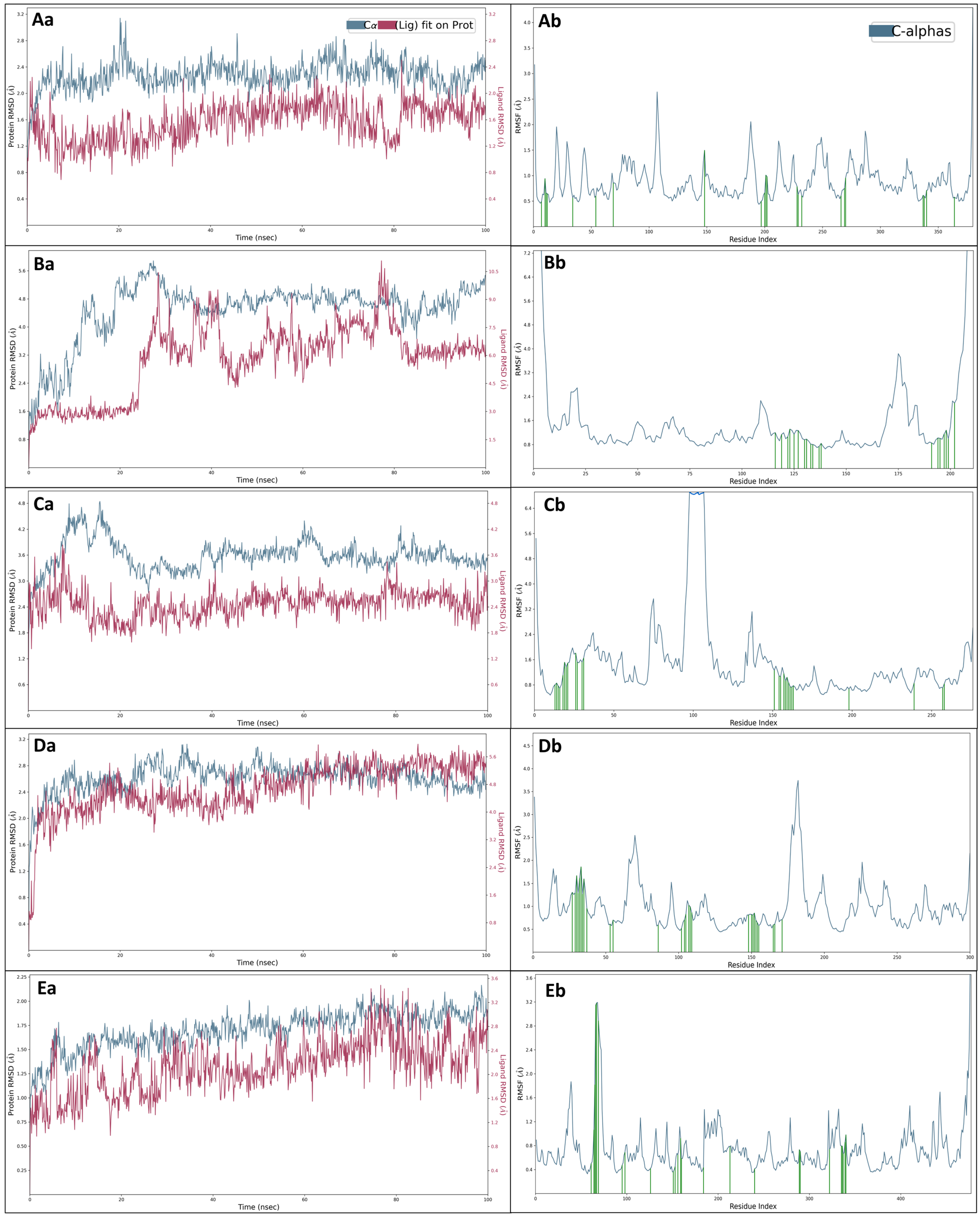
2.5. Molecular Dynamics Simulations
2.5.1. Root Mean Square Deviation (RMSD)
2.5.2. Root Mean Square Fluctuations (RMSF)
2.5.3. Simulation Interaction Diagrams (SID)
3. Discussion
4. Methods and Materials
4.1. Ligand and Protein Preparation and Structural Validation
4.2. Receptor Grid Generation (RGG) and Molecular Docking
4.3. Pharmacokinetics and QM-Based DFT Studies
4.4. Interaction Fingerprints
4.5. Molecular Dynamics (MD) Simulation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Birring, S.; Peake, M. Symptoms and the Early Diagnosis of Lung Cancer; BMJ Publishing Group Ltd.: Hoboken, NJ, USA, 2005; Volume 60, pp. 268–269. [Google Scholar]
- Ahmad, S.; Singh, V.; Gautam, H.K.; Raza, K. Multisampling-based docking reveals Imidazolidinyl urea as a multitargeted inhibitor for lung cancer: An optimisation followed multi-simulation and in-vitro study. J. Biomol. Struct. Dyn. 2023, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.K.; Ahmad, S.; Raza, K.; Kumar, S.; Eswaran, M.; Pasha KM, M. Predictive modeling and therapeutic repurposing of natural compounds against the receptor-binding domain of SARS-CoV-2. J. Biomol. Struct. Dyn. 2023, 41, 1527–1539. [Google Scholar] [CrossRef] [PubMed]
- Oser, M.G.; Niederst, M.J.; Sequist, L.V.; Engelman, J.A. Transformation from non-small-cell lung cancer to small-cell lung cancer: Molecular drivers and cells of origin. Lancet Oncol. 2015, 16, e165–e172. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Bhanu, P.; Kumar, J.; Pathak, R.K.; Mallick, D.; Uttarkar, A.; Niranjan, V.; Mishra, V. Molecular dynamics simulation and docking analysis of NF-κB protein binding with sulindac acid. Bioinformation 2022, 18, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Bano, N.; Qazi, S.; Yadav, M.K.; Ahmad, N.; Raza, K. Multitargeted molecular dynamic understanding of butoxypheser against SARS-CoV-2: An in silico study. Nat. Prod. Commun. 2022, 17, 1934578X221115499. [Google Scholar] [CrossRef]
- Abaza, A.; Idris, F.S.; Shaikh, H.A.; Vahora, I.; Moparthi, K.P.; Al Rushaidi, M.T.; Muddam, M.R.; Obajeun, O.A.; Jaramillo, A.P.; Khan, S. Programmed Cell Death Protein 1 (PD-1) and Programmed Cell Death Ligand 1 (PD-L1) Immunotherapy: A Promising Breakthrough in Cancer Therapeutics. Cureus 2023, 15, e44582. [Google Scholar] [CrossRef] [PubMed]
- Vanka, K.S.; Shukla, S.; Gomez, H.M.; James, C.; Palanisami, T.; Williams, K.; Chambers, D.C.; Britton, W.J.; Ilic, D.; Hansbro, P.M. Understanding the pathogenesis of occupational coal and silica dust-associated lung disease. Eur. Respir. Rev. 2022, 31, 210250. [Google Scholar] [CrossRef]
- Tripathi, M.K.; Ahmad, S.; Tyagi, R.; Dahiya, V.; Yadav, M.K. Fundamentals of molecular modeling in drug design. In Computer Aided Drug Design (CADD): From Ligand-Based Methods to Structure-Based Approaches; Elsevier: Amsterdam, The Netherlands, 2022; pp. 125–155. [Google Scholar]
- Tarique, M.; Ahmad, S.; Malik, A.; Ahmad, I.; Saeed, M.; Almatroudi, A.; Qadah, T.; Murad, M.A.; Mashraqi, M.; Alam, Q. Novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV2) and other coronaviruses: A genome-wide comparative annotation and analysis. Mol. Cell. Biochem. 2021, 476, 2203–2217. [Google Scholar] [CrossRef]
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo Clin. Proc. 2008, 83, 584–594. [Google Scholar] [CrossRef]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Ahmad, S.; Khan, F.N.; Ramlal, A.; Begum, S.; Qazi, S.; Raza, K. Nanoinformatics and nanomodeling: Recent developments in computational nanodrug design and delivery systems. In Emerging Nanotechnologies for Medical Applications; Elsevier: Amsterdam, The Netherlands, 2023; pp. 297–332. [Google Scholar]
- Ahmad, S.; Dahiya, V.; Vibhuti, A.; Pandey, R.P.; Tripathi, M.K.; Yadav, M.K. Therapeutic Protein-Based Vaccines. In Protein-Based Therapeutics; Springer: Berlin/Heidelberg, Germany, 2023; pp. 355–384. [Google Scholar]
- Ahmad, S.; Chitkara, P.; Khan, F.N.; Kishan, A.; Alok, V.; Ramlal, A.; Mehta, S. Mobile technology solution for COVID-19: Surveillance and prevention. In Computational Intelligence Methods in COVID-19: Surveillance, Prevention, Prediction and Diagnosis; Springer: Singapore, 2021; pp. 79–108. [Google Scholar]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.P.; Ahmad, S.; Raza, K.; Gautam, H.K. Computational screening and MM/GBSA-based MD simulation studies reveal the high binding potential of FDA-approved drugs against Cutibacterium acnes sialidase. J. Biomol. Struct. Dyn. 2023, 1–11. [Google Scholar] [CrossRef]
- Sheikh, K.; Sayeed, S.; Asif, A.; Siddiqui, M.F.; Rafeeq, M.M.; Sahu, A.; Ahmad, S. Consequential Innovations in Nature-Inspired Intelligent Computing Techniques for Biomarkers and Potential Therapeutics Identification. In Nature-Inspired Intelligent Computing Techniques in Bioinformatics; Springer Nature: Singapore, 2022; pp. 247–274. [Google Scholar]
- Shah, A.A.; Ahmad, S.; Yadav, M.K.; Raza, K.; Kamal, M.A.; Akhtar, S. Structure-based virtual screening, molecular docking, molecular dynamics simulation, and metabolic reactivity studies of quinazoline derivatives for their anti-EGFR activity against tumor angiogenesis. Curr. Med. Chem. 2024, 31, 595–619. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Brambilla, E.; Faivre-Finn, C.; Sage, J. Small-cell lung cancer. Nat. Rev. Dis. Primers 2021, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.C.; Kim, S.H.; Kim, T.-J.; Kim, H.K.; Moon, M.H.; Beck, K.S.; Suh, Y.-G.; Song, C.; Ahn, J.S.; Lee, J.E. Five-year overall survival and prognostic factors in patients with lung cancer: Results from the Korean Association of Lung Cancer Registry (KALC-R) 2015. Cancer Res. Treat. 2022, 55, 103–111. [Google Scholar]
- Karwasra, R.; Ahmad, S.; Bano, N.; Qazi, S.; Raza, K.; Singh, S.; Varma, S. Macrophage-targeted punicalagin nanoengineering to alleviate methotrexate-induced neutropenia: A molecular docking, DFT, and MD simulation analysis. Molecules 2022, 27, 6034. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Sheikh, K.; Bano, N.; Rafeeq, M.M.; Mohammed, M.R.S.; Yadav, M.K.; Raza, K. Illustrious Implications of Nature-Inspired Computing Methods in Therapeutics and Computer-Aided Drug Design. In Nature-Inspired Intelligent Computing Techniques in Bioinformatics; Springer Nature: Singapore, 2022; pp. 293–308. [Google Scholar]
- Ahmad, S.; Sayeed, S.; Bano, N.; Sheikh, K.; Raza, K. In-silico analysis reveals Quinic acid as a multitargeted inhibitor against cervical cancer. J. Biomol. Struct. Dyn. 2023, 41, 9770–9786. [Google Scholar] [CrossRef] [PubMed]
- Wisniewska, M.; Karlberg, T.; Lehtiö, L.; Johansson, I.; Kotenyova, T.; Moche, M.; Schüler, H. Crystal structures of the ATPase domains of four human Hsp70 isoforms: HSPA1L/Hsp70-hom, HSPA2/Hsp70-2, HSPA6/Hsp70B', and HSPA5/BiP/GRP78. PLoS ONE 2010, 5, e8625. [Google Scholar] [CrossRef]
- Kong, R.; Yi, F.; Wen, P.; Liu, J.; Chen, X.; Ren, J.; Li, X.; Shang, Y.; Nie, Y.; Wu, K. Myo9b is a key player in SLIT/ROBO-mediated lung tumor suppression. J. Clin. Investig. 2015, 125, 4407–4420. [Google Scholar] [CrossRef]
- Anantharajan, J.; Zhou, H.; Zhang, L.; Hotz, T.; Vincent, M.Y.; Blevins, M.A.; Jansson, A.E.; Kuan, J.W.L.; Ng, E.Y.; Yeo, Y.K. Structural and functional analyses of an allosteric EYA2 phosphatase inhibitor that has on-target effects in human lung cancer cells. Mol. Cancer Ther. 2019, 18, 1484–1496. [Google Scholar] [CrossRef]
- Chrysostomou, S.; Roy, R.; Prischi, F.; Thamlikitkul, L.; Chapman, K.L.; Mufti, U.; Peach, R.; Ding, L.; Hancock, D.; Moore, C. Repurposed floxacins targeting RSK4 prevent chemoresistance and metastasis in lung and bladder cancer. Sci. Transl. Med. 2021, 13, eaba4627. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-H.; Huang, S.-F.; Hsu, Y.-L.; Pan, S.-H.; Chen, Y.-J.; Lin, Y.-H. Structure of human collapsin response mediator protein 1: A possible role of its C-terminal tail. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2015, 71, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Alzamami, A.; Alturki, N.A.; Alghamdi, Y.S.; Ahmad, S.; Alshamrani, S.; Asiri, S.A.; Mashraqi, M.M. Hemi-Babim and fenoterol as potential inhibitors of MPro and papain-like protease against SARS-CoV-2: An in-silico study. Medicina 2022, 58, 515. [Google Scholar] [CrossRef] [PubMed]
- Alturki, N.A.; Mashraqi, M.M.; Alzamami, A.; Alghamdi, Y.S.; Alharthi, A.A.; Asiri, S.A.; Ahmad, S.; Alshamrani, S. In-silico screening and molecular dynamics simulation of drug bank experimental compounds against SARS-CoV-2. Molecules 2022, 27, 4391. [Google Scholar] [CrossRef] [PubMed]
- Alghamdi, Y.S.; Mashraqi, M.M.; Alzamami, A.; Alturki, N.A.; Ahmad, S.; Alharthi, A.A.; Alshamrani, S.; Asiri, S.A. Unveiling the multitargeted potential of N-(4-Aminobutanoyl)-S-(4-methoxybenzyl)-L-cysteinylglycine (NSL-CG) against SARS CoV-2: A virtual screening and molecular dynamics simulation study. J. Biomol. Struct. Dyn. 2023, 41, 6633–6642. [Google Scholar] [CrossRef]
- Release, S. LigPrep; Schrödinger, LLC: New York, NY, USA, 2022. [Google Scholar]
- Release, S. Maestro, Schrödinger, 2022; Maestro S, LLC: New York, NY, USA, 2022. [Google Scholar]
- Ramlal, A.; Ahmad, S.; Kumar, L.; Khan, F.N.; Chongtham, R. From molecules to patients: The clinical applications of biological databases and electronic health records. In Translational Bioinformatics in Healthcare and Medicine; Academic Press: Cambridge, MA, USA, 2021; pp. 107–125. [Google Scholar]
- Jorgensen, W.L.; Tirado-Rives, J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK a prediction and protonation state generation for drug-like molecules. J. Comput.-Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Berman, H.; Henrick, K.; Kleywegt, G.; Nakamura, H.; Markley, J. RCSB PDB. 2012. Available online: http://rcsb.org/ (accessed on 15 December 2023).
- Balasubramanian, B.; Ahmad, S.; Alok, V.; Khan, F.N.; Anand, K.; Mehta, S.; Easwaran, M.; Meyyazhagan, A.; Saravanan, M. Exosomes as an emerging nanoplatform for functional therapeutics. In Handbook on Nanobiomaterials for Therapeutics and Diagnostic Applications; Elsevier: Amsterdam, The Netherlands, 2021; pp. 483–498. [Google Scholar]
- Release, S. Protein Preparation Wizard; Epik, Schrödinger, LLC: New York, NY, USA, 2022. [Google Scholar]
- Rana, M.; Ahmedi, S.; Fatima, A.; Ahmad, S.; Siddiqui, N.; Raza, K.; Manzoor, N.; Javed, S. Synthesis, single crystal, TD-DFT, molecular dynamics simulation and DNA binding studies of carbothioamide analog. J. Mol. Struct. 2023, 1287, 135701. [Google Scholar] [CrossRef]
- Li, H.; Robertson, A.D.; Jensen, J.H. Very fast empirical prediction and rationalization of protein pKa values. Proteins: Struct. Funct. Bioinform. 2005, 61, 704–721. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein−ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Glide; Schrödinger LLC: New York, NY, USA, 2022.
- QikProp; Schrödinger LLC: New York, NY, USA, 2022.
- Lipinski, C.A. Lead-and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Raza, K. Identification of 5-nitroindazole as a multitargeted inhibitor for CDK and transferase kinase in lung cancer: A multisampling algorithm-based structural study. Mol. Divers. 2023, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Pasha Km, M.; Raza, K.; Rafeeq, M.M.; Habib, A.H.; Eswaran, M.; Yadav, M.K. Reporting dinaciclib and theodrenaline as a multitargeted inhibitor against SARS-CoV-2: An in-silico study. J. Biomol. Struct. Dyn. 2023, 41, 4013–4023. [Google Scholar] [CrossRef] [PubMed]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Famuyiwa, S.O.; Ahmad, S.; Fakola, E.G.; Olusola, A.J.; Adesida, S.A.; Obagunle, F.O.; Raza, K.; Ugwo, J.P.; Oyelekan, E.I.; Faloye, K.O. Comprehensive computational studies of naturally occurring kuguacins as antidiabetic agents by targeting visfatin. Chem. Afr. 2023, 6, 1415–1427. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006. [Google Scholar]
- Römer, F.; Lervik, A.; Bresme, F. Nonequilibrium molecular dynamics simulations of the thermal conductivity of water: A systematic investigation of the SPC/E and TIP4P/2005 models. J. Chem. Phys. 2012, 137, 074503. [Google Scholar] [CrossRef]
- Jorgensen, W.L. Convergence of Monte Carlo simulations of liquid water in the NPT ensemble. Chem. Phys. Lett. 1982, 92, 405–410. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNo | PDBID | Docking Score | MMGBSA dG Bind | Ligand Efficiency Sa | Ligand Efficiency Ln | Evdw | Ecoul |
|---|---|---|---|---|---|---|---|
| 1 | 3IUC | −9.02 | −39.68 | −2.05 | −9.09 | −42.64 | −26.63 |
| 2 | 5C5S | −5.83 | −36.30 | −1.88 | −8.31 | −31.05 | −14.89 |
| 3 | 5ZMA | −7.47 | −7.56 | −0.39 | −1.73 | −30.30 | −6.92 |
| 4 | 6G77 | −10.66 | −50.14 | −2.59 | −11.48 | −38.69 | −22.65 |
| 5 | 4B3Z | −9.71 | −43.56 | −2.25 | −9.97 | −21.72 | −19.85 |
| Descriptor | Standard Values | 3-1-BenCarMethInYlPro-Phosphonic Acid | Descriptor | Standard Values | 3-1-BenCarMethInYlPro-Phosphonic Acid |
|---|---|---|---|---|---|
| Type | N/A | Small | glob | 0.75–0.95 | 0.7929173 |
| #amidine | 0 | 0 | EA(eV) | −0.9–1.7 | −0.03 |
| HumanOralAbsorption | - | 1 | #metab | 1–8 | 5 |
| #ringatoms | - | 15 | dipole | 1.0–12.5 | 3.074 |
| #in34 | - | 0 | QPlogKhsa | −1.5–1.5 | −0.703 |
| #in56 | - | 15 | QPpolrz | 13.0–70.0 | 40.839 |
| #noncon | - | 0 | mol MW | 130.0–725.0 | 416.413 |
| #nonHatm | - | 29 | #NandO | 2–15 | 7 |
| Jm | - | 0.014479409 | CNS | −2 (inactive), +2 (active) | −2 |
| QPPCaco | <25 poor, >500 great | 1.58 | accptHB | 2.0–20.0 | 8.25 |
| QPPMDCK | <25 poor, >500 great | 1.55 | QPlogPo/w | −2.0–6.5 | 2.523 |
| %HumanOralAbsorption | >80% = high, <25% = poor | 45.275 | QPlogBB | −3.0–1.2 | −2.697 |
| #amine | 0–1 | 0 | SASA | 300.0–1000.0 | 722.945 |
| #acid | 0–1 | 2 | QPlogPC16 | 4.0–18.0 | 14.715 |
| #amide | 0–1 | 1 | QPlogPw | 4.0–45.0 | 18.956 |
| #rotor | 0–15 | 11 | volume | 500.0–2000.0 | 1290.483 |
| #rtvFG | 0–2 | 0 | QPlogS | −6.5–0.5 | −3.356 |
| #stars | 0–5 | 0 | CIQPlogS | −6.5–0.5 | −4.496 |
| AcxDN0.5/SA | 0.0–0.05 | 0.0228233 | PSA | 7.0–200.0 | 128.095 |
| dip2/V | 0.0–0.13 | 0.0073203 | FISA | 7.0–330.0 | 245.723 |
| SAfluorine | 0.0–100.0 | 0 | IP(eV) | 7.9–10.5 | 8.031 |
| WPSA | 0.0–175.0 | 3.379 | QPlogKp | −8.0–−1.0 | −4.103 |
| SAamideO | 0.0–35.0 | 34.445 | QPlogPoct | 8.0–35.0 | 24.13 |
| PISA | 0.0–450.0 | 252.499 | QPlogHERG | concern below −5 | −1.098 |
| donorHB | 0.0–6.0 | 4 | RuleOfThree | maximum is 3 | 1 |
| FOSA | 0.0–750.0 | 221.344 | RuleOfFive | maximum is 4 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hakami, M.A.; Hazazi, A.; Albloui, F.; Gharib, A.F.; Alsaeedi, F.A.; Abdulaziz, O.; Alhazmi, A.Y.; Alsaiari, A.A. Delineated 3-1-BenCarMethInYlPro-Phosphonic Acid’s Adroit Activity against Lung Cancer through Multitargeted Docking, MM\GBSA, QM-DFT and Multiscale Simulations. Int. J. Mol. Sci. 2024, 25, 592. https://doi.org/10.3390/ijms25010592
Hakami MA, Hazazi A, Albloui F, Gharib AF, Alsaeedi FA, Abdulaziz O, Alhazmi AY, Alsaiari AA. Delineated 3-1-BenCarMethInYlPro-Phosphonic Acid’s Adroit Activity against Lung Cancer through Multitargeted Docking, MM\GBSA, QM-DFT and Multiscale Simulations. International Journal of Molecular Sciences. 2024; 25(1):592. https://doi.org/10.3390/ijms25010592
Chicago/Turabian StyleHakami, Mohammed Ageeli, Ali Hazazi, Fawaz Albloui, Amal F. Gharib, Fouzeyyah Ali Alsaeedi, Osama Abdulaziz, Abdulfattah Y. Alhazmi, and Ahad Amer Alsaiari. 2024. "Delineated 3-1-BenCarMethInYlPro-Phosphonic Acid’s Adroit Activity against Lung Cancer through Multitargeted Docking, MM\GBSA, QM-DFT and Multiscale Simulations" International Journal of Molecular Sciences 25, no. 1: 592. https://doi.org/10.3390/ijms25010592
APA StyleHakami, M. A., Hazazi, A., Albloui, F., Gharib, A. F., Alsaeedi, F. A., Abdulaziz, O., Alhazmi, A. Y., & Alsaiari, A. A. (2024). Delineated 3-1-BenCarMethInYlPro-Phosphonic Acid’s Adroit Activity against Lung Cancer through Multitargeted Docking, MM\GBSA, QM-DFT and Multiscale Simulations. International Journal of Molecular Sciences, 25(1), 592. https://doi.org/10.3390/ijms25010592