
Replication Protein A, the Main Eukaryotic Single-Stranded DNA Binding Protein, a Focal Point in Cellular DNA Metabolism

 ,
,  and
and 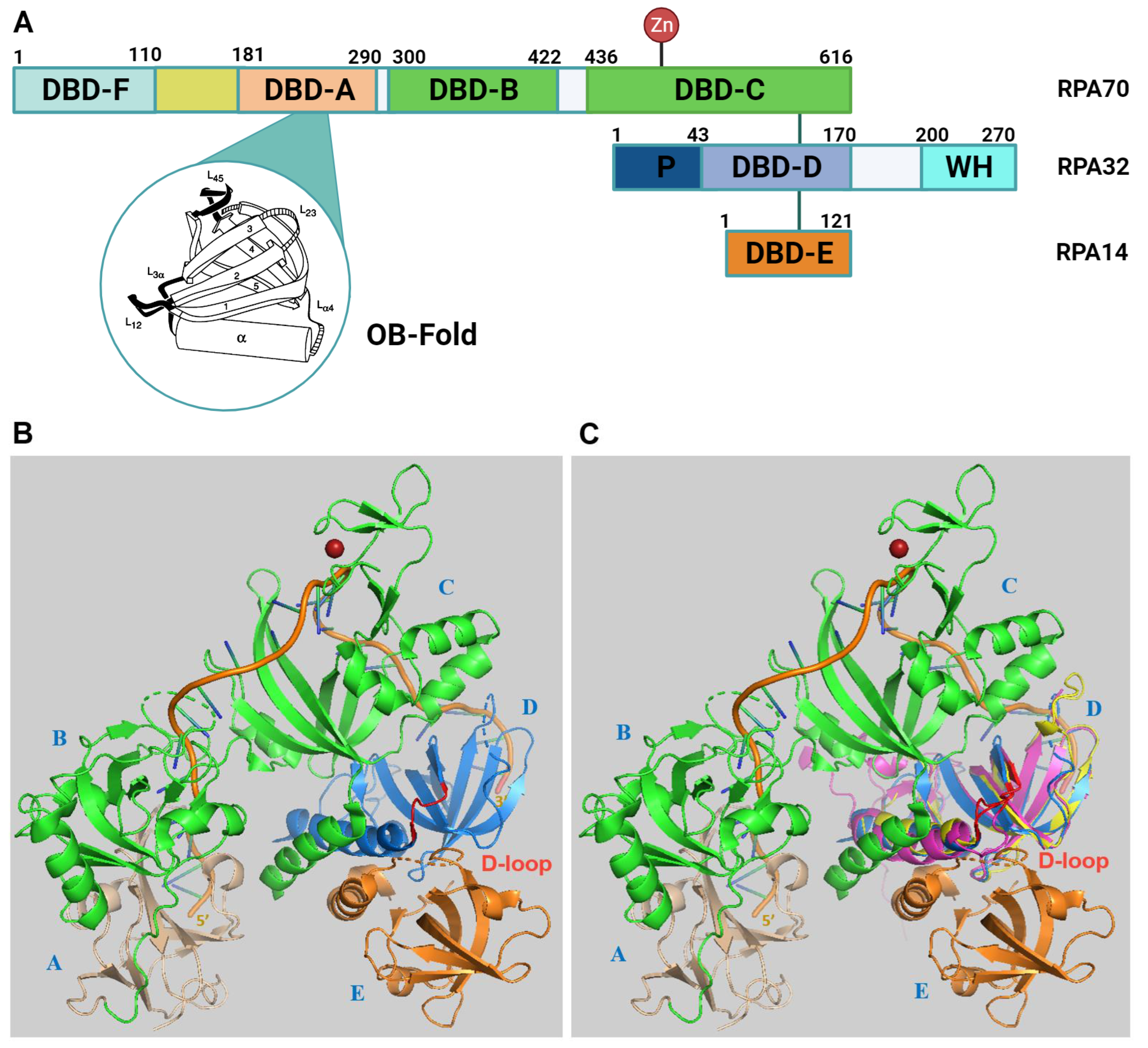{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
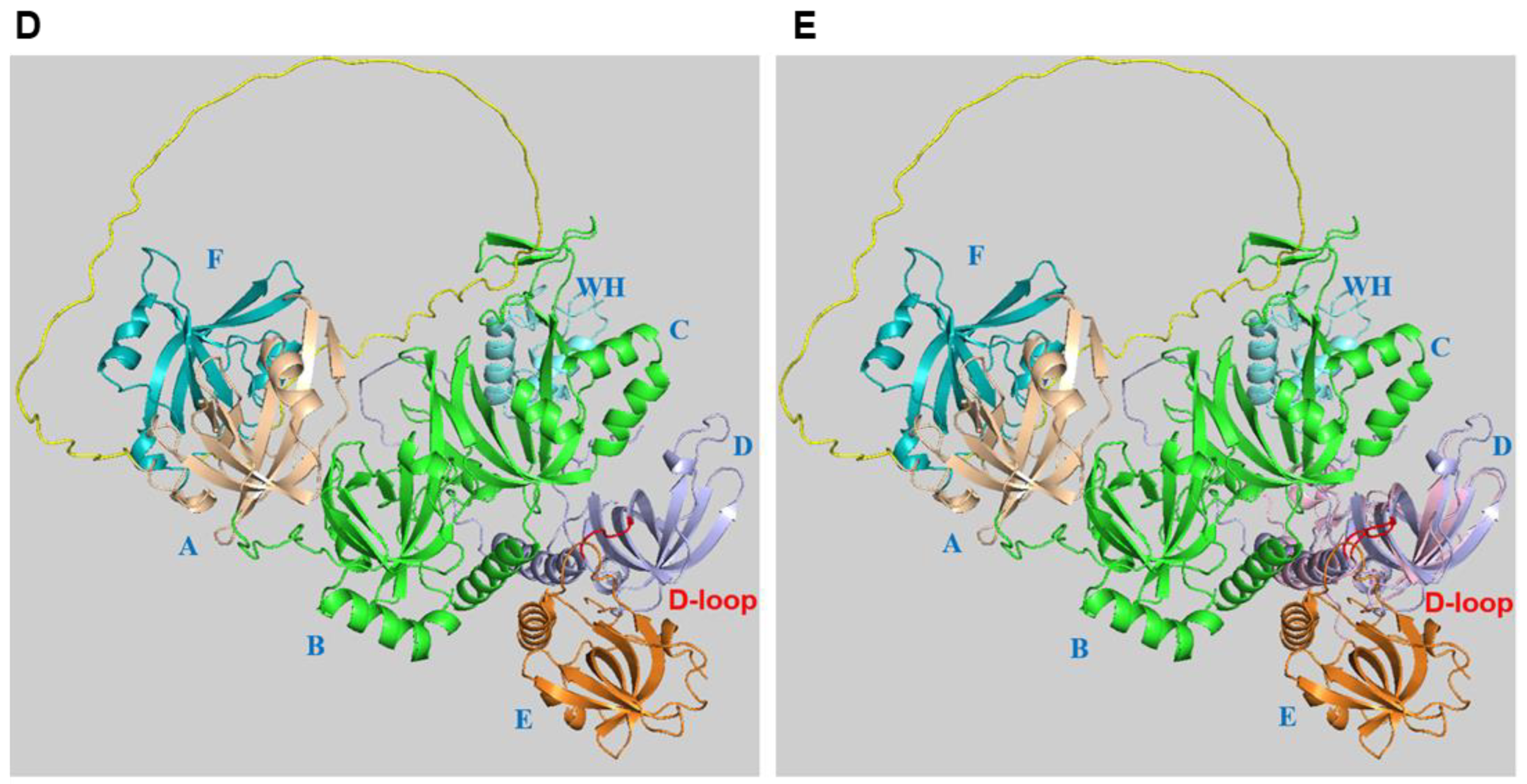
2. RPA Structure, ssDNA and Protein Interactions


3. Alternative RPA and Its Emerging Functions in Neurodegenerative Diseases
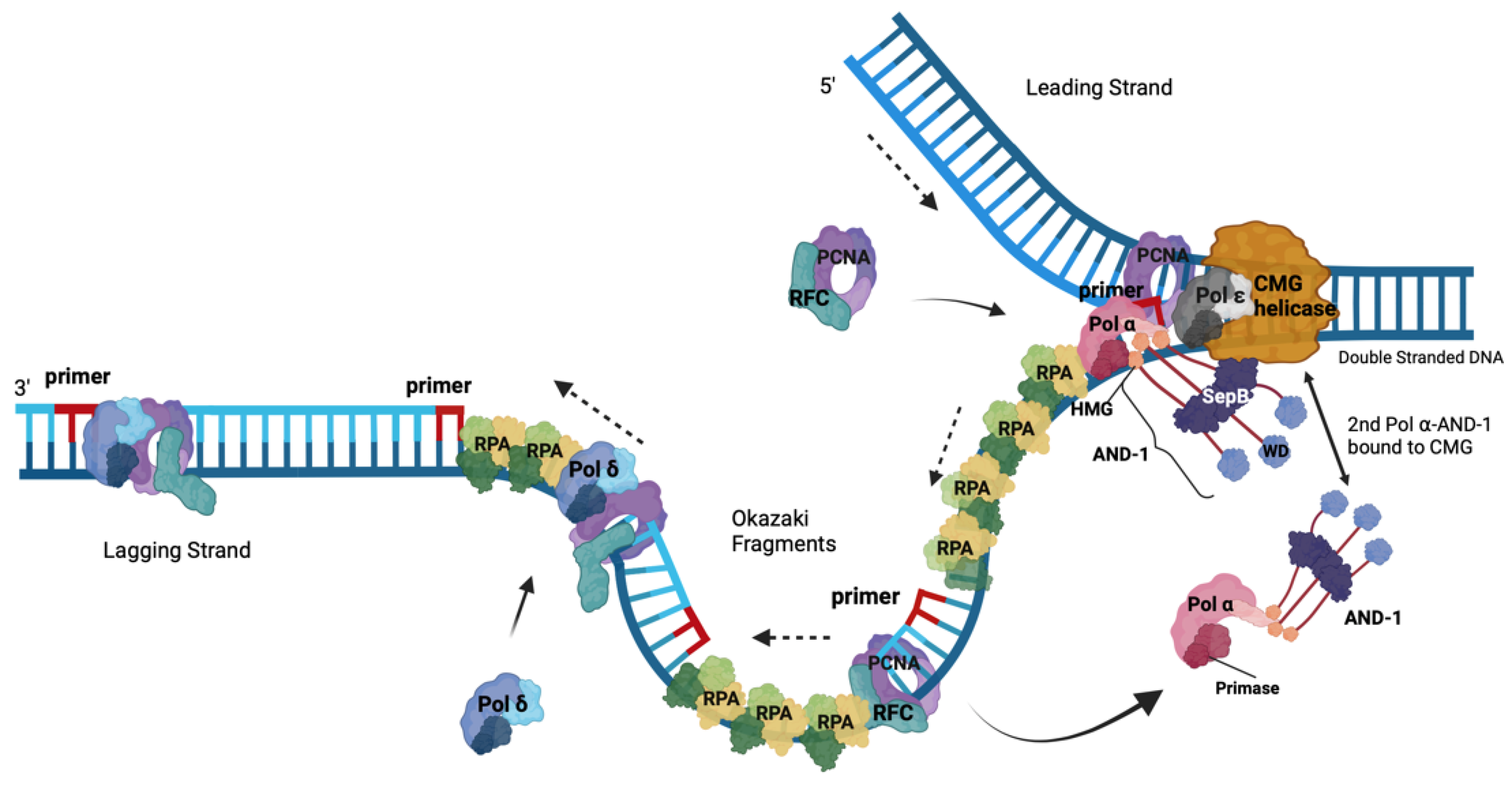
4. Roles of RPA in DNA Replication
5. The Role of the RPA-Related, ssDNA-Binding Protein Complex CST in the Initiation of Okazaki Fragment Synthesis
6. Functions of RPA in DNA Damage Response Pathways
- Double-strand breaks (DSBs) repair has evolved in eukaryotic cells as two main pathways, homologous recombination (HR) and non-homologous end joining (NHEJ). HR utilizes a homologous DNA template to allow for the accurate repair of DSBs whereas NHEJ only re-joins the broken ends, opening the possibility of the induction of insertions or deletions [65,90].
6.1. RPA in DNA Damage Signalling
6.2. RPA in DNA Repair Pathways
7. RPA and Health Prospects
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 9-1-1 | Rad 9-Hus 1-Rad 1 complex (fission yeast and human) equivalent to the |
| complex | Rad 17-Mec3-Ddc1 complex in budding yeast |
| 53BP1 | TP53-binding protein 1 |
| aa | amino acid |
| AND-1-CTF4- | acidic and nucleoplasmic DNA-binding protein-1/Chromosome - |
| WDHD1 | Transmission Fidelity 4/WD repeat and HMG-box DNA-binding protein 1 |
| A-T | ataxia telangiectasia |
| ATM | ataxia telangiectasia-mutated |
| ATR | ATM-Rad 3-related protein |
| ATRIP | ATR interacting protein |
| BER | base excision repair |
| BLM | Bloom’s helicase |
| BRCA1 | breast cancer type 1 susceptibility protein |
| BRCA2 | breast cancer type 1 susceptibility protein |
| CDC | cell division cycle |
| CDK | cyclin-dependent kinase |
| CMG | CDC45-MCM2-6-GINS |
| CST | CTC1-STN1-TEN1 |
| DBD | DNA-binding domain |
| DNA-PK | DNA-dependent protein kinase |
| DDR | DNA damage response |
| DSB | double-strand break |
| dsDNA | double-stranded DNA |
| G4 | G-quadruplexes |
| HR | homologous recombination |
| mt | mitochondrial |
| MCM | minichromosome maintenance |
| MDC1 | mediator of DNA damage checkpoint protein 1 |
| MMEJ | microhomology-mediated end joining |
| hPolprim1 | human primase-polymerase 1 |
| MMR | mismatch repair |
| NER | nucleotide excision repair |
| nt | nucleotide |
| NTD | N-terminal domain |
| OB-fold | oligosaccharide/oligonucleotide-binding fold |
| PCNA | proliferating cell nuclear antigen |
| PIKK | phosphotnositol-3 kinase-like protein kinase |
| Pol α | DNA polymerase α–primase |
| Pol δ | DNA polymerase δ |
| Pol ε | DNA polymerase ε |
| POT1 | protector of telomeres 1 |
| RAD | radiation-induced mutation |
| RFC | replication factor C |
| RFWD3 | RING finger and WD repeat |
| RPA | replication protein A |
| ROS | reactive oxygen species |
| SSB | single-stranded DNA-binding protein |
| ssDNA | single-stranded DNA |
| S. cerivisiae | Saccheromyces cerevisiae |
| SV40 | simian virus 40 |
| Tp53 | tumour suppressor protein p53 |
| TFIIH | transcription factor II H |
| TIP60 | Tat-interactive protein, 60 kD |
| TopBP1 | topoisomerase II-binding protein 1 |
References
- Wold, M.S. Replication protein A: A heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 1997, 66, 61–92. [Google Scholar] [CrossRef] [PubMed]
- Marechal, A.; Zou, L. RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res. 2015, 25, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Kenny, M.K.; Lee, S.H.; Hurwitz, J. Multiple functions of human single-stranded-DNA binding protein in simian virus 40 DNA replication: Single-strand stabilization and stimulation of DNA polymerases alpha and delta. Proc. Natl. Acad. Sci. USA 1989, 86, 9757–9761. [Google Scholar] [CrossRef] [PubMed]
- Fairman, M.P.; Stillman, B. Cellular factors required for multiple stages of SV40 replication in vitro. EMBO J. 1988, 7, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Wold, M.S.; Kelly, T.J. Purification and characterization of replication protein A, a cellular protein required for in vitro replication of simian virus 40 DNA. Proc. Natl. Acad. Sci. USA 1988, 85, 2523–2527. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Snyder, R.O.; Wold, M.S. Binding properties of replication protein A from human and yeast cells. Mol. Cell. Biol. 1992, 12, 3050–3059. [Google Scholar] [PubMed]
- Olson, C.L.; Barbour, A.T.; Wuttke, D.S. Filling in the blanks: How the C-strand catches up to the G-strand at replicating telomeres using CST. Nat. Struct. Mol. Biol. 2022, 29, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Onwubiko, N.O.; Borst, A.; Diaz, S.A.; Passkowski, K.; Scheffel, F.; Tessmer, I.; Nasheuer, H.P. SV40 T antigen interactions with ssDNA and replication protein A: A regulatory role of T antigen monomers in lagging strand DNA replication. Nucleic Acids Res. 2020, 48, 3657–3677. [Google Scholar] [CrossRef]
- Szambowska, A.; Tessmer, I.; Prus, P.; Schlott, B.; Pospiech, H.; Grosse, F. Cdc45-induced loading of human RPA onto single-stranded DNA. Nucleic Acids Res. 2017, 45, 3217–3230. [Google Scholar] [CrossRef]
- Wu, Y.; Fu, W.; Zang, N.; Zhou, C. Structural characterization of human RPA70N association with DNA damage response proteins. eLife 2023, 12, e81639. [Google Scholar] [CrossRef]
- Zhao, J.; Tian, S.; Guo, Q.; Bao, K.; Yu, G.; Wang, X.; Shen, X.; Zhang, J.; Chen, J.; Yang, Y.; et al. A PARylation-phosphorylation cascade promotes TOPBP1 loading and RPA-RAD51 exchange in homologous recombination. Mol. Cell 2022, 82, 2571–2587.e2579. [Google Scholar] [CrossRef] [PubMed]
- Bochkareva, E.; Korolev, S.; Lees-Miller, S.P.; Bochkarev, A. Structure of the RPA trimerization core and its role in the multistep DNA-binding mechanism of RPA. EMBO J. 2002, 21, 1855–1863. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Pavletich, N.P. Structure and conformational change of a replication protein A heterotrimer bound to ssDNA. Genes Dev. 2012, 26, 2337–2347. [Google Scholar] [CrossRef] [PubMed]
- Fanning, E.; Klimovich, V.; Nager, A.R. A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res. 2006, 34, 4126–4137. [Google Scholar] [CrossRef] [PubMed]
- Salas, T.R.; Petruseva, I.; Lavrik, O.; Saintome, C. Evidence for direct contact between the RPA3 subunit of the human replication protein A and single-stranded DNA. Nucleic Acids Res. 2009, 37, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Pestryakov, P.E.; Weisshart, K.; Schlott, B.; Khodyreva, S.N.; Kremmer, E.; Grosse, F.; Lavrik, O.I.; Nasheuer, H.P. Human replication protein A: The C-terminal RPA70 and the central RPA32 domains are involved in the interactions with the 3′-end of a primer-template DNA. J. Biol. Chem. 2003, 278, 17515–17524. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Wold, M.S. Replication protein A: Single-stranded DNA’s first responder: Dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. Bioessays 2014, 36, 1156–1161. [Google Scholar] [CrossRef] [PubMed]
- Pokhrel, N.; Caldwell, C.C.; Corless, E.I.; Tillison, E.A.; Tibbs, J.; Jocic, N.; Tabei, S.M.A.; Wold, M.S.; Spies, M.; Antony, E. Dynamics and selective remodeling of the DNA-binding domains of RPA. Nat. Struct. Mol. Biol. 2019, 26, 129–136. [Google Scholar] [CrossRef]
- Ding, J.; Li, X.; Shen, J.; Zhao, Y.; Zhong, S.; Lai, L.; Niu, H.; Qi, Z. ssDNA accessibility of Rad51 is regulated by orchestrating multiple RPA dynamics. Nat. Commun. 2023, 14, 3864. [Google Scholar] [CrossRef]
- Zaug, A.J.; Goodrich, K.J.; Song, J.J.; Sullivan, A.E.; Cech, T.R. Reconstitution of a telomeric replicon organized by CST. Nature 2022, 608, 819–825. [Google Scholar] [CrossRef]
- Barbour, A.T.; Wuttke, D.S. RPA-like single-stranded DNA-binding protein complexes including CST serve as specialized processivity factors for polymerases. Curr. Opin. Struct. Biol. 2023, 81, 102611. [Google Scholar] [CrossRef] [PubMed]
- Broderick, S.; Rehmet, K.; Concannon, C.; Nasheuer, H.P. Eukaryotic single-stranded DNA binding proteins: Central factors in genome stability. Subcell. Biochem. 2010, 50, 143–163. [Google Scholar] [PubMed]
- Liu, S.; Byrne, B.; Byrne, T.; Oakley, G. Role of RPA Phosphorylation in the ATR-Dependent G2 Cell Cycle Checkpoint. Genes 2023, 14, 2205. [Google Scholar] [CrossRef] [PubMed]
- Murzin, A.G. OB(oligonucleotide/oligosaccharide binding)-fold: Common structural and functional solution for non-homologous sequences. EMBO J. 1993, 12, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Dueva, R.; Iliakis, G. Replication protein A: A multifunctional protein with roles in DNA replication, repair and beyond. NAR Cancer 2020, 2, zcaa022. [Google Scholar] [CrossRef] [PubMed]
- Binz, S.K.; Wold, M.S. Regulatory Functions of the N-terminal Domain of the 70-kDa Subunit of Replication Protein A (RPA). J. Biol. Chem. 2008, 283, 21559–21570. [Google Scholar] [CrossRef]
- Taneja, P.; Boche, I.; Hartmann, H.; Nasheuer, H.P.; Grosse, F.; Fanning, E.; Weisshart, K. Different activities of the largest subunit of replication protein A cooperate during SV40 DNA replication. FEBS Lett. 2007, 581, 3973–3978. [Google Scholar] [CrossRef]
- Shorrocks, A.K.; Jones, S.E.; Tsukada, K.; Morrow, C.A.; Belblidia, Z.; Shen, J.; Vendrell, I.; Fischer, R.; Kessler, B.M.; Blackford, A.N. The Bloom syndrome complex senses RPA-coated single-stranded DNA to restart stalled replication forks. Nat. Commun. 2021, 12, 585. [Google Scholar] [CrossRef]
- Yates, L.A.; Tannous, E.A.; Morgan, R.M.; Burgers, P.M.; Zhang, X. A DNA damage-induced phosphorylation circuit enhances Mec1(ATR) Ddc2(ATRIP) recruitment to Replication Protein A. Proc. Natl. Acad. Sci. USA 2023, 120, e2300150120. [Google Scholar] [CrossRef]
- Dornreiter, I.; Erdile, L.F.; Gilbert, I.U.; von Winkler, D.; Kelly, T.J.; Fanning, E. Interaction of DNA polymerase alpha-primase with cellular replication protein A and SV40 T antigen. EMBO J. 1992, 11, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Braun, K.A.; Lao, Y.; He, Z.; Ingles, C.J.; Wold, M.S. Role of protein-protein interactions in the function of replication protein A (RPA): RPA modulates the activity of DNA polymerase a by multiple mechanisms. Biochemistry 1997, 36, 8443–8454. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Zhou, J.; Fan, J.; Shen, Z.; Chen, X. Streamline proteomic approach for characterizing protein-protein interaction network in a RAD52 protein complex. J. Proteome Res. 2009, 8, 2211–2217. [Google Scholar] [CrossRef] [PubMed]
- Weisshart, K.; Pestryakov, P.; Smith, R.W.; Hartmann, H.; Kremmer, E.; Lavrik, O.; Nasheuer, H.P. Coordinated regulation of replication protein A activities by its subunits p14 and p32. J. Biol. Chem. 2004, 279, 35368–35376. [Google Scholar] [CrossRef] [PubMed]
- Vaithiyalingam, S.; Warren, E.M.; Eichman, B.F.; Chazin, W.J. Insights into eukaryotic DNA priming from the structure and functional interactions of the 4Fe-4S cluster domain of human DNA primase. Proc. Natl. Acad. Sci. USA 2010, 107, 13684–13689. [Google Scholar] [CrossRef] [PubMed]
- Bainbridge, L.J.; Teague, R.; Doherty, A.J. Repriming DNA synthesis: An intrinsic restart pathway that maintains efficient genome replication. Nucleic Acids Res. 2021, 49, 4831–4847. [Google Scholar] [CrossRef] [PubMed]
- Broderick, R.; Rainey, M.D.; Santocanale, C.; Nasheuer, H.P. Cell cycle-dependent formation of Cdc45-Claspin complexes in human cells are compromized by UV-mediated DNA damage. FEBS J. 2013, 280, 4888–4902. [Google Scholar] [CrossRef]
- Wan, L.; Lou, J.; Xia, Y.; Su, B.; Liu, T.; Cui, J.; Sun, Y.; Lou, H.; Huang, J. hPrimpol1/CCDC111 is a human DNA primase-polymerase required for the maintenance of genome integrity. EMBO Rep. 2013, 14, 1104–1112. [Google Scholar] [CrossRef]
- Keshav, K.F.; Chen, C.; Dutta, A. Rpa4, a homolog of the 34-kilodalton subunit of the replication protein A complex. Mol. Cell. Biol. 1995, 15, 3119–3128. [Google Scholar] [CrossRef]
- Kemp, M.G.; Mason, A.C.; Carreira, A.; Reardon, J.T.; Haring, S.J.; Borgstahl, G.E.; Kowalczykowski, S.C.; Sancar, A.; Wold, M.S. An alternative form of replication protein A expressed in normal human tissues supports DNA repair. J. Biol. Chem. 2010, 285, 4788–4797. [Google Scholar] [CrossRef]
- Haring, S.J.; Humphreys, T.D.; Wold, M.S. A naturally occurring human RPA subunit homolog does not support DNA replication or cell-cycle progression. Nucleic Acids Res. 2010, 38, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Gall-Duncan, T.; Luo, J.; Jurkovic, C.M.; Fischer, L.A.; Fujita, K.; Deshmukh, A.L.; Harding, R.J.; Tran, S.; Mehkary, M.; Li, V.; et al. Antagonistic roles of canonical and Alternative-RPA in disease-associated tandem CAG repeat instability. Cell 2023, 186, 4898–4919.e4825. [Google Scholar] [CrossRef]
- Mason, A.C.; Haring, S.J.; Pryor, J.M.; Staloch, C.A.; Gan, T.F.; Wold, M.S. An alternative form of replication protein a prevents viral replication in vitro. J. Biol. Chem. 2009, 284, 5324–5331. [Google Scholar] [CrossRef] [PubMed]
- Mason, A.C.; Roy, R.; Simmons, D.T.; Wold, M.S. Functions of alternative replication protein A in initiation and elongation. Biochemistry 2010, 49, 5919–5928. [Google Scholar] [CrossRef] [PubMed]
- Möncke-Buchner, E.; Reich, S.; Mücke, M.; Reuter, M.; Messer, W.; Wanker, E.E.; Krüger, D.H. Counting CAG repeats in the Huntington’s disease gene by restriction endonuclease EcoP15I cleavage. Nucleic Acids Res. 2002, 30, e83. [Google Scholar] [CrossRef] [PubMed]
- Kenney, C.; Powell, S.; Jankovic, J. Autopsy-proven Huntington’s disease with 29 trinucleotide repeats. Mov. Disord. 2007, 22, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Cummings, D.M.; Alaghband, Y.; Hickey, M.A.; Joshi, P.R.; Hong, S.C.; Zhu, C.; Ando, T.K.; André, V.M.; Cepeda, C.; Watson, J.B.; et al. A critical window of CAG repeat-length correlates with phenotype severity in the R6/2 mouse model of Huntington’s disease. J. Neurophysiol. 2012, 107, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Morton, A.J.; Skillings, E.A.; Wood, N.I.; Zheng, Z. Antagonistic pleiotropy in mice carrying a CAG repeat expansion in the range causing Huntington’s disease. Sci. Rep. 2019, 9, 37. [Google Scholar] [CrossRef]
- Bleichert, F.; Botchan, M.R.; Berger, J.M. Mechanisms for initiating cellular DNA replication. Science 2017, 355, eaah6317. [Google Scholar] [CrossRef]
- Nasheuer, H.P.; Smith, R.; Bauerschmidt, C.; Grosse, F.; Weisshart, K. Initiation of eukaryotic DNA replication: Regulation and mechanisms. Prog. Nucleic Acid. Res. Mol. Biol. 2002, 72, 41–94. [Google Scholar]
- Vipat, S.; Gupta, D.; Jonchhe, S.; Anderspuk, H.; Rothenberg, E.; Moiseeva, T.N. The non-catalytic role of DNA polymerase epsilon in replication initiation in human cells. Nat. Commun. 2022, 13, 7099. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.L.; Aria, V.; Baris, Y.; Yeeles, J.T.P. How Pol α-primase is targeted to replisomes to prime eukaryotic DNA replication. Mol. Cell 2023, 83, 2911–2924.e16. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.L.; Baris, Y.; Taylor, M.R.G.; Yeeles, J.T.P. Structure of a human replisome shows the organisation and interactions of a DNA replication machine. EMBO J. 2021, 40, e108819. [Google Scholar] [CrossRef] [PubMed]
- Evrin, C.; Alvarez, V.; Ainsworth, J.; Fujisawa, R.; Alabert, C.; Labib, K.P. DONSON is required for CMG helicase assembly in the mammalian cell cycle. EMBO Rep. 2023, 24, e57677. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Sadano, K.; Miyata, N.; Ito, H.; Tanaka, H. Novel role of DONSON in CMG helicase assembly during vertebrate DNA replication initiation. EMBO J. 2023, 42, e114131. [Google Scholar] [CrossRef] [PubMed]
- Kingsley, G.; Skagia, A.; Passaretti, P.; Fernandez-Cuesta, C.; Reynolds-Winczura, A.; Koscielniak, K.; Gambus, A. DONSON facilitates Cdc45 and GINS chromatin association and is essential for DNA replication initiation. Nucleic Acids Res. 2023, 51, 9748–9763. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.; Tamayo-Orrego, L.; Schmid, E.; Tarnauskaite, Z.; Kochenova, O.V.; Gruar, R.; Muramatsu, S.; Lynch, L.; Schlie, A.V.; Carroll, P.L.; et al. In silico protein interaction screening uncovers DONSON’s role in replication initiation. Science 2023, 381, eadi3448. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Sonneville, R.; Jenkyn-Bedford, M.; Ji, L.; Alabert, C.; Hong, Y.; Yeeles, J.T.P.; Labib, K.P.M. DNSN-1 recruits GINS for CMG helicase assembly during DNA replication initiation in Caenorhabditis elegans. Science 2023, 381, eadi4932. [Google Scholar] [CrossRef]
- Lavrik, O.I.; Kolpashchikov, D.M.; Nasheuer, H.P.; Weisshart, K.; Favre, A. Alternative conformations of human replication protein A are detected by crosslinks with primers carrying a photoreactive group at the 3′-end. FEBS Lett. 1998, 441, 186–190. [Google Scholar] [CrossRef]
- Yuzhakov, A.; Kelman, Z.; Hurwitz, J.; O’Donnell, M. Multiple competition reactions for RPA order the assembly of the DNA polymerase delta holoenzyme. EMBO J. 1999, 18, 6189–6199. [Google Scholar] [CrossRef]
- Prindle, M.J.; Loeb, L.A. DNA polymerase delta in DNA replication and genome maintenance. Environ. Mol. Mutagen. 2012, 53, 666–682. [Google Scholar] [CrossRef] [PubMed]
- Baranovskiy, A.G.; Lisova, A.E.; Morstadt, L.M.; Babayeva, N.D.; Tahirov, T.H. Insight into RNA-DNA primer length counting by human primosome. Nucleic Acids Res. 2022, 50, 6264–6270. [Google Scholar] [CrossRef] [PubMed]
- Baranovskiy, A.G.; Babayeva, N.D.; Zhang, Y.; Gu, J.; Suwa, Y.; Pavlov, Y.I.; Tahirov, T.H. Mechanism of Concerted RNA-DNA Primer Synthesis by the Human Primosome. J. Biol. Chem. 2016, 291, 10006–10020. [Google Scholar] [CrossRef] [PubMed]
- Zerbe, L.K.; Kuchta, R.D. The p58 subunit of human DNA primase is important for primer initiation, elongation, and counting. Biochemistry 2002, 41, 4891–4900. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Aggarwal, A.K.; Rechkoblit, O. Eukaryotic DNA polymerases. Curr. Opin. Struct. Biol. 2018, 53, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Zabrady, K.; Li, A.W.H.; Doherty, A.J. Mechanism of primer synthesis by Primase-Polymerases. Curr. Opin. Struct. Biol. 2023, 82, 102652. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Stewart, J.A.; Kasbek, C.; Zhao, Y.; Wright, W.E.; Price, C.M. Human CST has independent functions during telomere duplex replication and C-strand fill-in. Cell Rep. 2012, 2, 1096–1103. [Google Scholar] [CrossRef]
- Jones, M.L.; Baris, Y.; Taylor, M.R.; Yeeles, J.T.P. Structure of a human replisome shows the organisation and interactions of a DNA replication machine. EMBO J. 2023, 42, e115685. [Google Scholar] [CrossRef]
- Maga, G.; Frouin, I.; Spadari, S.; Hübscher, U. Replication protein A as a “fidelity clamp” for DNA polymerase alpha. J. Biol. Chem. 2001, 276, 18235–18242. [Google Scholar] [CrossRef]
- Lujan, S.A.; Williams, J.S.; Kunkel, T.A. DNA Polymerases Divide the Labor of Genome Replication. Trends Cell Biol. 2016, 26, 640–654. [Google Scholar] [CrossRef]
- Jenkyn-Bedford, M.; Jones, M.L.; Baris, Y.; Labib, K.P.M.; Cannone, G.; Yeeles, J.T.P.; Deegan, T.D. A conserved mechanism for regulating replisome disassembly in eukaryotes. Nature 2021, 600, 743–747. [Google Scholar] [CrossRef] [PubMed]
- Burgers, P.M.J.; Kunkel, T.A. Eukaryotic DNA Replication Fork. Annu. Rev. Biochem. 2017, 86, 417–438. [Google Scholar] [CrossRef] [PubMed]
- Kapadia, N.; El-Hajj, Z.W.; Zheng, H.; Beattie, T.R.; Yu, A.; Reyes-Lamothe, R. Processive Activity of Replicative DNA Polymerases in the Replisome of Live Eukaryotic Cells. Mol. Cell 2020, 80, 114–126.e118. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.S.; Spenkelink, L.M.; Schauer, G.D.; Yurieva, O.; Mueller, S.H.; Natarajan, V.; Kaur, G.; Maher, C.; Kay, C.; O’Donnell, M.E.; et al. Tunability of DNA Polymerase Stability during Eukaryotic DNA Replication. Mol. Cell 2020, 77, 17–25.e15. [Google Scholar] [CrossRef] [PubMed]
- Nasheuer, H.P.; Onwubiko, N.O. Lagging Strand Initiation Processes in DNA Replication of Eukaryotes-Strings of Highly Coordinated Reactions Governed by Multiprotein Complexes. Genes 2023, 14, 1012. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Ma, L.; Tsai, Y.F.; Abeywardana, T.; Shen, B.; Zheng, L. Okazaki fragment maturation: DNA flap dynamics for cell proliferation and survival. Trends Cell Biol. 2023, 33, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Ganduri, S.; Lue, N.F. STN1-POLA2 interaction provides a basis for primase-pol α stimulation by human STN1. Nucleic Acids Res. 2017, 45, 9455–9466. [Google Scholar] [CrossRef] [PubMed]
- Arunkumar, A.I.; Klimovich, V.; Jiang, X.; Ott, R.D.; Mizoue, L.; Fanning, E.; Chazin, W.J. Insights into hRPA32 C-terminal domain--mediated assembly of the simian virus 40 replisome. Nat. Struct. Mol. Biol. 2005, 12, 332–339. [Google Scholar] [CrossRef]
- Melendy, T.; Stillman, B. An interaction between replication protein A and SV40 T antigen appears essential for primosome assembly during SV40 DNA replication. J. Biol. Chem. 1993, 268, 3389–3395. [Google Scholar] [CrossRef]
- Onwubiko, N.O.; Scheffel, F.; Tessmer, I.; Nasheuer, H.P. SV40 T antigen helicase domain regions responsible for oligomerisation regulate Okazaki fragment synthesis initiation. FEBS Open Bio 2022, 12, 649–663. [Google Scholar] [CrossRef]
- Weisshart, K.; Förster, H.; Kremmer, E.; Schlott, B.; Grosse, F.; Nasheuer, H.P. Protein-protein interactions of the primase subunits p58 and p48 with simian virus 40 T antigen are required for efficient primer synthesis in a cell-free system. J. Biol. Chem. 2000, 275, 17328–17337. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Weiner, B.E.; Zhang, H.; Fuller, B.E.; Gao, Y.; Wile, B.M.; Zhao, K.; Arnett, D.R.; Chazin, W.J.; Fanning, E. Structure of a DNA polymerase alpha-primase domain that docks on the SV40 helicase and activates the viral primosome. J. Biol. Chem. 2010, 285, 17112–17122. [Google Scholar] [CrossRef] [PubMed]
- Vaithiyalingam, S.; Arnett, D.R.; Aggarwal, A.; Eichman, B.F.; Fanning, E.; Chazin, W.J. Insights into eukaryotic primer synthesis from structures of the p48 subunit of human DNA primase. J. Mol. Biol. 2014, 426, 558–569. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Lin, X.; Chavez, B.L.; Agrawal, S.; Lusk, B.L.; Lim, C.J. Structures of the human CST-Polα–primase complex bound to telomere templates. Nature 2022, 608, 826–832. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Song, H.; Chan, H.; Liu, B.; Wang, Y.; Sušac, L.; Zhou, Z.H.; Feigon, J. Structure of Tetrahymena telomerase-bound CST with polymerase α-primase. Nature 2022, 608, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.W.; Zinder, J.C.; Svetlov, V.; Bush, M.W.; Nudler, E.; Walz, T.; de Lange, T. Cryo-EM structure of the human CST–Polα/primase complex in a recruitment state. Nat. Struct. Mol. Biol. 2022, 29, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Casteel, D.E.; Zhuang, S.; Zeng, Y.; Perrino, F.W.; Boss, G.R.; Goulian, M.; Pilz, R.B. A DNA Polymerase-α·Primase Cofactor with Homology to Replication Protein A-32 Regulates DNA Replication in Mammalian Cells. J. Biol. Chem. 2009, 284, 5807–5818. [Google Scholar] [CrossRef] [PubMed]
- Lue, N.F.; Chan, J.; Wright, W.E.; Hurwitz, J. The CDC13-STN1-TEN1 complex stimulates Pol alpha activity by promoting RNA priming and primase-to-polymerase switch. Nat. Commun. 2014, 5, 5762. [Google Scholar] [CrossRef]
- Schmid, E.; Walter, J.C. Predictomics Webpage. Available online: https://karolinskainnovations.ki.se/en/companies/predictomics/ (accessed on 28 November 2023).
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef]
- Gao, M.; Guo, G.; Huang, J.; Hou, X.; Ham, H.; Kim, W.; Zhao, F.; Tu, X.; Zhou, Q.; Zhang, C.; et al. DOCK7 protects against replication stress by promoting RPA stability on chromatin. Nucleic Acids Res. 2021, 49, 3322–3337. [Google Scholar] [CrossRef]
- Illuzzi, G.; Fouquerel, E.; Ame, J.C.; Noll, A.; Rehmet, K.; Nasheuer, H.P.; Dantzer, F.; Schreiber, V. PARG is dispensable for recovery from transient replicative stress but required to prevent detrimental accumulation of poly(ADP-ribose) upon prolonged replicative stress. Nucleic Acids Res. 2014, 42, 7776–7792. [Google Scholar] [CrossRef] [PubMed]
- Ball, H.L.; Myers, J.S.; Cortez, D. ATRIP binding to replication protein A-single-stranded DNA promotes ATR-ATRIP localization but is dispensable for Chk1 phosphorylation. Mol. Biol. Cell 2005, 16, 2372–2381. [Google Scholar] [CrossRef] [PubMed]
- Borgstahl, G.E.; Brader, K.; Mosel, A.; Liu, S.; Kremmer, E.; Goettsch, K.A.; Kolar, C.; Nasheuer, H.P.; Oakley, G.G. Interplay of DNA damage and cell cycle signaling at the level of human replication protein A. DNA Repair 2014, 21, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Nuss, J.E.; Patrick, S.M.; Oakley, G.G.; Alter, G.M.; Robison, J.G.; Dixon, K.; Turchi, J.J. DNA damage induced hyperphosphorylation of replication protein A. 1. Identification of novel sites of phosphorylation in response to DNA damage. Biochemistry 2005, 44, 8428–8437. [Google Scholar] [CrossRef] [PubMed]
- Han, S.S.; Wen, K.K.; García-Rubio, M.L.; Wold, M.S.; Aguilera, A.; Niedzwiedz, W.; Vyas, Y.M. WASp modulates RPA function on single-stranded DNA in response to replication stress and DNA damage. Nat. Commun. 2022, 13, 3743. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef]
- Friedberg, E.C.; Aguilera, A.; Gellert, M.; Hanawalt, P.C.; Hays, J.B.; Lehmann, A.R.; Lindahl, T.; Lowndes, N.; Sarasin, A.; Wood, R.D. DNA repair: From molecular mechanism to human disease. DNA Repair 2006, 5, 986–996. [Google Scholar] [CrossRef]
- Schärer, O.D. Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef]
- Kunkel, T.A.; Erie, D.A. DNA mismatch repair. Annu. Rev. Biochem. 2005, 74, 681–710. [Google Scholar] [CrossRef]
- Guo, S.; Zhang, Y.; Yuan, F.; Gao, Y.; Gu, L.; Wong, I.; Li, G.M. Regulation of replication protein a functions in DNA mismatch repair by phosphorylation. J. Biol. Chem. 2006, 281, 21607–21616. [Google Scholar] [CrossRef]
- Kieffer, S.R.; Lowndes, N.F. Immediate-Early, Early, and Late Responses to DNA Double Stranded Breaks. Front. Genet. 2022, 13, 793884. [Google Scholar] [CrossRef] [PubMed]
- Nimonkar, A.V.; Genschel, J.; Kinoshita, E.; Polaczek, P.; Campbell, J.L.; Wyman, C.; Modrich, P.; Kowalczykowski, S.C. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011, 25, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.; Rösner, H.; Zhao, W.; Selemenakis, P.; He, Z.; Kawale, A.S.; Katz, J.N.; Rogers, C.M.; Neal, F.E.; Badamchi Shabestari, A.; et al. DNA binding and RAD51 engagement by the BRCA2 C-terminus orchestrate DNA repair and replication fork preservation. Nat. Commun. 2023, 14, 432. [Google Scholar] [CrossRef] [PubMed]
- Soniat, M.M.; Myler, L.R.; Kuo, H.C.; Paull, T.T.; Finkelstein, I.J. RPA Phosphorylation Inhibits DNA Resection. Mol. Cell 2019, 75, 145–153.e145. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Vaithiyalingam, S.; San Filippo, J.; Maranon, D.G.; Jimenez-Sainz, J.; Fontenay, G.V.; Kwon, Y.; Leung, S.G.; Lu, L.; Jensen, R.B.; et al. Promotion of BRCA2-Dependent Homologous Recombination by DSS1 via RPA Targeting and DNA Mimicry. Mol. Cell 2015, 59, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Le, H.P.; Heyer, W.D.; Liu, J. Guardians of the Genome: BRCA2 and Its Partners. Genes 2021, 12, 1229. [Google Scholar] [CrossRef] [PubMed]
- Foertsch, F.; Kache, T.; Drube, S.; Biskup, C.; Nasheuer, H.P.; Melle, C. Determination of the number of RAD51 molecules in different human cell lines. Cell Cycle 2019, 18, 3581–3588. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.C.; Dombrowski, C.C.; Plank, J.L.; Jensen, R.B.; Kowalczykowski, S.C. BRCA2 chaperones RAD51 to single molecules of RPA-coated ssDNA. Proc. Natl. Acad. Sci. USA 2023, 120, e2221971120. [Google Scholar] [CrossRef]
- Špírek, M.; Mlcoušková, J.; Belán, O.; Gyimesi, M.; Harami, G.M.; Molnár, E.; Novacek, J.; Kovács, M.; Krejci, L. Human RAD51 rapidly forms intrinsically dynamic nucleoprotein filaments modulated by nucleotide binding state. Nucleic Acids Res. 2018, 46, 3967–3980. [Google Scholar] [CrossRef]
- Deng, S.K.; Gibb, B.; de Almeida, M.J.; Greene, E.C.; Symington, L.S. RPA antagonizes microhomology-mediated repair of DNA double-strand breaks. Nat. Struct. Mol. Biol. 2014, 21, 405–412. [Google Scholar] [CrossRef]
- Wang, H.; Xu, X. Microhomology-mediated end joining: New players join the team. Cell Biosci. 2017, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Donnianni, R.A.; Zhou, Z.X.; Lujan, S.A.; Al-Zain, A.; Garcia, V.; Glancy, E.; Burkholder, A.B.; Kunkel, T.A.; Symington, L.S. DNA Polymerase Delta Synthesizes Both Strands during Break-Induced Replication. Mol. Cell 2019, 76, 371–381.e374. [Google Scholar] [CrossRef] [PubMed]
- Paulson, H. Repeat expansion diseases. Handb. Clin. Neurol. 2018, 147, 105–123. [Google Scholar] [CrossRef] [PubMed]
- VanderVere-Carozza, P.S.; Gavande, N.S.; Jalal, S.I.; Pollok, K.E.; Ekinci, E.; Heyza, J.; Patrick, S.M.; Masters, A.; Turchi, J.J.; Pawelczak, K.S. In Vivo Targeting Replication Protein A for Cancer Therapy. Front. Oncol. 2022, 12, 826655. [Google Scholar] [CrossRef]
- Lim, C.J.; Cech, T.R. Shaping human telomeres: From shelterin and CST complexes to telomeric chromatin organization. Nat. Rev. Mol. Cell Biol. 2021, 22, 283–298. [Google Scholar] [CrossRef]
- Takasugi, T.; Gu, P.; Liang, F.; Staco, I.; Chang, S. Pot1b−/− tumors activate G-quadruplex-induced DNA damage to promote telomere hyper-elongation. Nucleic Acids Res. 2023, 51, 9227–9247. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nasheuer, H.P.; Meaney, A.M.; Hulshoff, T.; Thiele, I.; Onwubiko, N.O. Replication Protein A, the Main Eukaryotic Single-Stranded DNA Binding Protein, a Focal Point in Cellular DNA Metabolism. Int. J. Mol. Sci. 2024, 25, 588. https://doi.org/10.3390/ijms25010588
Nasheuer HP, Meaney AM, Hulshoff T, Thiele I, Onwubiko NO. Replication Protein A, the Main Eukaryotic Single-Stranded DNA Binding Protein, a Focal Point in Cellular DNA Metabolism. International Journal of Molecular Sciences. 2024; 25(1):588. https://doi.org/10.3390/ijms25010588
Chicago/Turabian StyleNasheuer, Heinz Peter, Anna Marie Meaney, Timothy Hulshoff, Ines Thiele, and Nichodemus O. Onwubiko. 2024. "Replication Protein A, the Main Eukaryotic Single-Stranded DNA Binding Protein, a Focal Point in Cellular DNA Metabolism" International Journal of Molecular Sciences 25, no. 1: 588. https://doi.org/10.3390/ijms25010588
APA StyleNasheuer, H. P., Meaney, A. M., Hulshoff, T., Thiele, I., & Onwubiko, N. O. (2024). Replication Protein A, the Main Eukaryotic Single-Stranded DNA Binding Protein, a Focal Point in Cellular DNA Metabolism. International Journal of Molecular Sciences, 25(1), 588. https://doi.org/10.3390/ijms25010588