
Optimization of Genome Knock-In Method: Search for the Most Efficient Genome Regions for Transgene Expression in Plants



Abstract
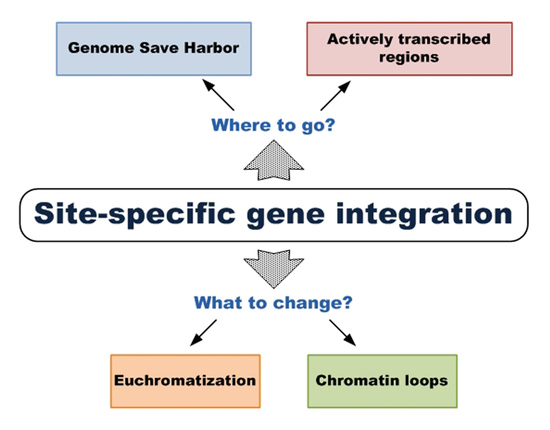
1. Introduction
2. Genomic Safe Harbors
3. Housekeeping Genes
3.1. 35S (45S) Pre-rRNA Genes
3.2. 5S rRNA Genes and tRNA Genes
3.3. Histone Genes
3.4. Genes of Actin, Tubulin, and Ubiquitin
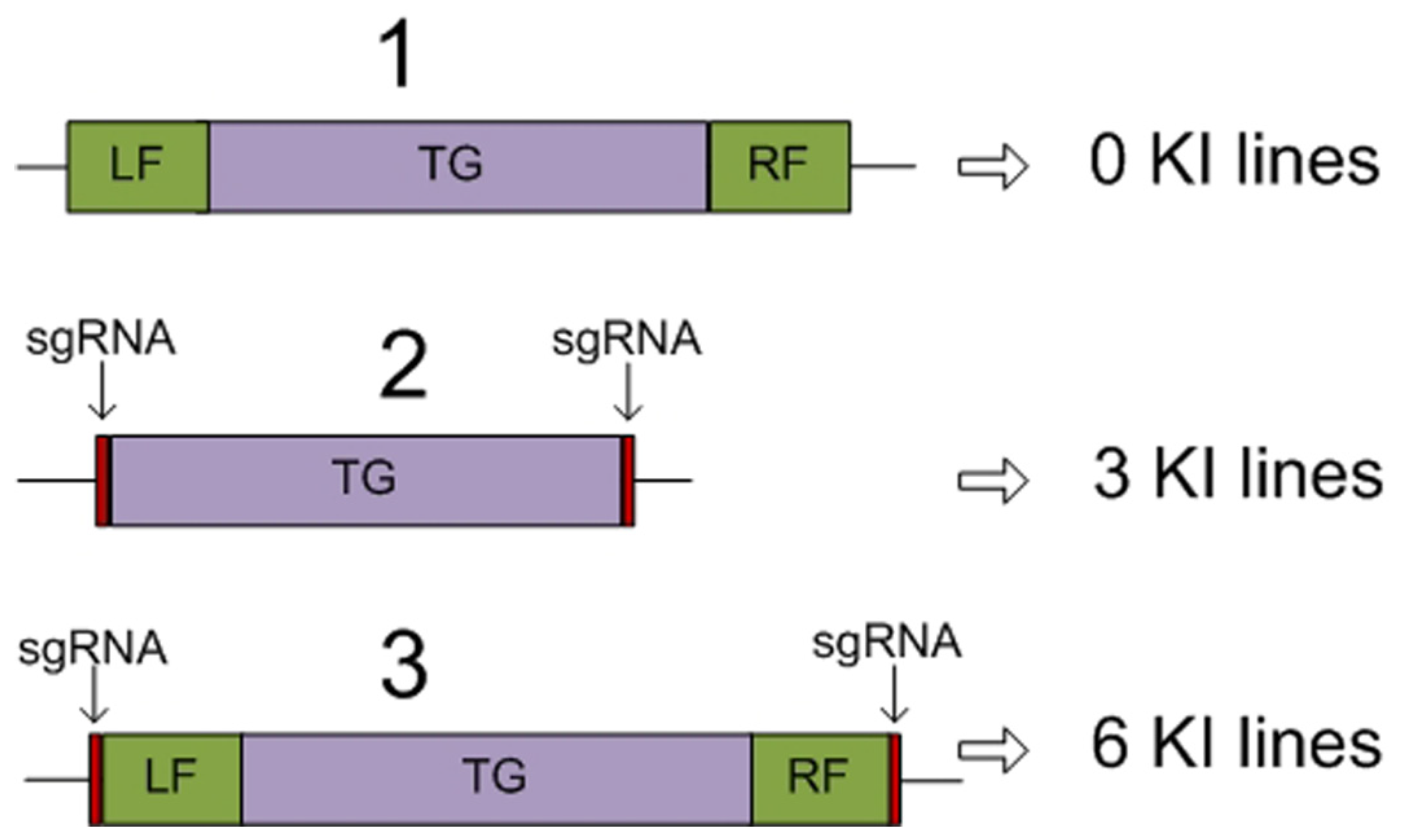
4. Other Approaches Providing Targeted Delivery of Transgenes to Previously Characterized Regions of the Plant Genome
5. Activation (Euchromatization) of Certain Genomic Regions
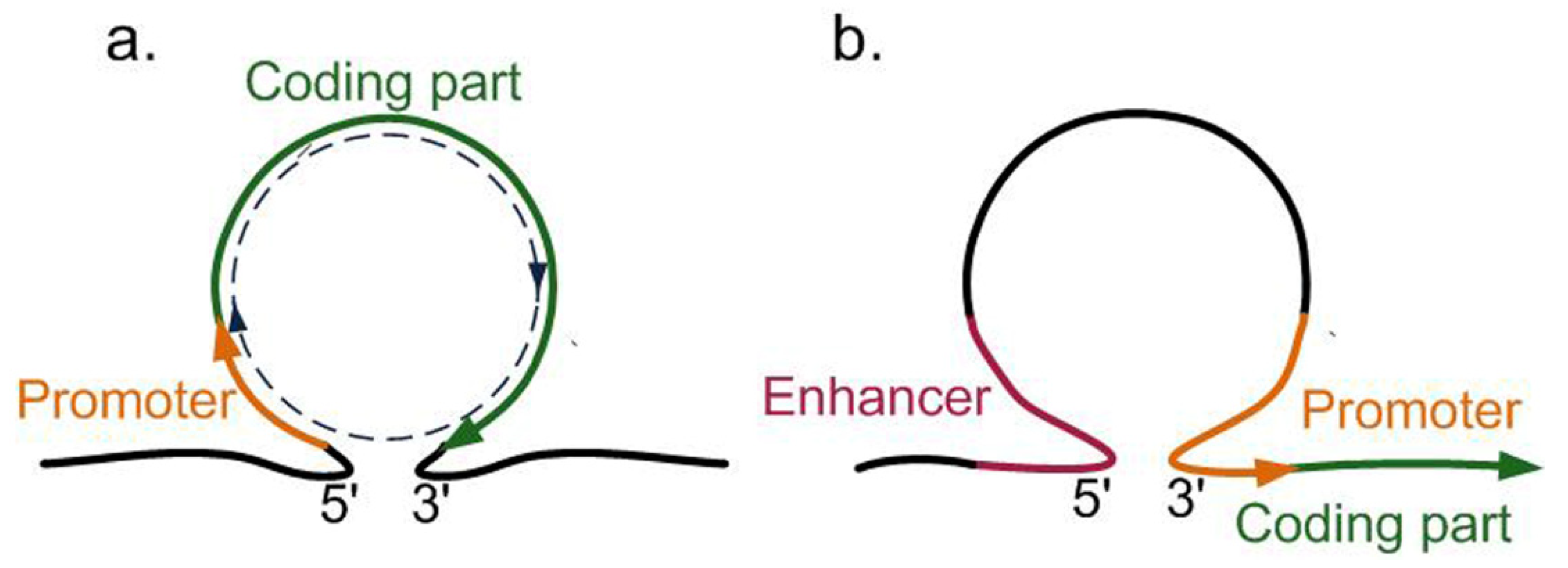
6. Spatial Organization of Chromatin and Transcriptional Activity
7. Conclusions
Funding
Conflicts of Interest
References
- Burnett, M.J.B.; Burnett, A.C. Therapeutic recombinant protein production in plants: Challenges and opportunities. Plants People Planet 2020, 2, 121–132. [Google Scholar] [CrossRef]
- Huebbers, J.W.; Buyel, J.F. On the verge of the market—Plant factories for the automated and standardized production of biopharmaceuticals. Biotechnol. Adv. 2021, 46, 107681. [Google Scholar] [CrossRef]
- Schillberg, S.; Finnern, R. Plant molecular farming for the production of valuable proteins—Critical evaluation of achievements and future challenges. J. Plant Physiol. 2021, 258–259, 153359. [Google Scholar] [CrossRef]
- Karki, U.; Fang, H.; Guo, W.; Unnold-Cofre, C.; Xu, J. Cellular engineering of plant cells for improved therapeutic protein production. Plant Cell Rep. 2021, 40, 1087–1099. [Google Scholar] [CrossRef] [PubMed]
- Rozov, S.M.; Deineko, E.V. Strategies for Optimizing Recombinant Protein Synthesis in Plant Cells: Classical Approaches and New Directions. Mol. Biol. 2019, 53, 179–199. [Google Scholar] [CrossRef]
- Buyel, J.F.; Stöger, E.; Bortesi, L. Targeted genome editing of plants and plant cells for biomanufacturing. Transgenic Res. 2021, 30, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Khatodia, S.; Bhatotia, K.; Passricha, N.; Khurana, S.M.P.; Tuteja, N. The CRISPR/Cas Genome-Editing Tool: Application in Improvement of Crops. Front. Plant Sci. 2016, 7, 506. [Google Scholar] [CrossRef] [PubMed]
- Demirci, Y.; Zhang, B.; Unver, T. CRISPR/Cas9: An RNA-guided highly precise synthetic tool for plant genome editing. J. Cell. Physiol. 2018, 233, 1844–1859. [Google Scholar] [CrossRef]
- Huang, T.K.; Puchta, H. CRISPR/Cas-mediated gene targeting in plants: Finally a turn for the better for homologous recombination. Plant Cell Rep. 2019, 38, 443–453. [Google Scholar] [CrossRef]
- Rozov, S.M.; Permyakova, N.V.; Deineko, E.V. The Problem of the Low Rates of CRISPR/Cas9-Mediated Knock-ins in Plants: Approaches and Solutions. Int. J. Mol. Sci. 2019, 20, 3371. [Google Scholar] [CrossRef]
- Collonnier, C.; Guyon-Debast, A.; Maclot, F.; Mara, K.; Charlot, F.; Nogué, F. Towards mastering CRISPR-induced gene knock-in in plants: Survey of key features and focus on the model Physcomitrella patens. Methods 2017, 121–122, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Dong, O.X.; Ronald, P.C. Targeted DNA insertion in plants. Proc. Natl. Acad. Sci. USA 2021, 118, e2004834117. [Google Scholar] [CrossRef] [PubMed]
- Rebetzke, G.J.; Jimenez-Berni, J.; Fischer, R.A.; Deery, D.M.; Smith, D.J. Review: High-throughput phenotyping to enhance the use of crop genetic resources. Plant Sci. 2019, 282, 40–48. [Google Scholar] [CrossRef]
- Yang, W.; Feng, H.; Zhang, X.; Zhang, J.; Doonan, J.H.; Batchelor, W.D.; Xiong, L.; Yan, J. Crop Phenomics and High-Throughput Phenotyping: Past Decades, Current Challenges, and Future Perspectives. Mol. Plant 2020, 13, 187–214. [Google Scholar] [CrossRef]
- Dong, O.X.; Yu, S.; Jain, R.; Zhang, N.; Duong, P.Q.; Butler, C.; Li, Y.; Lipzen, A.; Martin, J.A.; Barry, K.W.; et al. Marker-free carotenoid-enriched rice generated through targeted gene insertion using CRISPR-Cas9. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Permyakova, N.V.; Marenkova, T.V.; Belavin, P.A.; Zagorskaya, A.A.; Sidorchuk, Y.V.; Uvarova, E.A.; Kuznetsov, V.V.; Rozov, S.M.; Deineko, E.V. Assessment of the Level of Accumulation of the dIFN Protein Integrated by the Knock-In Method into the Region. Cells 2021, 10, 2137. [Google Scholar] [CrossRef]
- Belavin, P.A.; Permyakova, N.V.; Zagorskaya, A.A.; Marenkova, T.V.; Sidorchuk, Y.V.; Uvarova, E.A.; Rozov, S.M.; Deineko, E.V. Peculiarities in Creation of Genetic Engineering Constructions for Knock-In Variant of Genome Editing of Arabidopsis thaliana Cell Culture. Russ. J. Plant Physiol. 2020, 67, 855–866. [Google Scholar] [CrossRef]
- Joseph, J.T.; Poolakkalody, N.J.; Shah, J.M. Plant reference genes for development and stress response studies. J. Biosci. 2018, 43, 173–187. [Google Scholar] [CrossRef]
- Sadelain, M.; Papapetrou, E.P.; Bushman, F.D. Safe harbours for the integration of new DNA in the human genome. Nat. Rev. Cancer 2012, 12, 51–58. [Google Scholar] [CrossRef]
- Papapetrou, E.P.; Lee, G.; Malani, N.; Setty, M.; Riviere, I.; Tirunagari, L.M.S.; Kadota, K.; Roth, S.L.; Giardina, P.; Viale, A.; et al. Genomic safe harbors permit high β-globin transgene expression in thalassemia induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 73–81. [Google Scholar] [CrossRef]
- Papapetrou, E.P.; Schambach, A. Gene insertion into genomic safe harbors for human gene therapy. Mol. Ther. 2016, 24, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.J.; Saee-Marand, M.; Nyström, N.N.; Evans, M.M.; Chen, Y.; Martinez, F.M.; Hamilton, A.M.; Ronald, J.A. Safe harbor-targeted CRISPR-Cas9 homology-independent targeted integration for multimodality reporter gene-based cell tracking. Sci. Adv. 2021, 7, eabc3791. [Google Scholar] [CrossRef] [PubMed]
- Aznauryan, E.; Yermanos, A.; Kinzina, E.; Devaux, A.; Kapetanovic, E.; Milanova, D.; Church, G.M.; Reddy, S.T. Discovery and validation of human genomic safe harbor sites for gene and cell therapies. Cell Rep. Methods 2022, 2, 100154. [Google Scholar] [CrossRef]
- Pavani, G.; Amendola, M. Targeted Gene Delivery: Where to Land. Front. Genome Ed. 2021, 2, 36. [Google Scholar] [CrossRef]
- Kowalczyk, T.; Merecz-Sadowska, A.; Picot, L.; Brčić Karačonji, I.; Wieczfinska, J.; Śliwiński, T.; Sitarek, P. Genetic Manipulation and Bioreactor Culture of Plants as a Tool for Industry and Its Applications. Molecules 2022, 27, 795. [Google Scholar] [CrossRef]
- Iwase, A.; Ishii, H.; Aoyagi, H.; Ohme-Takagi, M.; Tanaka, H. Comparative analyses of the gene expression profiles of Arabidopsis intact plant and cultured cells. Biotechnol. Lett. 2005, 27, 1097–1103. [Google Scholar] [CrossRef]
- Tanurdzic, M.; Vaughn, M.W.; Jiang, H.; Lee, T.J.; Slotkin, R.K.; Sosinski, B.; Thompson, W.F.; Doerge, R.W.; Martienssen, R.A. Epigenomic consequences of immortalized plant cell suspension culture. PLoS Biol. 2008, 6, e302-95. [Google Scholar] [CrossRef]
- Gerlach, W.L.; Bedbrook, J.R. Cloning and characterization of ribosomal RNA genes from wheat and barley. Nucleic Acids Res. 1979, 7, 1869–1885. [Google Scholar] [CrossRef]
- Hemleben, V.; Zentgraf, U. Structural organization and regulation of transcription by RNA polymerase I of plant nuclear ribosomal RNA genes. Plant Promot. Transcr. Factors 1994, 20, 3–24. [Google Scholar]
- Garcia, S.; Cortes, P.P.; Fernàndez, X.; Garnatje, T.; Kovarik, A. Organisation, expression and evolution of rRNA genes in plant genomes. Recent Adv. Pharm. Sci. VI 2016, 49–75. Available online: http://diposit.ub.edu/dspace/handle/2445/103608 (accessed on 13 April 2022).
- Kalinina, N.O.; Makarova, S.; Makhotenko, A.; Love, A.J.; Taliansky, M. The multiple functions of the nucleolus in plant development, disease and stress responses. Front. Plant Sci. 2018, 9, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Stępiński, D. Functional ultrastructure of the plant nucleolus. Protoplasma 2014, 251, 1285–1306. [Google Scholar] [CrossRef] [PubMed]
- Trinkle-Mulcahy, L. Nucleolus: The Consummate Nuclear Body; Elsevier Inc.: Amsterdam, The Netherlands, 2018; ISBN 9780128034804. [Google Scholar]
- Pontvianne, F.; Carpentier, M.C.; Durut, N.; Pavlištová, V.; Jaške, K.; Schořová, Š.; Parrinello, H.; Rohmer, M.; Pikaard, C.S.; Fojtová, M.; et al. Identification of Nucleolus-Associated Chromatin Domains Reveals a Role for the Nucleolus in 3D Organization of the A. thaliana Genome. Cell Rep. 2016, 16, 1574–1587. [Google Scholar] [CrossRef] [PubMed]
- Xuan, J.; Gitareja, K.; Brajanovski, N.; Sanij, E. Harnessing the nucleolar dna damage response in cancer therapy. Genes 2021, 12, 1156. [Google Scholar] [CrossRef] [PubMed]
- Drouin, G.; de Sa, M.M. The concerted evolution of 5S ribosomal genes linked to the repeat units of other multigene families. Mol. Biol. Evol. 1995, 12, 481–493. [Google Scholar] [CrossRef][Green Version]
- Campell, B.R.; Song, Y.; Posch, T.E.; Cullis, C.A.; Town, C.D. Sequence and organization of 5S ribosomal RNA-encoding genes of Arabidopsis thaliana. Gene 1992, 112, 225–228. [Google Scholar] [CrossRef]
- Simon, L.; Rabanal, F.A.; Dubos, T.; Oliver, C.; Lauber, D.; Poulet, A.; Vogt, A.; Mandlbauer, A.; Le Goff, S.; Sommer, A.; et al. Genetic and epigenetic variation in 5S ribosomal RNA genes reveals genome dynamics in Arabidopsis thaliana. Nucleic Acids Res. 2018, 46, 3019–3033. [Google Scholar] [CrossRef]
- Benoit, M.; Layat, E.; Tourmente, S.; Probst, A.V. Heterochromatin dynamics during developmental transitions in Arabidopsis—A focus on ribosomal DNA loci. Gene 2013, 526, 39–45. [Google Scholar] [CrossRef]
- Mathieu, O.; Jasencakova, Z.; Vaillant, I.; Gendrel, A.V.; Colot, V.; Schubert, I.; Tourmente, S. Changes in 5S rDNA Chromatin Organization and Transcription during Heterochromatin Establishment in Arabidopsis. Plant Cell 2003, 15, 2929–2939. [Google Scholar] [CrossRef]
- Maréchal-Drouard, L.; Weil, J.H.; Dietrich, A. Transfer RNAs and transfer RNA genes in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1993, 44, 13–32. [Google Scholar] [CrossRef]
- Beier, D.; Stange, N.; Gross, H.J.; Beier, H. Nuclear tRNATyr genes are highly amplified at a single chromosomal site in the genome of Arabidopsis thaliana. MGG Mol. Gen. Genet. 1991, 225, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Arimbasseri, A.G.; Maraia, R.J. RNA Polymerase III Advances: Structural and tRNA Functional Views. Trends Biochem. Sci. 2016, 41, 546–559. [Google Scholar] [CrossRef] [PubMed]
- Eirín-López, J.M.; González-Romero, R.; Dryhurst, D.; Méndez, J.; Ausió, J. Long-Term Evolution of Histone Families: Old Notions and New Insights into Their Mechanisms of Diversification Across Eukaryotes. In Evolutionary Biology; Pontarott, P., Ed.; Springer: Berlin, Germany, 2009; pp. 139–162. ISBN 978-3-642-00952-5. [Google Scholar]
- Chaubet, N.; Philipps, G.; Gigot, C. Organization of the histone H3 and H4 multigenic families in maize and in related genomes. MGG Mol. Gen. Genet. 1989, 219, 404–412. [Google Scholar] [CrossRef]
- Li, M.; Fang, Y. Da Histone variants: The artists of eukaryotic chromatin. Sci. China Life Sci. 2015, 58, 232–239. [Google Scholar] [CrossRef][Green Version]
- Kanazin, V.; Blake, T.; Shoemaker, R.C. Organization of the histone H3 genes in soybean, barley and wheat. Mol. Gen. Genet. 1996, 250, 137–147. [Google Scholar] [CrossRef]
- Ingouff, M.; Berger, F. Histone3 variants in plants. Chromosoma 2010, 119, 27–33. [Google Scholar] [CrossRef]
- Kosterin, O.E.; Bogdanova, V.S.; Gorel, F.L.; Rozov, S.M.; Trusov, Y.A.; Berdnikov, V.A. Histone H1 of the garden pea (Pisum sativum L.); composition, developmental changes, allelic polymorphism and inheritance. Plant Sci. 1994, 101, 189–202. [Google Scholar] [CrossRef]
- Henikoff, S.; Ahmad, K. Assembly of variant histones into chromatin. Annu. Rev. Cell Dev. Biol. 2005, 21, 133–153. [Google Scholar] [CrossRef]
- Khadka, J.; Pesok, A.; Grafi, G. Plant histone htb (H2b) variants in regulating chromatin structure and function. Plants 2020, 9, 1435. [Google Scholar] [CrossRef]
- Okada, T.; Endo, M.; Singh, M.B.; Bhalla, P.L. Analysis of the histone H3 gene family in Arabidopsis and identification of the male-gamete-specific variant AtMGH3. Plant J. 2005, 44, 557–568. [Google Scholar] [CrossRef]
- Pathak, B.; Srivastava, V. Recombinase-mediated integration of a multigene cassette in rice leads to stable expression and inheritance of the stacked locus. Plant Direct 2020, 4, e00236. [Google Scholar] [CrossRef] [PubMed]
- Mcdowell, J.M.; Huang, S.; Mckinney, E.C.; An, Y.; Meaghed, R.B. Structure and Evolution of the Actin Gene Family in Arabidopsis thaliana. Genetics 1996, 154, 587–602. [Google Scholar] [CrossRef] [PubMed]
- Šlajcherová, K.; Fišerová, J.; Fischer, L.; Schwarzerová, K. Multiple actin isotypes in plants: Diverse genes for diverse roles? Front. Plant Sci. 2012, 3, 226. [Google Scholar] [CrossRef]
- Silflow, C.D.; Oppenheimer, D.G.; Kopozak, S.D.; Ploense, S.E.; Ludwig, S.R.; Haas, N.; Peter Snustad, D. Plant tubulin genes: Structure and differential expression during development. Dev. Genet. 1987, 8, 435–460. [Google Scholar] [CrossRef]
- Oakley, R.V.; Wang, Y.S.; Ramakrishna, W.; Harding, S.A.; Tsai, C.J. Differential expansion and expression of α- and β-tubulin gene families in Populus. Plant Physiol. 2007, 145, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Breviario, D. Plant Tubulin Genes: Regulatory and Evolutionary Aspects. In Plant Microtubules. Plant Cell Monographs; Nick, P., Ed.; Springer: Berlin, Germany, 2008; pp. 207–232. ISBN 978-3-540-77178-4. [Google Scholar]
- Callis, J.; Carpenter, T.; Sun, C.; Vierstrat, R.D. Structure and Evolution of Genes Encoding Polyubiquitin and Ubiquitin-Like Proteins in Arabidqpsis thalianu Ecotype Columbia. Genetics 1995, 139, 921–939. [Google Scholar] [CrossRef] [PubMed]
- Callis, J. The Ubiquitination Machinery of the Ubiquitin System. Arab. Book 2014, 12, e0174. [Google Scholar] [CrossRef]
- Radici, L.; Bianchi, M.; Crinelli, R.; Magnani, M. Ubiquitin C gene: Structure, function, and transcriptional regulation. Adv. Biosci. Biotechnol. 2013, 04, 1057–1062. [Google Scholar] [CrossRef]
- Bachmair, A.; Finley, D.; Varshavsky, A. In vivo half-life of a protein is a function of its amino-terminal residue. Science 1986, 234, 179–186. [Google Scholar] [CrossRef]
- Gonda, D.K.; Bachmair, A.; Wunning, I.; Tobias, J.W.; Lane, W.S.; Varshavsky, A. Universality and stucture of the N-end rule. J. Biol. Chem. 1989, 264, 16700–16712. [Google Scholar] [CrossRef]
- Hondred, D.; Walker, J.M.; Mathews, D.E.; Vierstra, R.D. Use of ubiquitin fusions to augment protein expression in transgenic plants. Plant Physiol. 1999, 119, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Hussain, B.; Lucas, S.J.; Budak, H. CRISPR/Cas9 in plants: At play in the genome and at work for crop improvement. Brief. Funct. Genomics 2018, 17, 319–328. [Google Scholar] [CrossRef]
- Shan, S.; Soltis, P.S.; Soltis, D.E.; Yang, B. Considerations in adapting CRISPR / Cas9 in nongenetic model plant systems. Appl. Plant Sci. 2020, 8, e11314. [Google Scholar] [CrossRef] [PubMed]
- Day, C.D.; Lee, E.; Kobayashi, J.; Holappa, L.D.; Albert, H.; Ow, D.W. Transgene integration into the same chromosome location can produce alleles that express at a predictable level, or alleles that are differentially silenced. Genes Dev. 2000, 14, 2869–2880. [Google Scholar] [CrossRef] [PubMed]
- Mumm, R.H.; Walters, D.S. Quality control in the development of transgenic crop seed products. Crop Sci. 2001, 41, 1381–1389. [Google Scholar] [CrossRef]
- Anand, A.; Jones, T.J. Advancing Agrobacterium-Based Crop Transformation and Genome Modification Technology for Agricultural Biotechnology. Curr. Top. Microbiol. Immunol. 2018, 418, 489–507. [Google Scholar]
- Schiml, S.; Fauser, F.; Puchta, H. The CRISPR/Cas system can be used as nuclease for in planta gene targeting and as paired nickases for directed mutagenesis in Arabidopsis resulting in heritable progeny. Plant J. 2014, 80, 1139–1150. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, C.; Liu, W.; Gao, W.; Liu, C.; Song, G.; Li, W.-X.; Mao, L.; Chen, B.; Xu, Y.; et al. An alternative strategy for targeted gene replacement in plants using a dual-sgRNA/Cas9 design. Sci. Rep. 2016, 6, 23890. [Google Scholar] [CrossRef]
- Miki, D.; Zhang, W.; Zeng, W.; Feng, Z.; Zhu, J.K. CRISPR/Cas9-mediated gene targeting in Arabidopsis using sequential transformation. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef]
- Dahan-Meir, T.; Filler-Hayut, S.; Melamed-Bessudo, C.; Bocobza, S.; Czosnek, H.; Aharoni, A.; Levy, A.A. Efficient in planta gene targeting in tomato using geminiviral replicons and the CRISPR/Cas9 system. Plant J. 2018, 95, 5–16. [Google Scholar] [CrossRef]
- Butler, N.M.; Baltes, N.J.; Voytas, D.F.; Douches, D.S. Geminivirus-Mediated Genome Editing in Potato (Solanum tuberosum L.) Using Sequence-Specific Nucleases. Front. Plant Sci. 2016, 7, 1045. [Google Scholar] [CrossRef] [PubMed]
- Gil-Humanes, J.; Wang, Y.; Liang, Z.; Shan, Q.; Ozuna, C.V.; Sánchez-León, S.; Baltes, N.J.; Starker, C.; Barro, F.; Gao, C.; et al. High-efficiency gene targeting in hexaploid wheat using DNA replicons and CRISPR/Cas9. Plant J. 2017, 89, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Lu, Y.; Botella, J.R.; Mao, Y.; Hua, K.; Zhu, J.-K. Gene Targeting by Homology-Directed Repair in Rice Using a Geminivirus-Based CRISPR/Cas9 System. Mol. Plant 2017, 10, 1007–1010. [Google Scholar] [CrossRef] [PubMed]
- Svitashev, S.; Young, J.K.; Schwartz, C.; Gao, H.; Falco, S.C.; Cigan, A.M. Targeted Mutagenesis, Precise Gene Editing, and Site-Specific Gene Insertion in Maize Using Cas9 and Guide RNA. Plant Physiol. 2015, 169, 931–945. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, Z.B.; Xing, A.; Moon, B.P.; Koellhoffer, J.P.; Huang, L.; Ward, R.T.; Clifton, E.; Falco, S.C.; Cigan, A.M. Cas9-guide RNA directed genome editing in soybean. Plant Physiol. 2015, 169, 960–970. [Google Scholar] [CrossRef]
- Li, J.; Meng, X.; Zong, Y.; Chen, K.; Zhang, H.; Liu, J.; Li, J.; Gao, C. Gene replacements and insertions in rice by intron targeting using CRISPR-Cas9. Nat. Plants 2016, 2, 16139. [Google Scholar] [CrossRef]
- Maeder, M.L.; Linder, S.J.; Cascio, V.M.; Fu, Y.; Ho, Q.H.; Joung, J.K. CRISPR RNA—Guided activation of endogenous human genes. Nat. Methods 2013, 10, 977. [Google Scholar] [CrossRef]
- Perez-Pinera, P.; Kocak, D.D.; Vockley, C.M.; Adler, A.F.; Kabadi, A.M.; Polstein, L.R.; Thakore, P.I.; Glass, K.A.; Ousterout, D.G.; Leong, K.W.; et al. RNA-guided gene activation by CRISPR- Cas9—Based transcription factors. Nat. Methods 2013, 10, 973. [Google Scholar] [CrossRef]
- Lowder, L.G.; Zhang, D.; Baltes, N.J.; Paul, J.W.; Tang, X.; Zheng, X.; Voytas, D.F.; Hsieh, T.-F.; Zhang, Y.; Qi, Y. A CRISPR/Cas9 Toolbox for Multiplexed Plant Genome Editing and Transcriptional Regulation. Plant Physiol. 2015, 169, 971–985. [Google Scholar] [CrossRef]
- Piatek, A.; Ali, Z.; Baazim, H.; Li, L.; Abulfaraj, A.; Al-Shareef, S.; Aouida, M.; Mahfouz, M.M. RNA-guided transcriptional regulation in planta via synthetic dCas9-based transcription factors. Plant Biotechnol. J. 2015, 13, 578–589. [Google Scholar] [CrossRef]
- Papikian, A.; Liu, W.; Gallego-Bartolomé, J.; Jacobsen, S.E. Site-specific manipulation of Arabidopsis loci using CRISPR-Cas9 SunTag systems. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Liang, J.; Li, Z.; Gong, B.Q.; Li, J.F. Multiplex and optimization of dCas9-TV-mediated gene activation in plants. J. Integr. Plant Biol. 2021, 63, 634–645. [Google Scholar] [CrossRef] [PubMed]
- Selma, S.; Bernabé-Orts, J.M.; Vazquez-Vilar, M.; Diego-Martin, B.; Ajenjo, M.; Garcia-Carpintero, V.; Granell, A.; Orzaez, D. Strong gene activation in plants with genome-wide specificity using a new orthogonal CRISPR/Cas9-based programmable transcriptional activator. Plant Biotechnol. J. 2019, 17, 1703–1705. [Google Scholar] [CrossRef] [PubMed]
- Lowder, L.G.; Zhou, J.; Zhang, Y.; Malzahn, A.; Zhong, Z.; Hsieh, T.F.; Voytas, D.F.; Zhang, Y.; Qi, Y. Robust Transcriptional Activation in Plants Using Multiplexed CRISPR-Act2.0 and mTALE-Act Systems. Mol. Plant 2018, 11, 245–256. [Google Scholar] [CrossRef]
- Pan, C.; Wu, X.; Markel, K.; Malzahn, A.A.; Kundagrami, N.; Sretenovic, S.; Zhang, Y.; Cheng, Y.; Shih, P.M.; Qi, Y. CRISPR–Act3.0 for highly efficient multiplexed gene activation in plants. Nat. Plants 2021, 7, 942–953. [Google Scholar] [CrossRef]
- Doğan, E.S.; Liu, C. Three-dimensional chromatin packing and positioning of plant genomes. Nat. Plants 2018, 4, 521–529. [Google Scholar] [CrossRef]
- Stam, M.; Tark-Dame, M.; Fransz, P. 3D genome organization: A role for phase separation and loop extrusion? Curr. Opin. Plant Biol. 2019, 48, 36–46. [Google Scholar] [CrossRef]
- Gibcus, J.H.; Dekker, J. The Hierarchy of the 3D Genome. Mol. Cell 2013, 49, 773–782. [Google Scholar] [CrossRef]
- Liu, C.; Wang, C.; Wang, G.; Becker, C.; Zaidem, M.; Weigel, D. Genome-wide analysis of chromatin packing in Arabidopsis thaliana at single-gene resolution. Genome Res. 2016, 26, 1057–1068. [Google Scholar] [CrossRef]
- Dekker, J.; Marti-Renom, M.A.; Mirny, L.A. Exploring the three-dimensional organization of genomes: Interpreting chromatin interaction data. Nat. Rev. Genet. 2013, 14, 390–403. [Google Scholar] [CrossRef]
- Chung, I.M.; Ketharnathan, S.; Kim, S.H.; Thiruvengadam, M.; Rani, M.K.; Rajakumar, G. Making sense of the tangle: Insights into chromatin folding and gene regulation. Genes 2016, 7, 71. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Granados, N.Y.; Ramirez-Prado, J.S.; Veluchamy, A.; Latrasse, D.; Raynaud, C.; Crespi, M.; Ariel, F.; Benhamed, M. Put your 3D glasses on: Plant chromatin is on show. J. Exp. Bot. 2016, 67, 3205–3221. [Google Scholar] [CrossRef] [PubMed]
- Pontvianne, F.; Liu, C. Chromatin domains in space and their functional implications. Curr. Opin. Plant Biol. 2020, 54, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, D.; Manavella, P.A. Short-range regulatory chromatin loops in plants. New Phytol. 2020, 228, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.D.; Cook, P.R.; Papantonis, A. Dynamic Reconfiguration of Long Human Genes during One Transcription Cycle. Mol. Cell. Biol. 2012, 32, 2738–2747. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cavalli, G.; Misteli, T. Functional implications of genome topology. Nat. Struct. Mol. Biol. 2013, 20, 290–299. [Google Scholar] [CrossRef]
- Gagliardi, D.; Cambiagno, D.A.; Arce, A.L.; Tomassi, A.H.; Giacomelli, J.I.; Ariel, F.D.; Manavella, P.A. Dynamic regulation of chromatin topology and transcription by inverted repeat-derived small RNAs in sunflower. Proc. Natl. Acad. Sci. USA 2019, 116, 17578–17583. [Google Scholar] [CrossRef]
- Crevillén, P.; Sonmez, C.; Wu, Z.; Dean, C. A gene loop containing the floral repressor FLC is disrupted in the early phase of vernalization. EMBO J. 2013, 32, 140–148. [Google Scholar] [CrossRef]
- Jégu, T.; Domenichini, S.; Blein, T.; Ariel, F.; Christ, A.; Kim, S.K.; Crespi, M.; Boutet-Mercey, S.; Mouille, G.; Bourge, M.; et al. A SWI/SNF chromatin remodelling protein controls cytokinin production through the regulation of chromatin architecture. PLoS ONE 2015, 10, e0138276. [Google Scholar] [CrossRef]
- Louwers, M.; Bader, R.; Haring, M.; Van Driel, R.; De Laat, W.; Stam, M. Tissue- and expression level-specific chromatin looping at maize b1 epialleles. Plant Cell 2009, 21, 832–842. [Google Scholar] [CrossRef]
- Cao, S.; Kumimoto, R.W.; Gnesutta, N.; Calogero, A.M.; Mantovani, R.; Holt, B.F. A distal CCAAT/NUCLEAR FACTOR Y complex promotes chromatin looping at the FLOWERING LOCUS T promoter and regulates the timing of flowering in Arabidopsis. Plant Cell 2014, 26, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Cao, X.; Liu, Y.; Li, J.; Li, Y.; Li, D.; Zhang, K.; Gao, C.; Dong, A.; Liu, X. A chromatin loop represses WUSCHEL expression in Arabidopsis. Plant J. 2018, 94, 1083–1097. [Google Scholar] [CrossRef] [PubMed]
- Ariel, F.; Jegu, T.; Latrasse, D.; Romero-Barrios, N.; Christ, A.; Benhamed, M.; Crespi, M. Noncoding transcription by alternative rna polymerases dynamically regulates an auxin-driven chromatin loop. Mol. Cell 2014, 55, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Ariel, F.; Lucero, L.; Christ, A.; Mammarella, M.F.; Jegu, T.; Veluchamy, A.; Mariappan, K.; Latrasse, D.; Blein, T.; Liu, C.; et al. R-Loop Mediated trans Action of the APOLO Long Noncoding RNA. Mol. Cell 2020, 77, 1055–1065.e4. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Housekeeping Genes | Copy Number in Haploid Genome | Arrangement in Genome | Distribution over Chromosomes (Loci) | Transcribed by * | Phase of Activity |
|---|---|---|---|---|---|
| 35S rRNA genes | 750 | Tandem clusters 18S, 5.8S, and 25S rRNAs | Two loci in two chromosomes | RPol I | Constitutively active in the interphase |
| 5S rRNA genes | 1000 | Tandem repeats | Four loci in three chromosomes | RPol III | Constitutively active in the interphase |
| tRNA genes | 10–20 (of each tRNA) | In part, form tandem repeats; genes coding for different tRNAs are dispersed over the genome | All chromosomes | RPol III | Constitutively active in the interphase |
| Histone genes | 15 (of each of the five histones) | Mainly not clustered and do not form tandem repeats | In four chromosomes (histone H3) | RPol II | S phase; part of copies is constitutively active in the interphase |
| Actin genes | 8–10 | Dispersed over the genome | In four chromosomes | RPol II | Constitutively active in the interphase |
| Tubulin genes | 12 | Dispersed over the genome | In four chromosomes | RPol II | Constitutively active in the interphase |
| Ubiquitin genes | 12; 5 of them, code for polyubiquitin (3–6 repeats) | Dispersed over the genome | All chromosomes | RPol II | Constitutively active in the interphase |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rozov, S.M.; Permyakova, N.V.; Sidorchuk, Y.V.; Deineko, E.V. Optimization of Genome Knock-In Method: Search for the Most Efficient Genome Regions for Transgene Expression in Plants. Int. J. Mol. Sci. 2022, 23, 4416. https://doi.org/10.3390/ijms23084416
Rozov SM, Permyakova NV, Sidorchuk YV, Deineko EV. Optimization of Genome Knock-In Method: Search for the Most Efficient Genome Regions for Transgene Expression in Plants. International Journal of Molecular Sciences. 2022; 23(8):4416. https://doi.org/10.3390/ijms23084416
Chicago/Turabian StyleRozov, Sergey M., Natalya V. Permyakova, Yuriy V. Sidorchuk, and Elena V. Deineko. 2022. "Optimization of Genome Knock-In Method: Search for the Most Efficient Genome Regions for Transgene Expression in Plants" International Journal of Molecular Sciences 23, no. 8: 4416. https://doi.org/10.3390/ijms23084416
APA StyleRozov, S. M., Permyakova, N. V., Sidorchuk, Y. V., & Deineko, E. V. (2022). Optimization of Genome Knock-In Method: Search for the Most Efficient Genome Regions for Transgene Expression in Plants. International Journal of Molecular Sciences, 23(8), 4416. https://doi.org/10.3390/ijms23084416