Figure 1.
The pharmacological or genetic manipulation of DAGLα decreases the level of 2-AG in bEnD.3 endothelial cells. bEnD.3 endothelial cells were treated with LEI-106 (1.3 mM) or vehicle (0.9% DMSO in media) for 15 min, then subjected to LC-MS/MS to measure 2-AG and AEA levels. In a separate set, the level of DAG was quantified by ELISA. In the siRNA transfection experiments, bEnD.3 cells were transiently transfected with siRNA targeting DAGLα or a scrambled, non-targeting control, then harvested 72 h post-transfection, followed by a Western blot to detect DAGLα expression. In a separate set of experiments, the cell pellet was lysed and subjected to LC-MS to measure 2-AG and AEA levels. (A) The schema of the experimental setting. (B) The pharmacological blockade of DAGLα with LEI-106 significantly reduced the 2-AG level in endothelial cells (LEI-106 vs. vehicle, p = 0.001, t(12) = 5.447 as assessed by unpaired t-test). Data are shown as mean ± SEM in nmol/g pellet (n = 4-10 in each group). *** denotes significantly different (p < 0.001). (C) After LEI-106 treatment, the level of AEA was unchanged (LEI-106 vs. vehicle, p = 0.7578, t(13) = 0.3149 as assessed by unpaired t-test). Data are shown as mean ± SEM in pmol/g pellet (n = 5–10 in each group). ns = non-significant. (D) The LEI-106 treatment did not significantly change the level of DAG compared to vehicle control (LEI-106, 650 µM vs. vehicle: p = 0.4364, t(14) = 0.8011; LEI-106, 1.3 mM vs. vehicle: p = 0.4626, t(14) = 0.7553 as assessed by unpaired t-test). Data are presented as % of media-treated ± SEM (n = 8/group). ns = non-significant. (E) Representative image showing the expression of DAGLα with α-tubulin as loading control in bEnd.3 cell lysate harvested 72 h after siRNA transfection. The transfection of DAGLα-targeting siRNA significantly reduced the detection of DAGLα protein compared to the non-targeting control (DAGLα siRNA vs. non-targeting control siRNA: p < 0.0001, t(14) = 7.227, as assessed by unpaired t-test). Data are shown as % of non-targeting control ± SEM (n = 8/group). **** denotes significantly different (p < 0.0001). (F) siRNA knockdown of DAGLα in bEnD.3 cells significantly reduced 2-AG levels compared to non-targeting control (DAGLα siRNA vs. non-targeting control siRNA: p = 0.0266, t(6) = 2.919, as assessed by unpaired t-test). Data are shown as mean ± SEM in nmol/g pellet (n = 4/group). * denotes significantly different (p < 0.05). (G) The AEA level of bEnd.3 cells was significantly increased after DAGLα siRNA transfection compared to non-targeting control, suggesting the presence of a compensatory mechanism after the loss of 2-AG (DAGLα siRNA vs. non-targeting control siRNA: p = 0.031, t(6) = 2.804, as assessed by unpaired t-test). Data are shown as mean ± SEM in pmol/g pellet (n = 4/group). * denotes significantly different (p < 0.05).
Figure 1.
The pharmacological or genetic manipulation of DAGLα decreases the level of 2-AG in bEnD.3 endothelial cells. bEnD.3 endothelial cells were treated with LEI-106 (1.3 mM) or vehicle (0.9% DMSO in media) for 15 min, then subjected to LC-MS/MS to measure 2-AG and AEA levels. In a separate set, the level of DAG was quantified by ELISA. In the siRNA transfection experiments, bEnD.3 cells were transiently transfected with siRNA targeting DAGLα or a scrambled, non-targeting control, then harvested 72 h post-transfection, followed by a Western blot to detect DAGLα expression. In a separate set of experiments, the cell pellet was lysed and subjected to LC-MS to measure 2-AG and AEA levels. (A) The schema of the experimental setting. (B) The pharmacological blockade of DAGLα with LEI-106 significantly reduced the 2-AG level in endothelial cells (LEI-106 vs. vehicle, p = 0.001, t(12) = 5.447 as assessed by unpaired t-test). Data are shown as mean ± SEM in nmol/g pellet (n = 4-10 in each group). *** denotes significantly different (p < 0.001). (C) After LEI-106 treatment, the level of AEA was unchanged (LEI-106 vs. vehicle, p = 0.7578, t(13) = 0.3149 as assessed by unpaired t-test). Data are shown as mean ± SEM in pmol/g pellet (n = 5–10 in each group). ns = non-significant. (D) The LEI-106 treatment did not significantly change the level of DAG compared to vehicle control (LEI-106, 650 µM vs. vehicle: p = 0.4364, t(14) = 0.8011; LEI-106, 1.3 mM vs. vehicle: p = 0.4626, t(14) = 0.7553 as assessed by unpaired t-test). Data are presented as % of media-treated ± SEM (n = 8/group). ns = non-significant. (E) Representative image showing the expression of DAGLα with α-tubulin as loading control in bEnd.3 cell lysate harvested 72 h after siRNA transfection. The transfection of DAGLα-targeting siRNA significantly reduced the detection of DAGLα protein compared to the non-targeting control (DAGLα siRNA vs. non-targeting control siRNA: p < 0.0001, t(14) = 7.227, as assessed by unpaired t-test). Data are shown as % of non-targeting control ± SEM (n = 8/group). **** denotes significantly different (p < 0.0001). (F) siRNA knockdown of DAGLα in bEnD.3 cells significantly reduced 2-AG levels compared to non-targeting control (DAGLα siRNA vs. non-targeting control siRNA: p = 0.0266, t(6) = 2.919, as assessed by unpaired t-test). Data are shown as mean ± SEM in nmol/g pellet (n = 4/group). * denotes significantly different (p < 0.05). (G) The AEA level of bEnd.3 cells was significantly increased after DAGLα siRNA transfection compared to non-targeting control, suggesting the presence of a compensatory mechanism after the loss of 2-AG (DAGLα siRNA vs. non-targeting control siRNA: p = 0.031, t(6) = 2.804, as assessed by unpaired t-test). Data are shown as mean ± SEM in pmol/g pellet (n = 4/group). * denotes significantly different (p < 0.05).
![Ijms 25 00531 g001]()
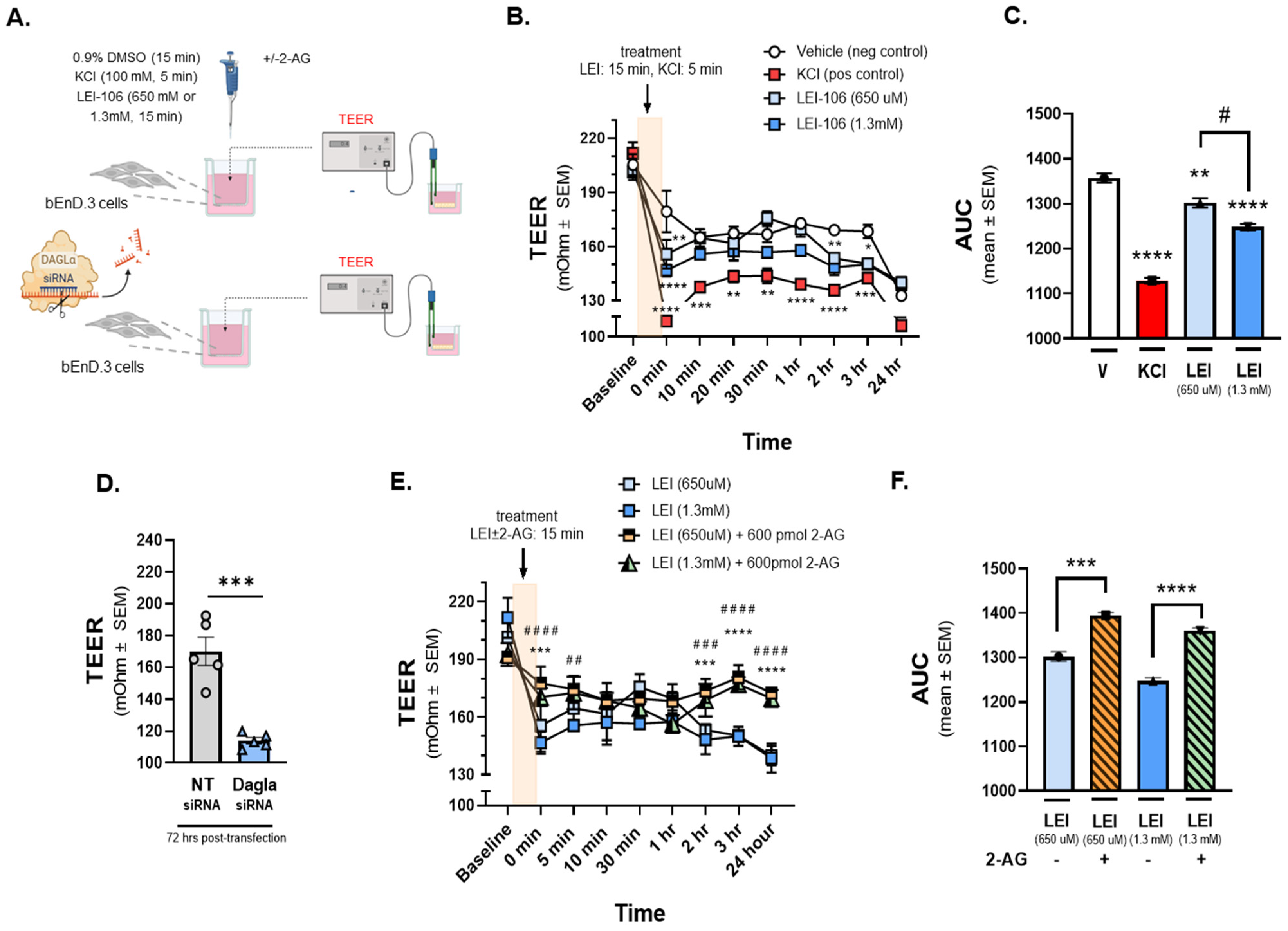
Figure 2.
The depletion of 2-AG decreased Trans-Endothelial Electrical Resistance (TEER) of bEnD.3 cells, indicating the loss of barrier integrity. The bEnD.3 endothelial cells were cultured on a trans-well insert, then treated with LEI-106 (650 uM or 1.3 mM) or vehicle (0.9% DMSO in media) with or without 2-AG (600 pmol) for 15 min. KCl pulse (100 mM, 5 min) was used as a positive control. TEER was measured at baseline (before any treatment), right after the treatment (0 min), and then 10, 20, 30 min, 1, 2, 3, and 24 h after post-treatment. In the transfection experiments, TEER was measured 72 h post-transfection. (A) This panel shows the schematic experimental setting. (B) The application of LEI-106 in either dose significantly decreased the TEER value, compared to vehicle control at 0 min time-point, suggesting the loss of barrier integrity (LEI-106-650 uM vs. vehicle: p = 0.0017; LEI-106-1.3 mM vs. vehicle: p < 0.0001, as assessed by two-way ANOVA, F(24,72) = 3.723). At later time-points, the TEER was then normalized, followed by an additional significant decrease at 2 and 3 h (2 h time-point: LEI-106-1.3 uM vs. vehicle: p = 0.0075; 3 h time-point: LEI-106-650 uM vs. vehicle: p = 0.0254 LEI-106-1.3 mM vs vehicle: p < 0.022, as assessed by two-way ANOVA, F(24,72) = 3.723). All data represent the mean ± SEM of three independent experiments performed in triplicate. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 compared to vehicle control. (C) The area under the curve (AUC) of the corresponding panel B. The AUC analysis further confirmed the significant reduction in TEER induced by LEI-106 treatment (LEI-106-650 uM vs. vehicle: p = 0.009; LEI-106-1.3 mM vs. vehicle: p < 0.0001; LEI-106-650 uM vs.LEI-106-1.3 mM: p = 0.01, as assessed by one-way ANOVA with Tukey post-test, F(3,8) = 125.2). Data are shown mean ± SEM. ** p < 0.01, **** p < 0.0001 compared to vehicle control. # p < 0.05: LEI-106-650 uM vs. LEI-106-1.3 mM. (D) bEnD.3 cells were transfected either with non-targeting control siRNA or siRNA targeting DAGLα. TEER was assessed 72 h post-transfection. A significant decrease in TEER was observed in DAGLα siRNA-transfected cells, compared to non-targeting control, indicating disruption of the barrier integrity after silencing of DAGLα (DAGLα siRNA vs. non-targeting siRNA: p = 0.0003, t(8) = 6.203 as assessed by unpaired t-test, n = 5). Data are shown mean ± SEM. *** p < 0.001, compared to the non-targeting control. (E) The application of 2-AG diminished the effect of LEI-106 on BBB integrity. Significant differences were observed between LEI-106 vs. LEI-106 + 2-AG groups at 0 min time-point (LEI-106-650 uM vs. LEI-106-650 uM + 2-AG: p = 0.0002; hjnnnoLEI-106-1.3 mM vs. LEI-106-1.3 mM + 2-AG: p < 0.0001, as assessed by two-way ANOVA, F(24,90) = 7.318). All data represent the mean ± SEM of three independent experiments performed in triplicate. LEI-106-650 uM vs. LEI-106-650 uM + 2-AG: *** p < 0.001, **** p < 0.0001. LEI-106-1.3 mM vs. LEI-106 1.3 + 2-AG: ## p < 0.01, ### p < 0.001, #### p < 0.0001. (F) The area under the curve (AUC) for panel E. The AUC analysis also showed that 2-AG can mitigate the loss of barrier integrity caused by the inhibition of DAGLα (LEI-106-650 uM vs. LEI-106-650 uM + 2-AG: p = 0.0001; LEI-106-1.3 mM vs. LEI-106-1.3 mM + 2-AG: p < 0.0001, as assessed by one-way ANOVA, F(3,8) = 67.39). Data are shown mean ± SEM. *** p < 0.001, **** p < 0.0001 compared to the corresponding 2-AG treatment.
Figure 2.
The depletion of 2-AG decreased Trans-Endothelial Electrical Resistance (TEER) of bEnD.3 cells, indicating the loss of barrier integrity. The bEnD.3 endothelial cells were cultured on a trans-well insert, then treated with LEI-106 (650 uM or 1.3 mM) or vehicle (0.9% DMSO in media) with or without 2-AG (600 pmol) for 15 min. KCl pulse (100 mM, 5 min) was used as a positive control. TEER was measured at baseline (before any treatment), right after the treatment (0 min), and then 10, 20, 30 min, 1, 2, 3, and 24 h after post-treatment. In the transfection experiments, TEER was measured 72 h post-transfection. (A) This panel shows the schematic experimental setting. (B) The application of LEI-106 in either dose significantly decreased the TEER value, compared to vehicle control at 0 min time-point, suggesting the loss of barrier integrity (LEI-106-650 uM vs. vehicle: p = 0.0017; LEI-106-1.3 mM vs. vehicle: p < 0.0001, as assessed by two-way ANOVA, F(24,72) = 3.723). At later time-points, the TEER was then normalized, followed by an additional significant decrease at 2 and 3 h (2 h time-point: LEI-106-1.3 uM vs. vehicle: p = 0.0075; 3 h time-point: LEI-106-650 uM vs. vehicle: p = 0.0254 LEI-106-1.3 mM vs vehicle: p < 0.022, as assessed by two-way ANOVA, F(24,72) = 3.723). All data represent the mean ± SEM of three independent experiments performed in triplicate. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 compared to vehicle control. (C) The area under the curve (AUC) of the corresponding panel B. The AUC analysis further confirmed the significant reduction in TEER induced by LEI-106 treatment (LEI-106-650 uM vs. vehicle: p = 0.009; LEI-106-1.3 mM vs. vehicle: p < 0.0001; LEI-106-650 uM vs.LEI-106-1.3 mM: p = 0.01, as assessed by one-way ANOVA with Tukey post-test, F(3,8) = 125.2). Data are shown mean ± SEM. ** p < 0.01, **** p < 0.0001 compared to vehicle control. # p < 0.05: LEI-106-650 uM vs. LEI-106-1.3 mM. (D) bEnD.3 cells were transfected either with non-targeting control siRNA or siRNA targeting DAGLα. TEER was assessed 72 h post-transfection. A significant decrease in TEER was observed in DAGLα siRNA-transfected cells, compared to non-targeting control, indicating disruption of the barrier integrity after silencing of DAGLα (DAGLα siRNA vs. non-targeting siRNA: p = 0.0003, t(8) = 6.203 as assessed by unpaired t-test, n = 5). Data are shown mean ± SEM. *** p < 0.001, compared to the non-targeting control. (E) The application of 2-AG diminished the effect of LEI-106 on BBB integrity. Significant differences were observed between LEI-106 vs. LEI-106 + 2-AG groups at 0 min time-point (LEI-106-650 uM vs. LEI-106-650 uM + 2-AG: p = 0.0002; hjnnnoLEI-106-1.3 mM vs. LEI-106-1.3 mM + 2-AG: p < 0.0001, as assessed by two-way ANOVA, F(24,90) = 7.318). All data represent the mean ± SEM of three independent experiments performed in triplicate. LEI-106-650 uM vs. LEI-106-650 uM + 2-AG: *** p < 0.001, **** p < 0.0001. LEI-106-1.3 mM vs. LEI-106 1.3 + 2-AG: ## p < 0.01, ### p < 0.001, #### p < 0.0001. (F) The area under the curve (AUC) for panel E. The AUC analysis also showed that 2-AG can mitigate the loss of barrier integrity caused by the inhibition of DAGLα (LEI-106-650 uM vs. LEI-106-650 uM + 2-AG: p = 0.0001; LEI-106-1.3 mM vs. LEI-106-1.3 mM + 2-AG: p < 0.0001, as assessed by one-way ANOVA, F(3,8) = 67.39). Data are shown mean ± SEM. *** p < 0.001, **** p < 0.0001 compared to the corresponding 2-AG treatment.
![Ijms 25 00531 g002]()
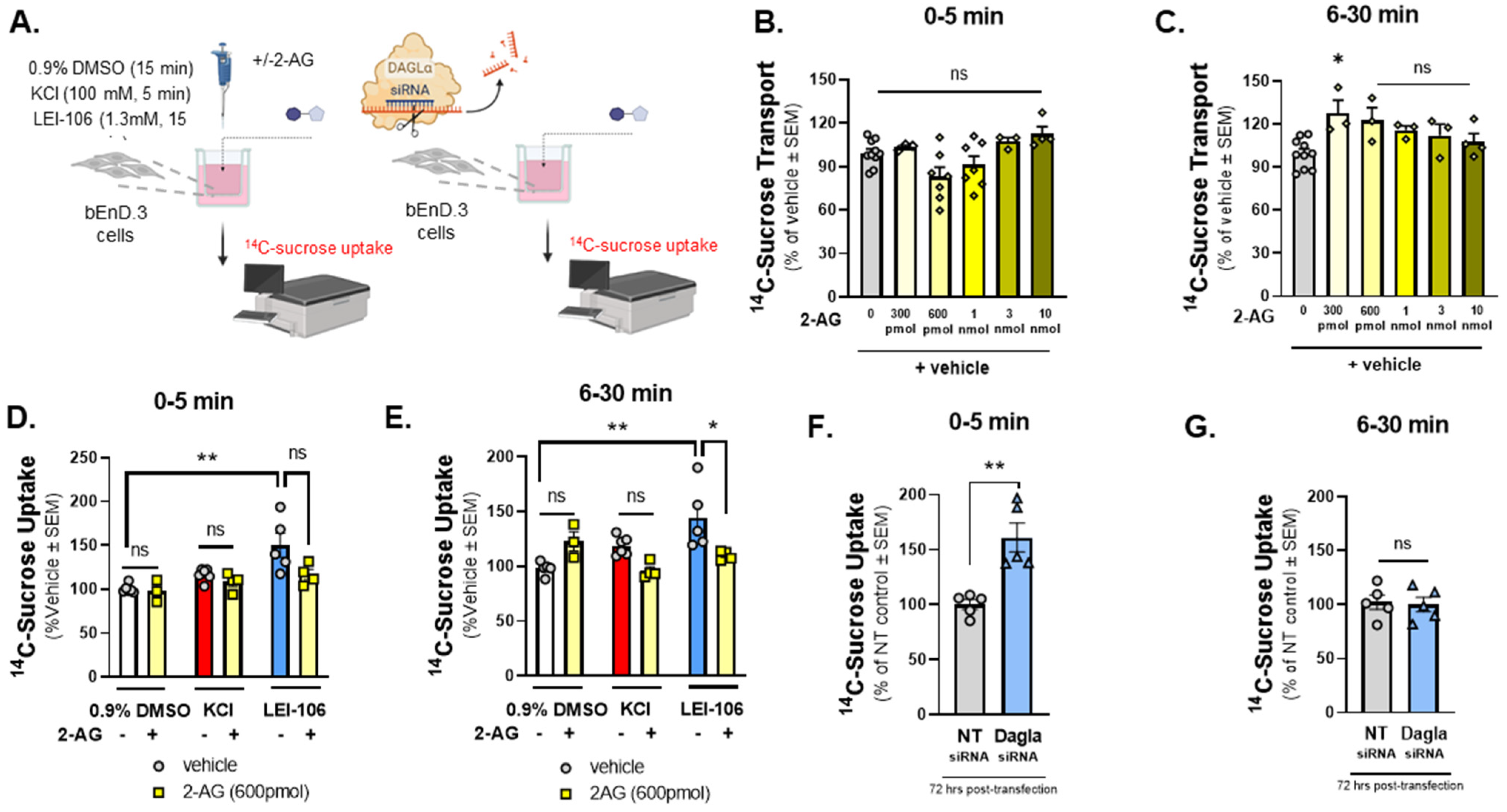
Figure 3.
14C-Sucrose transport through the bEnd.3 monolayer was increased after the blockade of DAGLα. The bEnD.3 endothelial cells were cultured on a trans-well insert, then treated with an increasing amount of 2-AG (0–10 nmole). In a separate experiment, cells were treated with LEI-106 (1.3 mM) or vehicle (0.9% DMSO in media) with or without 2-AG (600 pmol) for 15 min. KCl pulse (100 mM, 5 min) was used as a positive control. Following treatment, luminal media was replaced with fresh media containing 14C-sucrose (0.25 µCi/mL). Abluminal media was collected at 5 min and 30 min post-treatment and subjected to measure radioactivity with a liquid scintillation counter. 14C-sucrose uptake assay was performed 72 h after transfection of DAGLα siRNA or non-targeting control. 14C-sucrose (0.25 µCi/mL) was added to the luminal side, abluminal media was collected at two time-points (5 min and 30 min), and then radioactivity was measured. (A) Schema of experimental setting. (B) 2-AG treatment at any dose did not cause a significant change in 14C-sucrose uptake at 5 min time-point compared to vehicle control (2-AG at any dose vs. vehicle: p > 0.05, F(5,28) = 4.010, as assessed by one-way ANOVA with Bartlett’s test) All data represent the % of vehicle-treated ± SEM (n = 3–10/group). ns = non-significant. (C) A significant increase in 14C-sucrose uptake after 300 pmol of 2-AG treatment was observed at 6–30 min time-point compared to vehicle control (2-AG-300 pmol vs. vehicle: p = 0.0172, 2-AG at other doses vs. vehicle: p > 0.05, F(5,20) = 3.879, as assessed by one-way ANOVA with Bartlett’s test) All data represent the % of vehicle-treated ± SEM (n = 3–10/group). ns = non-significant, * p < 0.05. (D) LEI-106 (1.3 mM) increased the 14C-sucrose uptake at 5 min time-point compared to vehicle control, indicating the presence of paracellular leak (LEI-106 vs. vehicle: p = 0.0033, t(9) = 3.965 as assessed by unpaired t-test). The application of 2-AG did not significantly reduce the elevated sucrose uptake induced by LEI-106 (LEI-106 vs. LEI-106 + 2-AG: p = 0.0797, t(7) = 2.049 as assessed by unpaired t-test). All data represent the % of vehicle-treated ± SEM from three-four independent experiments using 4 trans-well inserts/group. ** p < 0.01 compared to vehicle control. ns = non-significant. (E) Increased 14C-sucrose uptake was also observed at the 30 min time-point after LEI-106 treatment (LEI-106 vs. vehicle: p = 0.0092, t(8) = 3.415 as assessed by unpaired t-test). 2-AG treatment significantly mitigated the increase in sucrose uptake caused by DAGLα inhibition (LEI-106 vs. LEI-106 + 2-AG: p = 0.05, t(7) = 2.273 as assessed by unpaired t-test). All data represent the % of vehicle-treated ± SEM from three-four independent experiments using 4 trans-well inserts/group. * p < 0.05, ** p < 0.01 compared to corresponding controls. (F) Silencing of DAGLα significantly increased the sucrose uptake in the first 5 min, compared to non-targeting control, suggesting reduced barrier integrity caused by genetic inhibition of DAGLα (DAGLα siRNA vs. non-targeting siRNA: p = 0.0022, t(8) = 4.420 as assessed by unpaired t-test, n = 5). Data are shown mean ± SEM. ** p < 0.01, compared to the non-targeting control. (G) No significant difference between DAGLα siRNA and control was observed at the later time-point (6–30 min) (DAGLα siRNA vs. non-targeting siRNA: p = 0.8204, t(8) = 0.2347 as assessed by unpaired t-test, n = 5). Data are shown mean ± SEM. ns = non-significant, compared to the non-targeting control.
Figure 3.
14C-Sucrose transport through the bEnd.3 monolayer was increased after the blockade of DAGLα. The bEnD.3 endothelial cells were cultured on a trans-well insert, then treated with an increasing amount of 2-AG (0–10 nmole). In a separate experiment, cells were treated with LEI-106 (1.3 mM) or vehicle (0.9% DMSO in media) with or without 2-AG (600 pmol) for 15 min. KCl pulse (100 mM, 5 min) was used as a positive control. Following treatment, luminal media was replaced with fresh media containing 14C-sucrose (0.25 µCi/mL). Abluminal media was collected at 5 min and 30 min post-treatment and subjected to measure radioactivity with a liquid scintillation counter. 14C-sucrose uptake assay was performed 72 h after transfection of DAGLα siRNA or non-targeting control. 14C-sucrose (0.25 µCi/mL) was added to the luminal side, abluminal media was collected at two time-points (5 min and 30 min), and then radioactivity was measured. (A) Schema of experimental setting. (B) 2-AG treatment at any dose did not cause a significant change in 14C-sucrose uptake at 5 min time-point compared to vehicle control (2-AG at any dose vs. vehicle: p > 0.05, F(5,28) = 4.010, as assessed by one-way ANOVA with Bartlett’s test) All data represent the % of vehicle-treated ± SEM (n = 3–10/group). ns = non-significant. (C) A significant increase in 14C-sucrose uptake after 300 pmol of 2-AG treatment was observed at 6–30 min time-point compared to vehicle control (2-AG-300 pmol vs. vehicle: p = 0.0172, 2-AG at other doses vs. vehicle: p > 0.05, F(5,20) = 3.879, as assessed by one-way ANOVA with Bartlett’s test) All data represent the % of vehicle-treated ± SEM (n = 3–10/group). ns = non-significant, * p < 0.05. (D) LEI-106 (1.3 mM) increased the 14C-sucrose uptake at 5 min time-point compared to vehicle control, indicating the presence of paracellular leak (LEI-106 vs. vehicle: p = 0.0033, t(9) = 3.965 as assessed by unpaired t-test). The application of 2-AG did not significantly reduce the elevated sucrose uptake induced by LEI-106 (LEI-106 vs. LEI-106 + 2-AG: p = 0.0797, t(7) = 2.049 as assessed by unpaired t-test). All data represent the % of vehicle-treated ± SEM from three-four independent experiments using 4 trans-well inserts/group. ** p < 0.01 compared to vehicle control. ns = non-significant. (E) Increased 14C-sucrose uptake was also observed at the 30 min time-point after LEI-106 treatment (LEI-106 vs. vehicle: p = 0.0092, t(8) = 3.415 as assessed by unpaired t-test). 2-AG treatment significantly mitigated the increase in sucrose uptake caused by DAGLα inhibition (LEI-106 vs. LEI-106 + 2-AG: p = 0.05, t(7) = 2.273 as assessed by unpaired t-test). All data represent the % of vehicle-treated ± SEM from three-four independent experiments using 4 trans-well inserts/group. * p < 0.05, ** p < 0.01 compared to corresponding controls. (F) Silencing of DAGLα significantly increased the sucrose uptake in the first 5 min, compared to non-targeting control, suggesting reduced barrier integrity caused by genetic inhibition of DAGLα (DAGLα siRNA vs. non-targeting siRNA: p = 0.0022, t(8) = 4.420 as assessed by unpaired t-test, n = 5). Data are shown mean ± SEM. ** p < 0.01, compared to the non-targeting control. (G) No significant difference between DAGLα siRNA and control was observed at the later time-point (6–30 min) (DAGLα siRNA vs. non-targeting siRNA: p = 0.8204, t(8) = 0.2347 as assessed by unpaired t-test, n = 5). Data are shown mean ± SEM. ns = non-significant, compared to the non-targeting control.
![Ijms 25 00531 g003]()
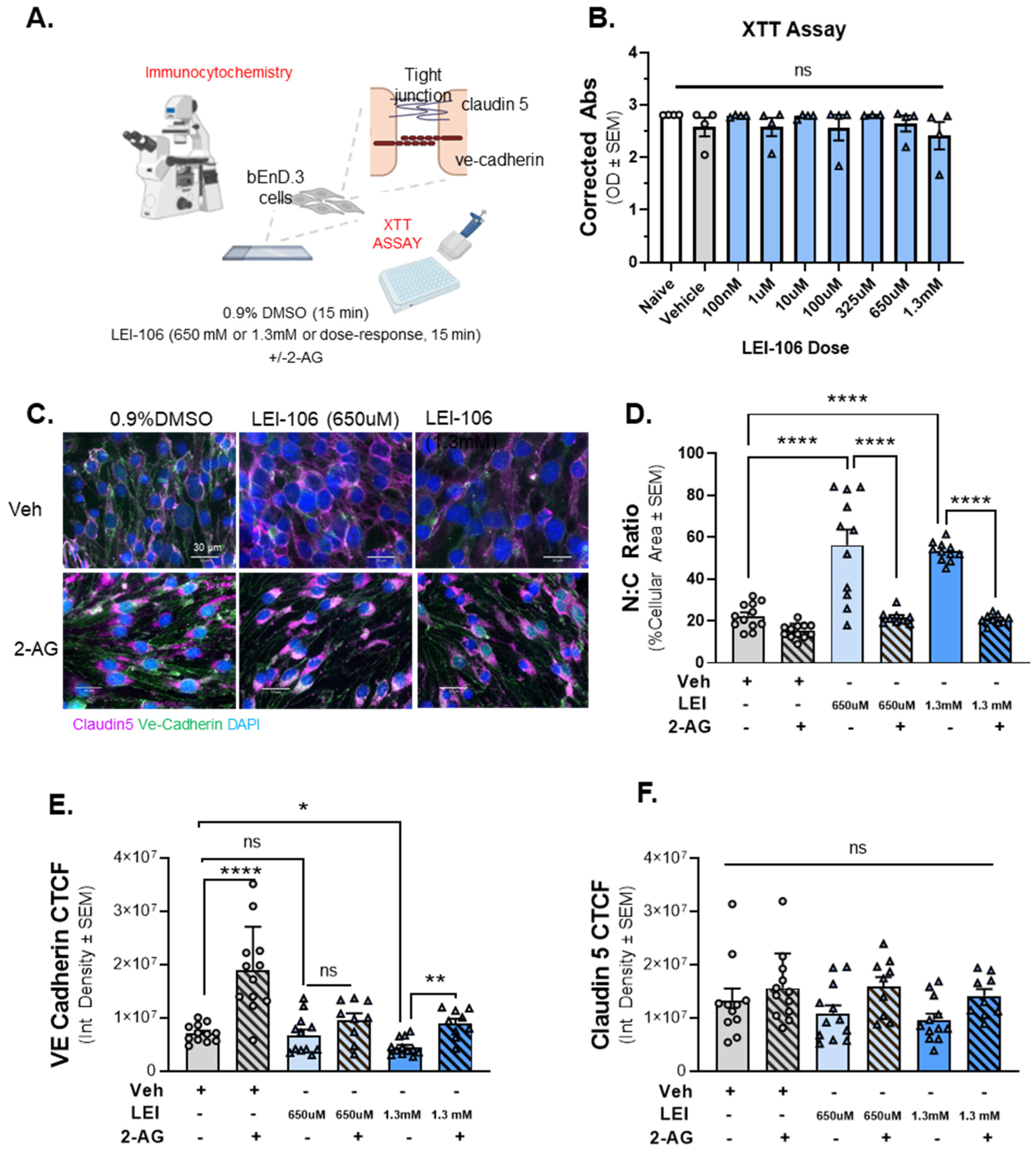
Figure 4.
The inhibition of DAGLα induced morphological changes in bEnD.3 cells accompanied with reduced expression of VE-cadherin. Cell viability assay on bEnD.3 cells was performed using increasing doses of LEI-106 (100 nM–1.3 mM). For ICC experiments, the bEnD.3 cells were treated with two doses of LEI-106 (650 uM and 1.3 mM) for 15 min, followed by 2-AG or vehicle application. ICC was performed to detect possible changes in the expression of two tight junction proteins, claudin 5 and VE-cadherin. (A) The panel represents the setting of the experiments. (B) LEI-106 did not cause significant changes in the cell viability at any doses applied in the study. (LEI-106 at any doses vs. vehicle: p > 0.05, as assessed by one-way ANOVA with Bartlett post-test, F(8,26) = 0.7738). All data are expressed as the mean of optical density ± SEM (n = 4 in each group). ns = non-significant. (C) Representative ICC images of bEnd.3 cells treated with LEI-106 ±2-AG application. (D) Significant changes in the nuclear: cytoplasmic ratio were observed following LEI-106 treatment, suggesting possible morphological alteration caused by 2-AG depletion (LEI-106-650 µM vs. vehicle: p < 0.0001, LEI-106-1.3 mM vs. vehicle: p < 0.0001, as assessed by one-way ANOVA with Tukey post-test, F(8,85) = 30.50). 2-AG treatment following LEI-106 application significantly reversed these effects (LEI-106-650 µM vs. LEI-106-650 µM + 2-AG: p < 0.0001, LEI-106-1.3 mM vs. LEI-106-1.3 mM + 2-AG: p < 0.0001, as assessed by one-way ANOVA with Tukey post-test, F(8,85) = 30.50). All data are shown as the mean % of cellular area ± SEM (n = 9–12/condition). **** p < 0.0001, compared to corresponding controls. (E) The higher dose of LEI-106 (1.3 mM) significantly reduced VE-cadherin CTCF; however, no significant change was detected at the lower dose (650 µM) compared to vehicle control (LEI-106-650 µM vs. vehicle: p = 0.9052, LEI-106-1.3 mM vs. vehicle: p = 0.0233, as assessed by one-way ANOVA with Bartlett post-test, F(2,33) = 4.100). The administration of 2-AG increased the VE-cadherin CTCF, moderating the loss of VE-cadherin expression after LEI-106 treatment (LEI-106-1.3 mM vs. LEI-106-1.3 mM + 2-AG: p = 0.005, as assessed by one-way ANOVA with Tukey post-test, F(4,49) = 5.568). All values are expressed as the mean of corrected total cell fluorescence (CTCF) ± SEM (n = 9–12/condition). * p < 0.05, ** p < 0.01, **** p < 0.0001, compared to the corresponding controls. ns = non-significant. (F) Neither dose of LEI-106 treatment significantly influenced the CTCF of claudin-5 compared to vehicle control (LEI-106-650 µM vs. vehicle: p = 0.8982, LEI-106-1.3 mM vs. vehicle: p = 0.6161, as assessed by one-way ANOVA with Tukey post-test, F(5,59) = 2.276). All values are expressed as the mean of corrected total cell fluorescence (CTCF) ± SEM (n = 9–12/condition). ns = non-significant.
Figure 4.
The inhibition of DAGLα induced morphological changes in bEnD.3 cells accompanied with reduced expression of VE-cadherin. Cell viability assay on bEnD.3 cells was performed using increasing doses of LEI-106 (100 nM–1.3 mM). For ICC experiments, the bEnD.3 cells were treated with two doses of LEI-106 (650 uM and 1.3 mM) for 15 min, followed by 2-AG or vehicle application. ICC was performed to detect possible changes in the expression of two tight junction proteins, claudin 5 and VE-cadherin. (A) The panel represents the setting of the experiments. (B) LEI-106 did not cause significant changes in the cell viability at any doses applied in the study. (LEI-106 at any doses vs. vehicle: p > 0.05, as assessed by one-way ANOVA with Bartlett post-test, F(8,26) = 0.7738). All data are expressed as the mean of optical density ± SEM (n = 4 in each group). ns = non-significant. (C) Representative ICC images of bEnd.3 cells treated with LEI-106 ±2-AG application. (D) Significant changes in the nuclear: cytoplasmic ratio were observed following LEI-106 treatment, suggesting possible morphological alteration caused by 2-AG depletion (LEI-106-650 µM vs. vehicle: p < 0.0001, LEI-106-1.3 mM vs. vehicle: p < 0.0001, as assessed by one-way ANOVA with Tukey post-test, F(8,85) = 30.50). 2-AG treatment following LEI-106 application significantly reversed these effects (LEI-106-650 µM vs. LEI-106-650 µM + 2-AG: p < 0.0001, LEI-106-1.3 mM vs. LEI-106-1.3 mM + 2-AG: p < 0.0001, as assessed by one-way ANOVA with Tukey post-test, F(8,85) = 30.50). All data are shown as the mean % of cellular area ± SEM (n = 9–12/condition). **** p < 0.0001, compared to corresponding controls. (E) The higher dose of LEI-106 (1.3 mM) significantly reduced VE-cadherin CTCF; however, no significant change was detected at the lower dose (650 µM) compared to vehicle control (LEI-106-650 µM vs. vehicle: p = 0.9052, LEI-106-1.3 mM vs. vehicle: p = 0.0233, as assessed by one-way ANOVA with Bartlett post-test, F(2,33) = 4.100). The administration of 2-AG increased the VE-cadherin CTCF, moderating the loss of VE-cadherin expression after LEI-106 treatment (LEI-106-1.3 mM vs. LEI-106-1.3 mM + 2-AG: p = 0.005, as assessed by one-way ANOVA with Tukey post-test, F(4,49) = 5.568). All values are expressed as the mean of corrected total cell fluorescence (CTCF) ± SEM (n = 9–12/condition). * p < 0.05, ** p < 0.01, **** p < 0.0001, compared to the corresponding controls. ns = non-significant. (F) Neither dose of LEI-106 treatment significantly influenced the CTCF of claudin-5 compared to vehicle control (LEI-106-650 µM vs. vehicle: p = 0.8982, LEI-106-1.3 mM vs. vehicle: p = 0.6161, as assessed by one-way ANOVA with Tukey post-test, F(5,59) = 2.276). All values are expressed as the mean of corrected total cell fluorescence (CTCF) ± SEM (n = 9–12/condition). ns = non-significant.
![Ijms 25 00531 g004]()
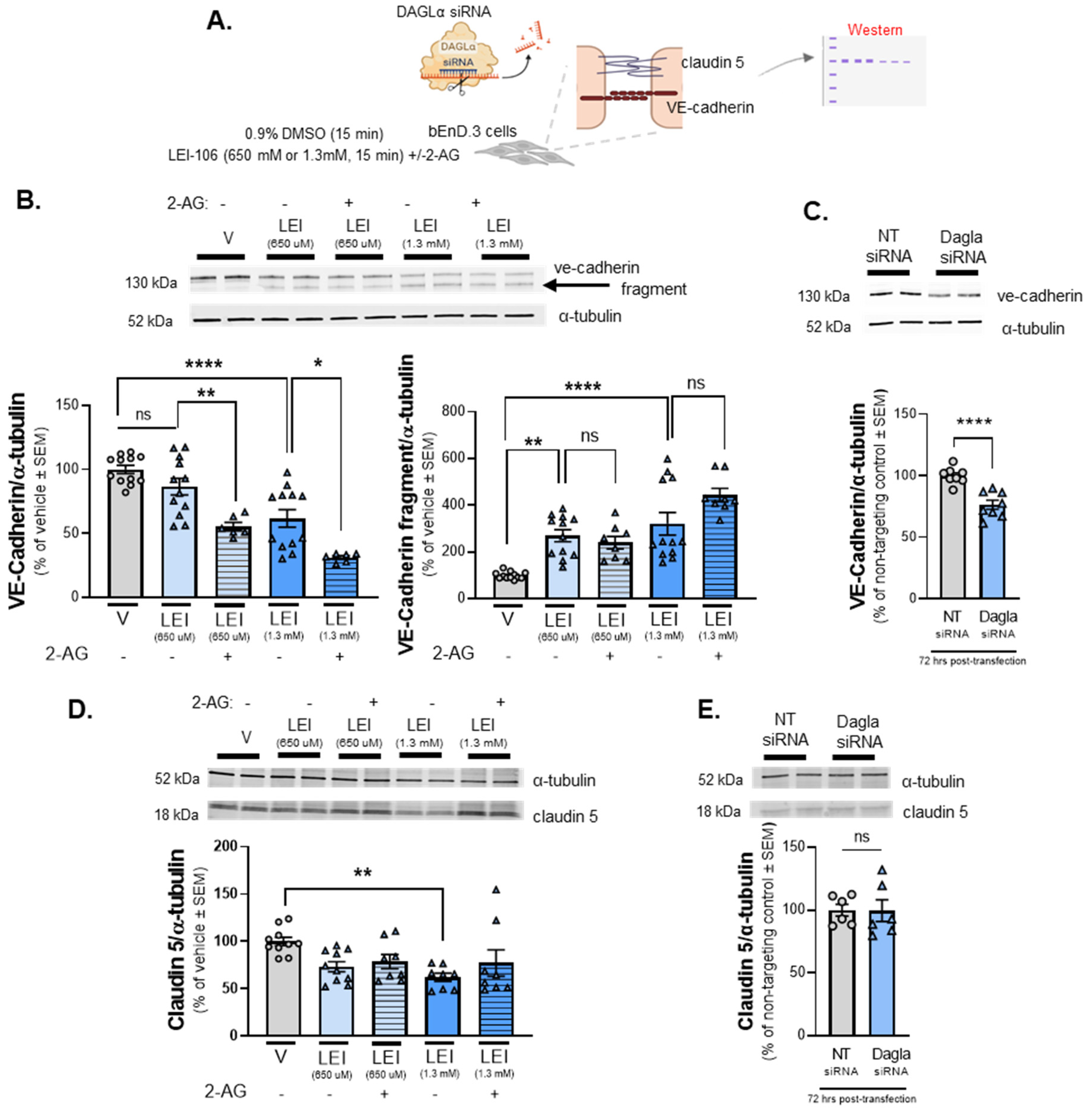
Figure 5.
The detection of VE-cadherin was altered after DAGLα blockade in bEnd.3 cells. The bEnD.3 cells were treated with LEI-106 (650 µM or 1.3 mM) for 15 min with or without 2-AG (600 pmol) application. The cell lysate was subjected to Western to detect the expression of VE-cadherin and claudin 5. In the siRNA transfection experiment, samples were harvested 72 h post-transfection and subjected to Western to detect the expression of VE-cadherin and claudin 5. (A) Scheme of experimental outline. (B) Representative image showing the expression of VE-cadherin with α-tubulin used as loading control. The pharmacological blockade of DAGLα induced fragmentation of VE-cadherin. The treatment of LEI-106 at 650 µM dose did not cause significant change in the detection of VE-cadherin main band compared to vehicle control (vehicle vs. LEI-106, 650 µM: p = 0.3565 as assessed by one-way ANOVA with Bartlett’s test, F(4,43) = 19.62), but it significantly increased the detection of VE-cadherin fragment (vehicle vs. LEI-106, 650 µM: p = 0.0014 as assessed by one-way ANOVA with Bartlett’s test, F(4,47) = 15.19). The higher dose of LEI-106 (1.3 mM) significantly reduced the detection of VE-cadherin main band and increased the detection of the fragmented one compared to vehicle control (main band: vehicle vs. LEI-106-1.3 mM: p < 0.0001 as assessed by one-way ANOVA with Bartlett’s test, F(4,43) = 19.62; fragment: vehicle vs. LEI-106, 1.3 mM: p < 0.0001 as assessed by one-way ANOVA with Bartlett’s test, F(4,47) = 15.19). The application of 2-AG further decreased the detection of main VE-cadherin band, but it did not significantly influence the fragmentation compared to corresponding controls (main band: LEI-106, 650 µM vs. LEI-106, 650 µM + 2-AG: p = 0.0092, LEI-106, 1.3 mM vs. LEI-106, 1.3 mM + 2-AG: p = 0.011 as assessed by one-way ANOVA with Bartlett’s test, F(4,43) = 19.62; fragment: LEI-106-650 µM vs. LEI-106, 650 µM + 2-AG: p = 0.9665, LEI-106-1.3 mM vs. LEI-106-1.3 mM + 2-AG: p = 0.0769 as assessed by one-way ANOVA with Bartlett’s test, F(4,47) = 15.19). All data presented as % of vehicle-treated ± SEM (n = 8–12/condition). * p < 0.05, ** p < 0.01, **** p < 0.0001, compared to the corresponding controls. ns = non-significant. (C) Representative image showing the expression of VE-cadherin with α-tubulin as loading control in transfected cell samples. The transfection of DAGLα-specific siRNA significantly reduced the detection of VE-cadherin compared to the non-targeting control (DAGLα siRNA vs. non-targeting control siRNA: p < 0.0001, t(14) = 5.376, as assessed by unpaired t-test). Data are shown as % of non-targeting control ± SEM (n = 8/group). **** denotes significantly different (p < 0.0001). (D) Representative image displaying the expression level of claudin 5. α-tubulin was used as a loading control. The lower dose of LEI-106 (650 µM) did not significantly influence the detection of claudin 5 compared to vehicle control (vehicle vs. LEI-106-650 µM: p = 0.0759 as assessed by one-way ANOVA with Tukey’s test, F(4,39) = 3.559). The treatment of LEI-106 at 1.3 mM dose significantly reduced the detection of claudin 5 compared to vehicle control (vehicle vs. LEI-106-1.3 mM: p = 0.0076 as assessed by one-way ANOVA with Tukey’s test, F(4,39) = 3.559). The application of 2-AG did not cause significant changes compared to the corresponding controls (LEI-106-650 µM vs. LEI-106-650 µM + 2-AG: p = 0.9870, LEI-106-1.3 mM vs. LEI-106-1.3 mM + 2-AG: p = 0.6535 as assessed by one-way ANOVA with Tukey’s test, F(4,39) = 3.559). All data presented as % of vehicle-treated ± SEM (n = 8-10/condition). ** p < 0.01, compared to vehicle control. (E) Representative image of the expression of claudin 5 along with α-tubulin as loading control in transfected cell samples. No significant difference in the detection of claudin 5 was observed after the transfection of DAGLα-specific siRNA (DAGLα siRNA vs. non-targeting control siRNA: p = 0.9666, t(10) = 0.04292, as assessed by unpaired t-test). Data are shown as % of non-targeting control ± SEM (n = 6/group). ns = non-significant.
Figure 5.
The detection of VE-cadherin was altered after DAGLα blockade in bEnd.3 cells. The bEnD.3 cells were treated with LEI-106 (650 µM or 1.3 mM) for 15 min with or without 2-AG (600 pmol) application. The cell lysate was subjected to Western to detect the expression of VE-cadherin and claudin 5. In the siRNA transfection experiment, samples were harvested 72 h post-transfection and subjected to Western to detect the expression of VE-cadherin and claudin 5. (A) Scheme of experimental outline. (B) Representative image showing the expression of VE-cadherin with α-tubulin used as loading control. The pharmacological blockade of DAGLα induced fragmentation of VE-cadherin. The treatment of LEI-106 at 650 µM dose did not cause significant change in the detection of VE-cadherin main band compared to vehicle control (vehicle vs. LEI-106, 650 µM: p = 0.3565 as assessed by one-way ANOVA with Bartlett’s test, F(4,43) = 19.62), but it significantly increased the detection of VE-cadherin fragment (vehicle vs. LEI-106, 650 µM: p = 0.0014 as assessed by one-way ANOVA with Bartlett’s test, F(4,47) = 15.19). The higher dose of LEI-106 (1.3 mM) significantly reduced the detection of VE-cadherin main band and increased the detection of the fragmented one compared to vehicle control (main band: vehicle vs. LEI-106-1.3 mM: p < 0.0001 as assessed by one-way ANOVA with Bartlett’s test, F(4,43) = 19.62; fragment: vehicle vs. LEI-106, 1.3 mM: p < 0.0001 as assessed by one-way ANOVA with Bartlett’s test, F(4,47) = 15.19). The application of 2-AG further decreased the detection of main VE-cadherin band, but it did not significantly influence the fragmentation compared to corresponding controls (main band: LEI-106, 650 µM vs. LEI-106, 650 µM + 2-AG: p = 0.0092, LEI-106, 1.3 mM vs. LEI-106, 1.3 mM + 2-AG: p = 0.011 as assessed by one-way ANOVA with Bartlett’s test, F(4,43) = 19.62; fragment: LEI-106-650 µM vs. LEI-106, 650 µM + 2-AG: p = 0.9665, LEI-106-1.3 mM vs. LEI-106-1.3 mM + 2-AG: p = 0.0769 as assessed by one-way ANOVA with Bartlett’s test, F(4,47) = 15.19). All data presented as % of vehicle-treated ± SEM (n = 8–12/condition). * p < 0.05, ** p < 0.01, **** p < 0.0001, compared to the corresponding controls. ns = non-significant. (C) Representative image showing the expression of VE-cadherin with α-tubulin as loading control in transfected cell samples. The transfection of DAGLα-specific siRNA significantly reduced the detection of VE-cadherin compared to the non-targeting control (DAGLα siRNA vs. non-targeting control siRNA: p < 0.0001, t(14) = 5.376, as assessed by unpaired t-test). Data are shown as % of non-targeting control ± SEM (n = 8/group). **** denotes significantly different (p < 0.0001). (D) Representative image displaying the expression level of claudin 5. α-tubulin was used as a loading control. The lower dose of LEI-106 (650 µM) did not significantly influence the detection of claudin 5 compared to vehicle control (vehicle vs. LEI-106-650 µM: p = 0.0759 as assessed by one-way ANOVA with Tukey’s test, F(4,39) = 3.559). The treatment of LEI-106 at 1.3 mM dose significantly reduced the detection of claudin 5 compared to vehicle control (vehicle vs. LEI-106-1.3 mM: p = 0.0076 as assessed by one-way ANOVA with Tukey’s test, F(4,39) = 3.559). The application of 2-AG did not cause significant changes compared to the corresponding controls (LEI-106-650 µM vs. LEI-106-650 µM + 2-AG: p = 0.9870, LEI-106-1.3 mM vs. LEI-106-1.3 mM + 2-AG: p = 0.6535 as assessed by one-way ANOVA with Tukey’s test, F(4,39) = 3.559). All data presented as % of vehicle-treated ± SEM (n = 8-10/condition). ** p < 0.01, compared to vehicle control. (E) Representative image of the expression of claudin 5 along with α-tubulin as loading control in transfected cell samples. No significant difference in the detection of claudin 5 was observed after the transfection of DAGLα-specific siRNA (DAGLα siRNA vs. non-targeting control siRNA: p = 0.9666, t(10) = 0.04292, as assessed by unpaired t-test). Data are shown as % of non-targeting control ± SEM (n = 6/group). ns = non-significant.
![Ijms 25 00531 g005]()
Figure 6.
Increased intracellular calcium level and elevated PKC activity after DAGLα inhibition in bEnD.3 cells. The bEnd.3 cells plated on collagen-coated coverslips were subjected to calcium imaging with the dye Fura-2. After 2-min baseline observation in buffer, cells were treated with LEI-106 (650 µM or 1.3 mM) or vehicle (0.9% DMSO) for 2 min, followed by a subsequent 2-min washout phase. In a separate experiment, cells received buffer spiked with 2-AG (600 pmol/coverslip) during the washout phase. Images used for analysis were captured at the beginning of the treatment application and after removal of the treatment, approximately 2 min apart. In a separate experiment, protein kinase C (PKC) activity was measured by ELISA in bEnD.3 cells after LEI-106 treatment. bEnD.3 cells were treated with calpain inhibitor, calpeptin (10 µM) or Rho-kinase inhibitor, SR3677 (100 nM) 30 min prior to the application of LEI-106 (650 µM), then subjected to Western blot to detect changes in the fragmentation of VE-cadherin caused by DAGLα inhibition. (A) Schema of experimental setting. (B) Representative calcium images of bEnD.3 cells at baseline, then treated with two different doses of LEI-106 (650 µM, 1.3 mM). LEI-106 at both doses significantly increased the intracellular calcium levels compared to vehicle control (LEI-106-650 µM vs. vehicle: p < 0.0001, LEI-106-1.3 mM vs. vehicle: p < 0.0001, as assessed by one-way ANOVA with Tukey post-test, F(3,3295) = 482.1). All values are % of baseline obtained from three independent experiments using three coverslips in each. In each coverslip, 100 cells were analyzed by FIJI Image J software 1.54. **** p < 0.0001 compared to vehicle control. Red dashed line represents the baseline. (C) Representative images showing intracellular calcium levels in bEnD.3 cells treated with LEI-106 (650 µM or 1.3 mM) with or without 2-AG. The application of 2-AG significantly reduced the elevation of intracellular calcium caused by LEI-106 treatment (LEI-106-650 µM vs. LEI-106-650 µM + 2-AG: p < 0.0001, LEI-106-1.3 mM vs. LEI-106-1.3 mM + 2-AG: p < 0.0001, as assessed by one-way ANOVA with Tukey post-test, F(3,2700) = 889.9). All data are shown as % of baseline obtained from three independent experiments using three coverslips in each. In each coverslip, 100 cells were analyzed by Image J software. **** p < 0.0001 compared to vehicle control. Red dashed line represents the baseline. (D) Representative image of Western blot showing VE-cadherin expression in bEnD.3 cells treated with LEI-106 with or without calpain inhibitor, calpeptin (10 µM). α-tubulin was used as a loading control. The calpain inhibitor did not significantly change the fragmentation of VE-cadherin caused by LEI-106 treatment (LEI-106-650 µM vs. LEI-106-650 µM + calpeptin: p > 0.9999, as assessed by one-way ANOVA with Tukey post-test, F(4,20) = 84.40). All data are shown as % of vehicle-treated ± SEM (n = 5–6/condition). **** p < 0.0001, ns = non-significant. (E) The assessment of PKC activity by ELISA showed that LEI-106 treatment (15 min) significantly increased the activity level of PKC compared to vehicle control (LEI-106-650 µM vs. vehicle: * p = 0.0289, t(8) = 2.658; LEI-106-1.3 mM vs. vehicle: ** p = 0.0099, t(8) = 3.362 as assessed by unpaired t-test). Data are presented as the mean of PKC in ng/µg of cell lysate ± SEM (n = 5/group). (F) Representative image of immunoblot targeting VE-cadherin and α-tubulin as loading control in bEnD.3 cells treated with LEI-106 (650 µM) with or without Rho-kinase inhibitor, SR3677 (100 nM). The pretreatment (30 min) with Rho-kinase inhibitor significantly mitigated the fragmentation of VE-cadherin caused by LEI-106 (LEI-106-650 µM vs. LEI-106-650 µM + SR3677: p < 0.0001, as assessed by one-way ANOVA with Tukey post-test, F(2,23) = 110.3). All data are shown as % of vehicle-treated ± SEM (n = 8–10/condition). **** p < 0.0001, compared to the LEI-106 (650 µM) treatment.
Figure 6.
Increased intracellular calcium level and elevated PKC activity after DAGLα inhibition in bEnD.3 cells. The bEnd.3 cells plated on collagen-coated coverslips were subjected to calcium imaging with the dye Fura-2. After 2-min baseline observation in buffer, cells were treated with LEI-106 (650 µM or 1.3 mM) or vehicle (0.9% DMSO) for 2 min, followed by a subsequent 2-min washout phase. In a separate experiment, cells received buffer spiked with 2-AG (600 pmol/coverslip) during the washout phase. Images used for analysis were captured at the beginning of the treatment application and after removal of the treatment, approximately 2 min apart. In a separate experiment, protein kinase C (PKC) activity was measured by ELISA in bEnD.3 cells after LEI-106 treatment. bEnD.3 cells were treated with calpain inhibitor, calpeptin (10 µM) or Rho-kinase inhibitor, SR3677 (100 nM) 30 min prior to the application of LEI-106 (650 µM), then subjected to Western blot to detect changes in the fragmentation of VE-cadherin caused by DAGLα inhibition. (A) Schema of experimental setting. (B) Representative calcium images of bEnD.3 cells at baseline, then treated with two different doses of LEI-106 (650 µM, 1.3 mM). LEI-106 at both doses significantly increased the intracellular calcium levels compared to vehicle control (LEI-106-650 µM vs. vehicle: p < 0.0001, LEI-106-1.3 mM vs. vehicle: p < 0.0001, as assessed by one-way ANOVA with Tukey post-test, F(3,3295) = 482.1). All values are % of baseline obtained from three independent experiments using three coverslips in each. In each coverslip, 100 cells were analyzed by FIJI Image J software 1.54. **** p < 0.0001 compared to vehicle control. Red dashed line represents the baseline. (C) Representative images showing intracellular calcium levels in bEnD.3 cells treated with LEI-106 (650 µM or 1.3 mM) with or without 2-AG. The application of 2-AG significantly reduced the elevation of intracellular calcium caused by LEI-106 treatment (LEI-106-650 µM vs. LEI-106-650 µM + 2-AG: p < 0.0001, LEI-106-1.3 mM vs. LEI-106-1.3 mM + 2-AG: p < 0.0001, as assessed by one-way ANOVA with Tukey post-test, F(3,2700) = 889.9). All data are shown as % of baseline obtained from three independent experiments using three coverslips in each. In each coverslip, 100 cells were analyzed by Image J software. **** p < 0.0001 compared to vehicle control. Red dashed line represents the baseline. (D) Representative image of Western blot showing VE-cadherin expression in bEnD.3 cells treated with LEI-106 with or without calpain inhibitor, calpeptin (10 µM). α-tubulin was used as a loading control. The calpain inhibitor did not significantly change the fragmentation of VE-cadherin caused by LEI-106 treatment (LEI-106-650 µM vs. LEI-106-650 µM + calpeptin: p > 0.9999, as assessed by one-way ANOVA with Tukey post-test, F(4,20) = 84.40). All data are shown as % of vehicle-treated ± SEM (n = 5–6/condition). **** p < 0.0001, ns = non-significant. (E) The assessment of PKC activity by ELISA showed that LEI-106 treatment (15 min) significantly increased the activity level of PKC compared to vehicle control (LEI-106-650 µM vs. vehicle: * p = 0.0289, t(8) = 2.658; LEI-106-1.3 mM vs. vehicle: ** p = 0.0099, t(8) = 3.362 as assessed by unpaired t-test). Data are presented as the mean of PKC in ng/µg of cell lysate ± SEM (n = 5/group). (F) Representative image of immunoblot targeting VE-cadherin and α-tubulin as loading control in bEnD.3 cells treated with LEI-106 (650 µM) with or without Rho-kinase inhibitor, SR3677 (100 nM). The pretreatment (30 min) with Rho-kinase inhibitor significantly mitigated the fragmentation of VE-cadherin caused by LEI-106 (LEI-106-650 µM vs. LEI-106-650 µM + SR3677: p < 0.0001, as assessed by one-way ANOVA with Tukey post-test, F(2,23) = 110.3). All data are shown as % of vehicle-treated ± SEM (n = 8–10/condition). **** p < 0.0001, compared to the LEI-106 (650 µM) treatment.
![Ijms 25 00531 g006]()
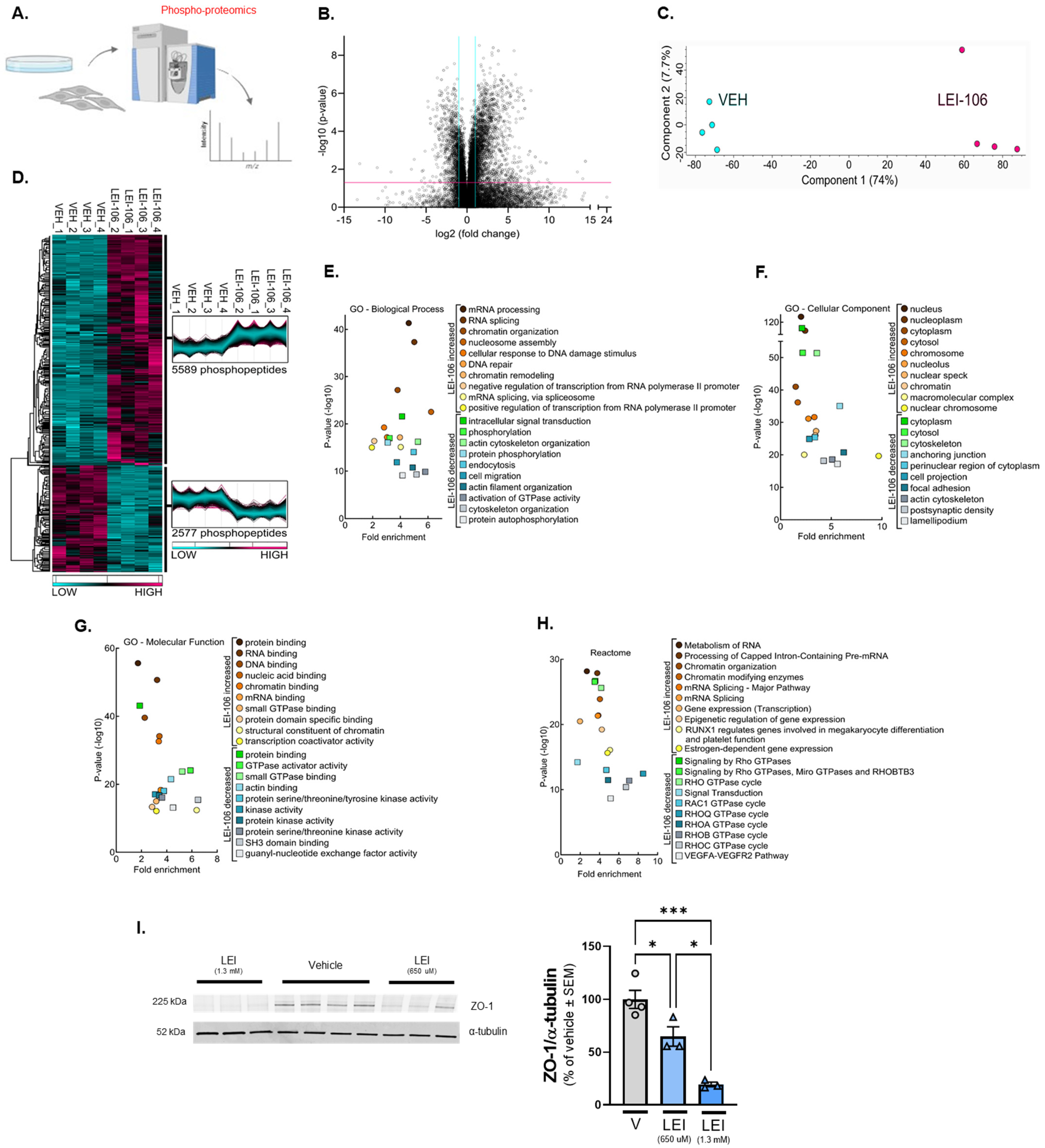
Figure 7.
Quantitative phospho-proteomics after DAGLα inhibition in bEnD.3 cells. bEnd.3 cells cultured on 150 mm plates were treated with vehicle (0.9% DMSO in media) or LEI-106 (650 µM) for 15 min, then subjected to quantitative phospho-proteomics. (A) Schema of the experiment. (B) Volcano plot of bEnd.3 of detected phospho-peptides showing a greater than 2-fold change after LEI-106 compared to vehicle treatment. (C) Principal Component Analysis (PCA) of cells treated with vehicle or LEI106 (650 µM). (D) Heatmap of phospho-peptides increased by LEI-106 treatment compared to vehicle or decreased compared to vehicle. (E) GO Biological process functional analysis showing the top 10 processes altered in each condition. (F) GO cellular compartment functional analysis showing the top 10 compartments in which phospho-peptides were impacted in each condition. (G) GO molecular function functional analysis shows the top 10 in which phospho-peptides were impacted in each condition. (H) Top 10 Reactome Pathways associated with changes in phospho-peptides in each outcome. n = 4 biological replicates per condition. (I) bEnd.3 cells were treated with LEI-106 (650 µM or 1.03 mM) or vehicle for 15 min, then subjected to Western to detect ZO-1. Representative image showing the expression of ZO-1 along with α-tubulin as a loading control. The blockade of DAGLα significantly decreased the detection of ZO-1, compared to vehicle control (vehicle vs. LEI-106-650 µM: p = 0.034, vehicle vs. LEI-106-1.3 mM: p = 0.0004, LEI-106-650 µM vs. LEI-106-1.3 mM: p = 0.0142 as assessed by one-way ANOVA with Tukey post-test, F(2,7) = 27.41). All data presented as % of vehicle-treated ± SEM (n = 3–4/condition). * p < 0.05, *** p < 0.001.
Figure 7.
Quantitative phospho-proteomics after DAGLα inhibition in bEnD.3 cells. bEnd.3 cells cultured on 150 mm plates were treated with vehicle (0.9% DMSO in media) or LEI-106 (650 µM) for 15 min, then subjected to quantitative phospho-proteomics. (A) Schema of the experiment. (B) Volcano plot of bEnd.3 of detected phospho-peptides showing a greater than 2-fold change after LEI-106 compared to vehicle treatment. (C) Principal Component Analysis (PCA) of cells treated with vehicle or LEI106 (650 µM). (D) Heatmap of phospho-peptides increased by LEI-106 treatment compared to vehicle or decreased compared to vehicle. (E) GO Biological process functional analysis showing the top 10 processes altered in each condition. (F) GO cellular compartment functional analysis showing the top 10 compartments in which phospho-peptides were impacted in each condition. (G) GO molecular function functional analysis shows the top 10 in which phospho-peptides were impacted in each condition. (H) Top 10 Reactome Pathways associated with changes in phospho-peptides in each outcome. n = 4 biological replicates per condition. (I) bEnd.3 cells were treated with LEI-106 (650 µM or 1.03 mM) or vehicle for 15 min, then subjected to Western to detect ZO-1. Representative image showing the expression of ZO-1 along with α-tubulin as a loading control. The blockade of DAGLα significantly decreased the detection of ZO-1, compared to vehicle control (vehicle vs. LEI-106-650 µM: p = 0.034, vehicle vs. LEI-106-1.3 mM: p = 0.0004, LEI-106-650 µM vs. LEI-106-1.3 mM: p = 0.0142 as assessed by one-way ANOVA with Tukey post-test, F(2,7) = 27.41). All data presented as % of vehicle-treated ± SEM (n = 3–4/condition). * p < 0.05, *** p < 0.001.
![Ijms 25 00531 g007]()
Figure 8.
2-AG depletion via inhibition of DAGLα disrupted integrity of bEnd.3 endothelial cells. The inhibition of DAGLα either with LEI-106 or siRNA led to loss of 2-AG and induced paracellular leak manifesting by increased C14-sucrose transport through the monolayer of bEnd.3 cells. Disruption of 2-AG homeostasis was connected to increased intracellular calcium signaling, enhanced activity of protein kinase C (PKC), and fragmentation of tight junction protein, VE-cadherin. Inhibition of Rho-kinase mitigated the fragmentation of VE-cadherin induced by the blockade of DAGLα.
Figure 8.
2-AG depletion via inhibition of DAGLα disrupted integrity of bEnd.3 endothelial cells. The inhibition of DAGLα either with LEI-106 or siRNA led to loss of 2-AG and induced paracellular leak manifesting by increased C14-sucrose transport through the monolayer of bEnd.3 cells. Disruption of 2-AG homeostasis was connected to increased intracellular calcium signaling, enhanced activity of protein kinase C (PKC), and fragmentation of tight junction protein, VE-cadherin. Inhibition of Rho-kinase mitigated the fragmentation of VE-cadherin induced by the blockade of DAGLα.


 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}