
Establishment of a Triple Quadrupole HPLC-MS Quantitation Method for Dystrophin Protein in Mouse and Human Skeletal Muscle

 ,
, 
 , , and
, , and
Abstract
:1. Introduction
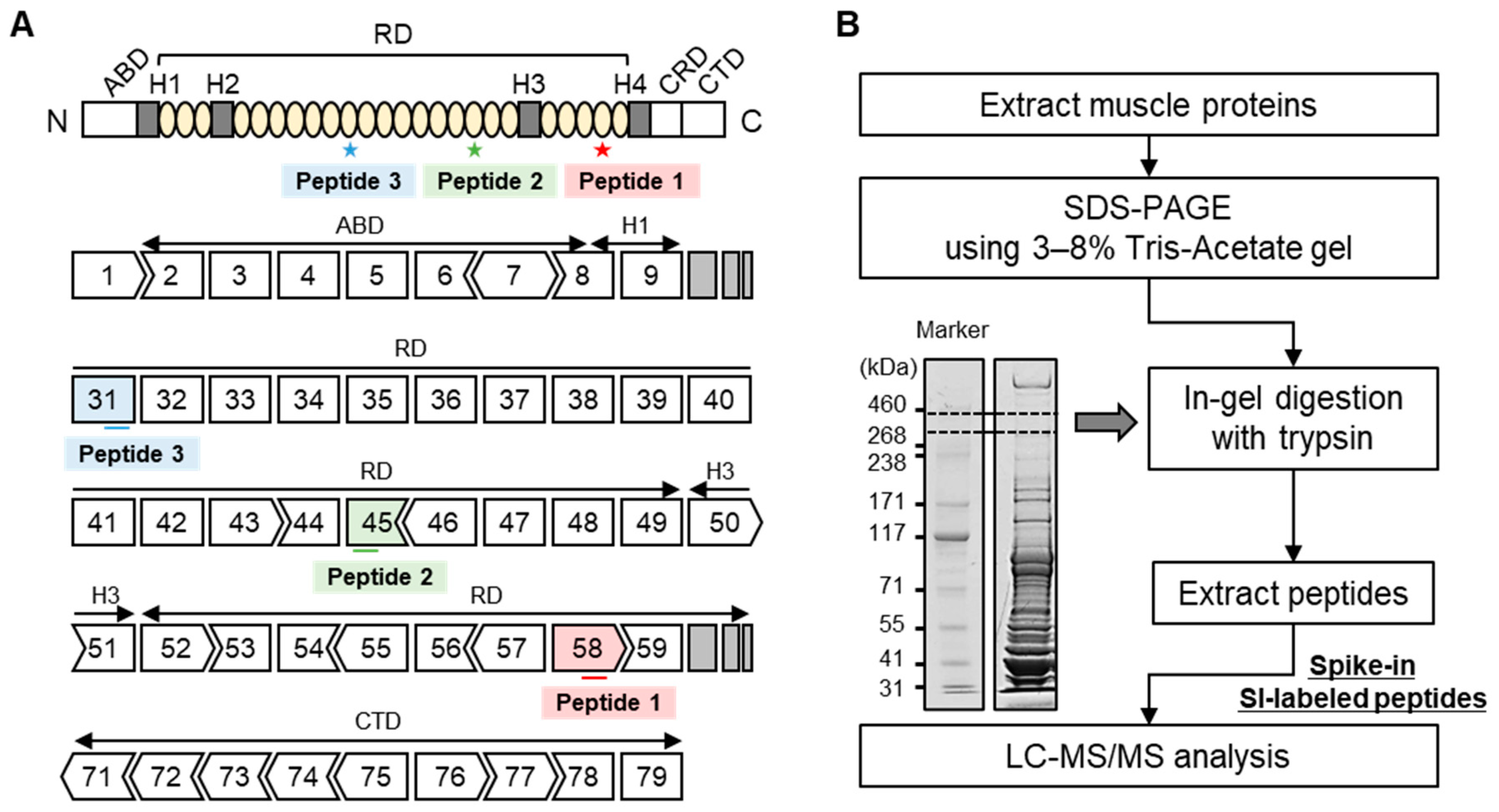
2. Results
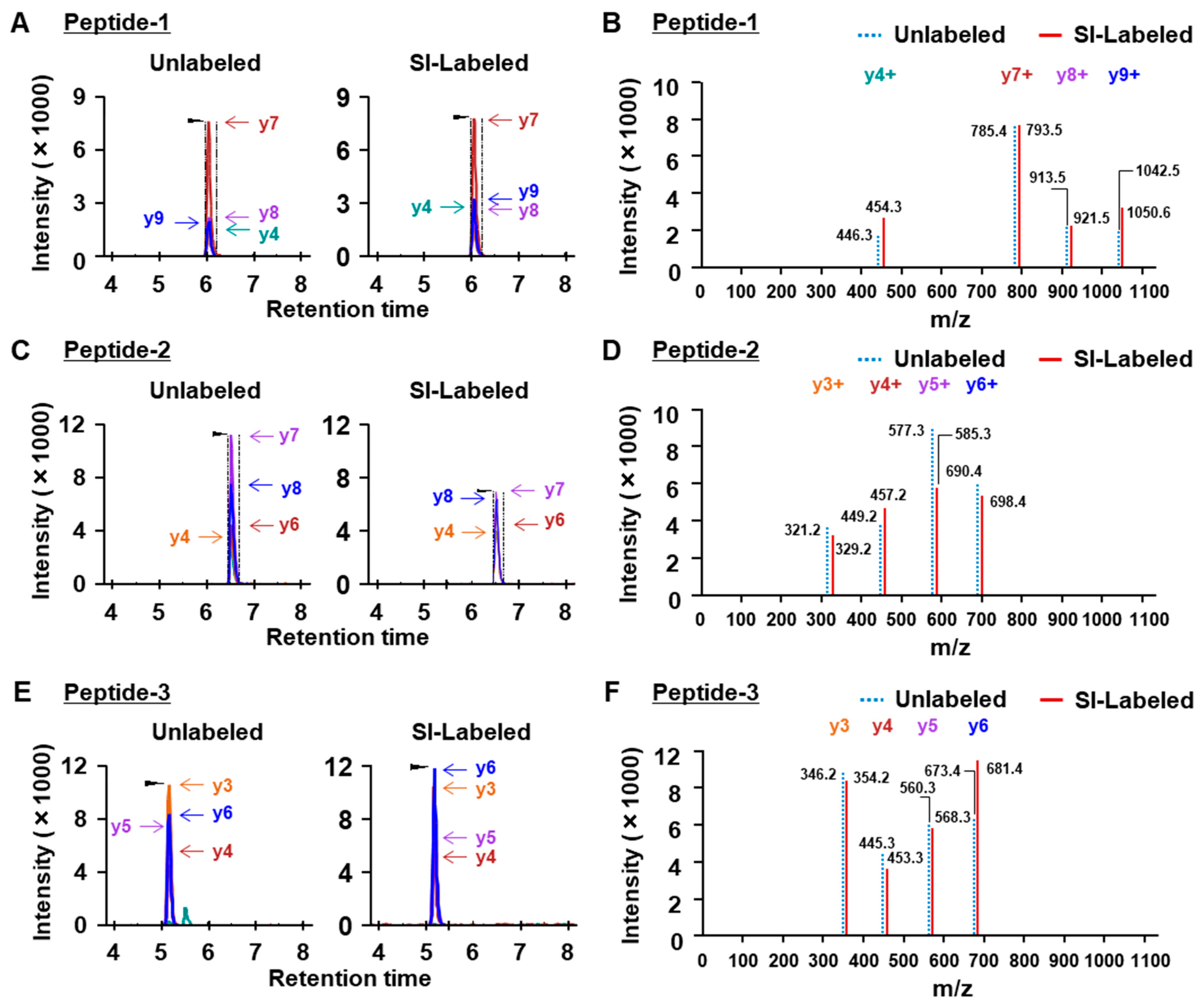
2.1. Optimization of LC-MS Conditions Using the Analysis of Dystrophin Peptides
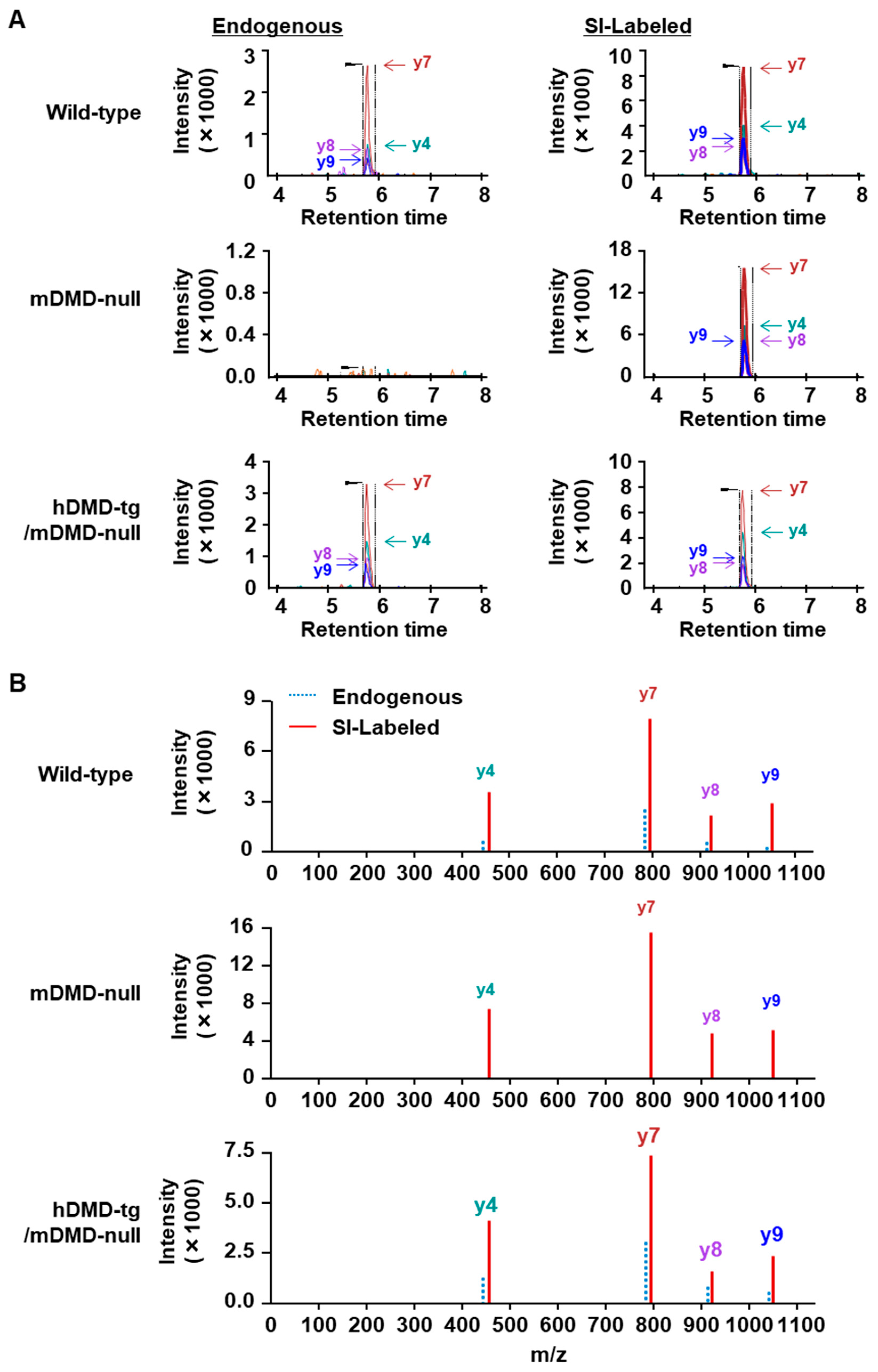
2.2. Protein Quantitation of Dystrophin in Mouse Skeletal Muscle
2.3. Protein Quantitation of Dystrophin in Human Muscle Biopsy Specimens
3. Discussion
4. Materials and Methods
4.1. Skeletal Muscles
4.2. Stable Isotope-Labeled Dystrophin Peptides
4.3. LC-MS Analysis
Total Protein Extraction from Mouse Skeletal Muscles
4.4. SDS-PAGE Using 3–8% Tris-Acetate Gel
4.5. In-Gel Tryptic Digestion
4.6. Protein Quantitation of Dystrophin Protein Using an LC-MS
LC-MS Analysis for MRM-Based Protein Quantitation
4.7. Optimization of LC-MS Conditions for the Analysis of Synthetic Dystrophin Peptides
4.8. Quantitation of Dystrophin in Mouse and Human Skeletal Muscle
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hoffman, E.P.; Brown, R.H.; Kunkel, L.M. Dystrophin: The Protein Product of the Duchenne Muscular Dystrophy Locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P.; Fischbeck, K.H.; Brown, R.H.; Johnson, M.; Medori, R.; Loire, J.D.; Harris, J.B.; Waterston, R.; Brooke, M.; Specht, L.; et al. Characterization of Dystrophin in Muscle-Biopsy Specimens from Patients with Duchenne’s or Becker’s Muscular Dystrophy. N. Engl. J. Med. 1988, 318, 1363–1368. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, C.; Wong, S.; Elson, E.L. Mechanical Function of Dystrophin in Muscle Cells. J. Cell Biol. 1995, 128, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Klingler, W.; Jurkat-Rott, K.; Lehmann-Horn, F.; Schleip, R. The Role of Fibrosis in Duchenne Muscular Dystrophy. Acta Myol. 2012, 31, 184–195. [Google Scholar] [PubMed]
- Petrof, B.J.; Shrager, J.B.; Stedman, H.H.; Kelly, A.M.; Sweeney, H.L. Dystrophin Protects the Sarcolemma from Stresses Developed during Muscle Contraction. Proc. Natl. Acad. Sci. USA 1993, 90, 3710–3714. [Google Scholar] [CrossRef] [PubMed]
- Tominari, T.; Aoki, Y. Clinical Development of Novel Therapies for Duchenne Muscular Dystrophy—Current and Future. Neurol. Clin. Neurosci. 2023, 11, 111–118. [Google Scholar] [CrossRef]
- Zhao, M.; Tazumi, A.; Takayama, S.; Takenaka-Ninagawa, N.; Nalbandian, M.; Nagai, M.; Nakamura, Y.; Nakasa, M.; Watanabe, A.; Ikeya, M.; et al. Induced Fetal Human Muscle Stem Cells with High Therapeutic Potential in a Mouse Muscular Dystrophy Model. Stem Cell Rep. 2020, 15, 80–94. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.M.; Campbell, C.; Torricelli, R.E.; Finkel, R.S.; Flanigan, K.M.; Goemans, N.; Heydemann, P.; Kaminska, A.; Kirschner, J.; Muntoni, F.; et al. Ataluren in Patients with Nonsense Mutation Duchenne Muscular Dystrophy (ACT DMD): A Multicentre, Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2017, 390, 1489–1498. [Google Scholar] [CrossRef]
- Min, Y.-L.; Li, H.; Rodriguez-Caycedo, C.; Mireault, A.A.; Huang, J.; Shelton, J.M.; McAnally, J.R.; Amoasii, L.; Mammen, P.P.A.; Bassel-Duby, R.; et al. CRISPR-Cas9 Corrects Duchenne Muscular Dystrophy Exon 44 Deletion Mutations in Mice and Human Cells. Sci. Adv. 2019, 5, eaav4324. [Google Scholar] [CrossRef]
- Moretti, A.; Fonteyne, L.; Giesert, F.; Hoppmann, P.; Meier, A.B.; Bozoglu, T.; Baehr, A.; Schneider, C.M.; Sinnecker, D.; Klett, K.; et al. Somatic Gene Editing Ameliorates Skeletal and Cardiac Muscle Failure in Pig and Human Models of Duchenne Muscular Dystrophy. Nat. Med. 2020, 26, 207–214. [Google Scholar] [CrossRef]
- Komaki, H.; Nagata, T.; Saito, T.; Masuda, S.; Takeshita, E.; Sasaki, M.; Tachimori, H.; Nakamura, H.; Aoki, Y.; Takeda, S. Systemic Administration of the Antisense Oligonucleotide NS-065/NCNP-01 for Skipping of Exon 53 in Patients with Duchenne Muscular Dystrophy. Sci. Transl. Med. 2018, 10, eaan0713. [Google Scholar] [CrossRef] [PubMed]
- Drachman, D.B.; Toyka, K.V.; Myer, E. Predonisone in Duchenne muscular dystrophy. Lancet 1974, 304, 1409–1412. [Google Scholar] [CrossRef] [PubMed]
- Angelini, C.; Peterle, E. Old and New Therapeutic Developments in Steroid Treatment in Duchenne Muscular Dystrophy. Acta Myol. 2012, 31, 9–15. [Google Scholar] [PubMed]
- Komaki, H.; Maegaki, Y.; Matsumura, T.; Shiraishi, K.; Awano, H.; Nakamura, A.; Kinoshita, S.; Ogata, K.; Ishigaki, K.; Saitoh, S.; et al. Early Phase 2 Trial of TAS-205 in Patients with Duchenne Muscular Dystrophy. Ann. Clin. Transl. Neurol. 2020, 7, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Mohri, I.; Aritake, K.; Taniguchi, H.; Sato, Y.; Kamauchi, S.; Nagata, N.; Maruyama, T.; Taniike, M.; Urade, Y. Inhibition of Prostaglandin D Synthase Suppresses Muscular Necrosis. Am. J. Pathol. 2009, 174, 1735–1744. [Google Scholar] [CrossRef] [PubMed]
- Echigoya, Y.; Nakamura, A.; Nagata, T.; Urasawa, N.; Lim, K.R.Q.; Trieu, N.; Panesar, D.; Kuraoka, M.; Moulton, H.M.; Saito, T.; et al. Effects of Systemic Multiexon Skipping with Peptide-Conjugated Morpholinos in the Heart of a Dog Model of Duchenne Muscular Dystrophy. Proc. Natl. Acad. Sci. USA 2017, 114, 4213–4218. [Google Scholar] [CrossRef]
- Cirak, S.; Arechavala-Gomeza, V.; Guglieri, M.; Feng, L.; Torelli, S.; Anthony, K.; Abbs, S.; Garralda, M.E.; Bourke, J.; Wells, D.J.; et al. Exon Skipping and Dystrophin Restoration in Patients with Duchenne Muscular Dystrophy after Systemic Phosphorodiamidate Morpholino Oligomer Treatment: An Open-Label, Phase 2, Dose-Escalation Study. Lancet 2011, 378, 595–605. [Google Scholar] [CrossRef]
- Aoki, Y.; Nakamura, A.; Yokota, T.; Saito, T.; Okazawa, H.; Nagata, T.; Takeda, S. In-Frame Dystrophin Following Exon 51-Skipping Improves Muscle Pathology and Function in the Exon 52–Deficient Mdx Mouse. Mol. Ther. 2010, 18, 1995–2005. [Google Scholar] [CrossRef]
- Shimizu, R.; Ohata, M.; Tachimori, H.; Kimura, E.; Harada, Y.; Takeshita, E.; Tamaura, A.; Takeda, S.; Komaki, H. Expectations and Anxieties of Duchenne Muscular Dystrophy Patients and Their Families during the First-in-Human Clinical Trial of NS-065/NCNP-01. Brain Dev. 2020, 42, 348–356. [Google Scholar] [CrossRef]
- Iftikhar, M.; Frey, J.; Shohan, M.J.; Malek, S.; Mousa, S.A. Current and Emerging Therapies for Duchenne Muscular Dystrophy and Spinal Muscular Atrophy. Pharmacol. Ther. 2021, 220, 107719. [Google Scholar] [CrossRef]
- Uaesoontrachoon, K.; Srinivassane, S.; Warford, J.; Mekhssian, K.; Montpetit, H.; Beauvois, R.; Keyhani, A.; Hathout, Y.; Yamashita, T.; Satou, Y.; et al. Orthogonal Analysis of Dystrophin Protein and MRNA as a Surrogate Outcome for Drug Development. Biomark. Med. 2019, 13, 1209–1225. [Google Scholar] [CrossRef]
- Harding, P.L.; Fall, A.M.; Honeyman, K.; Fletcher, S.; Wilton, S.D. The Influence of Antisense Oligonucleotide Length on Dystrophin Exon Skipping. Mol. Ther. 2007, 15, 157–166. [Google Scholar] [CrossRef]
- Brown, K.J.; Marathi, R.; Fiorillo, A.A.; Ciccimaro, E.F.; Sharma, S.; Rowlands, D.S.; Rayavarapu, S.; Nagaraju, K.; Hoffman, E.P.; Hathout, Y. Accurate Quantitation of Dystrophin Protein in Human Skeletal Muscle Using Mass Spectrometry. J. Bioanal. Biomed. 2012, S7. [Google Scholar] [CrossRef]
- Canessa, E.H.; Goswami, M.V.; Alayi, T.D.; Hoffman, E.P.; Hathout, Y. Absolute Quantification of Dystrophin Protein in Human Muscle Biopsies Using Parallel Reaction Monitoring (PRM). J. Mass Spectrom. 2020, 55, e4437. [Google Scholar] [CrossRef]
- Kudoh, H.; Ikeda, H.; Kakitani, M.; Ueda, A.; Hayasaka, M.; Tomizuka, K.; Hanaoka, K. A New Model Mouse for Duchenne Muscular Dystrophy Produced by 2.4 Mb Deletion of Dystrophin Gene Using Cre-LoxP Recombination System. Biochem. Biophys. Res. Commun. 2005, 328, 507–516. [Google Scholar] [CrossRef]
- AC’t Hoen, P.; de Meijer, E.J.; Boer, J.M.; Vossen, R.H.; Turk, R.; Maatman, R.G.; Davies, K.E.; van Ommen, G.J.; van Deutekom, J.C.; den Dunnen, J.T. Generation and Characterization of Transgenic Mice with the Full-Length Human DMD Gene. J. Biol. Chem. 2008, 283, 5899–5907. [Google Scholar] [CrossRef]
- Echigoya, Y.; Lim, K.R.Q.; Trieu, N.; Bao, B.; Nichols, B.M.; Vila, M.C.; Novak, J.S.; Hara, Y.; Lee, J.; Touznik, A.; et al. Quantitative Antisense Screening and Optimization for Exon 51 Skipping in Duchenne Muscular Dystrophy. Mol. Ther. 2017, 25, 2561–2572. [Google Scholar] [CrossRef]
- Roshmi, R.R.; Yokota, T. Viltolarsen for the Treatment of Duchenne Muscular Dystrophy. Drugs Today 2019, 55, 627. [Google Scholar] [CrossRef]
- Neubert, H.; Fan, Y.-Y.; Ocaa, M.F. Quantification of Protein Biomarkers in Tissues: New Capabilities with Pellet Digestion Peptide Immunoaffinity LCMS/MS. Bioanalysis 2016, 8, 1551–1555. [Google Scholar] [CrossRef]
- Razavi, M.; Anderson, N.L.; Pope, M.E.; Yip, R.; Pearson, T.W. High Precision Quantification of Human Plasma Proteins Using the Automated SISCAPA Immuno-MS Workflow. New Biotechnol. 2016, 33, 494–502. [Google Scholar] [CrossRef]
- Farrokhi, V.; Walsh, J.; Palandra, J.; Brodfuehrer, J.; Caiazzo, T.; Owens, J.; Binks, M.; Neelakantan, S.; Yong, F.; Dua, P.; et al. Dystrophin and Mini-Dystrophin Quantification by Mass Spectrometry in Skeletal Muscle for Gene Therapy Development in Duchenne Muscular Dystrophy. Gene Ther. 2022, 29, 608–615. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence | SI-Labeled M.W. | Unlabeled M.W. |
|---|---|---|---|
| Peptide 1 | IFLTEQPLEGLEK | 1524.7 | 1516.7 |
| Peptide 2 | TLNATGEEIIQQSSK | 1626.7 | 1618.7 |
| Peptide 3 | VLSQIDVAQK | 1108.2 | 1100.2 |
| Peptide 1 | Peptide 2 | Peptide 3 | |
|---|---|---|---|
| Precursor Ion | [IFLTEQPLEGLEK] ++ | [TLNATGEEIIQQSSK] +++ | [VLSQIDVAQK] ++ |
| Fragment ion 1 | y9: [EQPLEGLEK] + | y6: [IQQSSK] + | y6: [IDVAQK] + |
| Fragment ion 2 | y8: [QPLEGLEK] + | y5: [QQSSK] + | y5: [DVAQK] + |
| Fragment ion 3 | y7: [PLEGLEK] + | y4: [QSSK] + | y4: [VAQK] + |
| Fragment ion 4 | y4: [GLEK] + | y3: [SSK] + | y3: [AQK] + |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tominari, T.; Takatoya, M.; Matsubara, T.; Matsunobe, M.; Arai, D.; Matsumoto, C.; Hirata, M.; Yoshinouchi, S.; Miyaura, C.; Itoh, Y.; et al. Establishment of a Triple Quadrupole HPLC-MS Quantitation Method for Dystrophin Protein in Mouse and Human Skeletal Muscle. Int. J. Mol. Sci. 2024, 25, 303. https://doi.org/10.3390/ijms25010303
Tominari T, Takatoya M, Matsubara T, Matsunobe M, Arai D, Matsumoto C, Hirata M, Yoshinouchi S, Miyaura C, Itoh Y, et al. Establishment of a Triple Quadrupole HPLC-MS Quantitation Method for Dystrophin Protein in Mouse and Human Skeletal Muscle. International Journal of Molecular Sciences. 2024; 25(1):303. https://doi.org/10.3390/ijms25010303
Chicago/Turabian StyleTominari, Tsukasa, Masaru Takatoya, Toshiya Matsubara, Michio Matsunobe, Daichi Arai, Chiho Matsumoto, Michiko Hirata, Shosei Yoshinouchi, Chisato Miyaura, Yoshifumi Itoh, and et al. 2024. "Establishment of a Triple Quadrupole HPLC-MS Quantitation Method for Dystrophin Protein in Mouse and Human Skeletal Muscle" International Journal of Molecular Sciences 25, no. 1: 303. https://doi.org/10.3390/ijms25010303
APA StyleTominari, T., Takatoya, M., Matsubara, T., Matsunobe, M., Arai, D., Matsumoto, C., Hirata, M., Yoshinouchi, S., Miyaura, C., Itoh, Y., Komaki, H., Takeda, S., Aoki, Y., & Inada, M. (2024). Establishment of a Triple Quadrupole HPLC-MS Quantitation Method for Dystrophin Protein in Mouse and Human Skeletal Muscle. International Journal of Molecular Sciences, 25(1), 303. https://doi.org/10.3390/ijms25010303