
Docosahexaenoic Acid Suppresses Oxidative Stress-Induced Autophagy and Cell Death via the AMPK-Dependent Signaling Pathway in Immortalized Fischer Rat Schwann Cells 1

 , , ,
, , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. DHA Protects tBHP-Induced Cytotoxicity in the IFRS1 Cell Line
2.2. DHA Suppresses tBHP-Induced ROS Production in the IFRS1 Cell Line
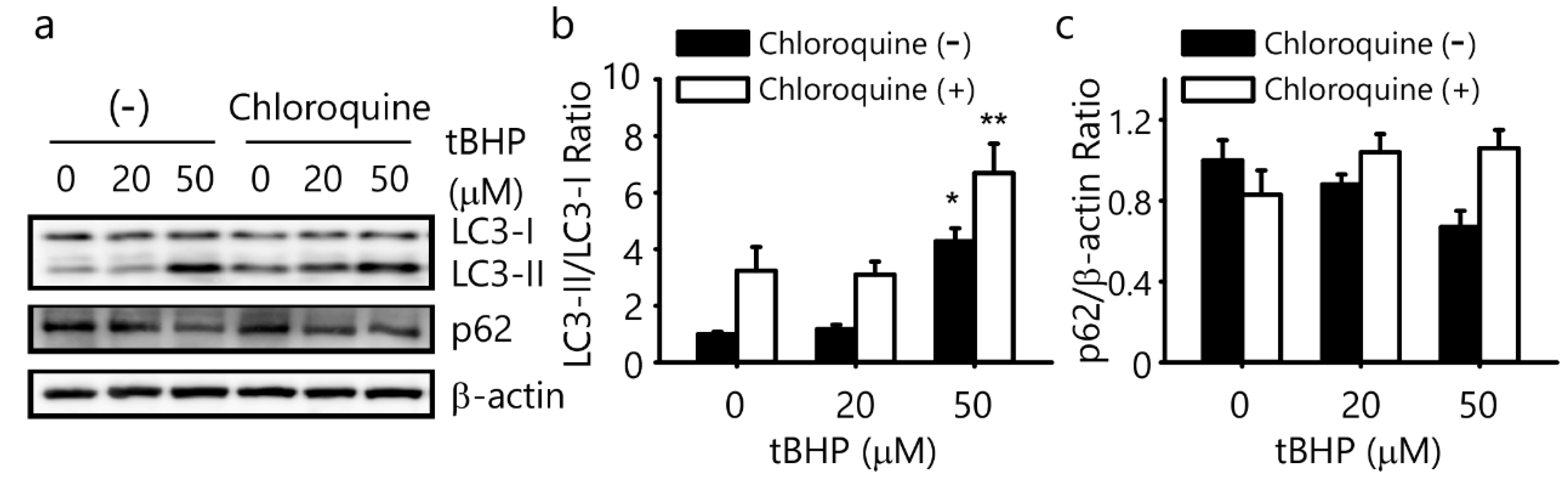
2.3. tBHP Induces Autophagy in the IFRS1 Cell Line
2.4. DHA Suppresses tBHP-Induced Autophagy in the IFRS1 Cell Line
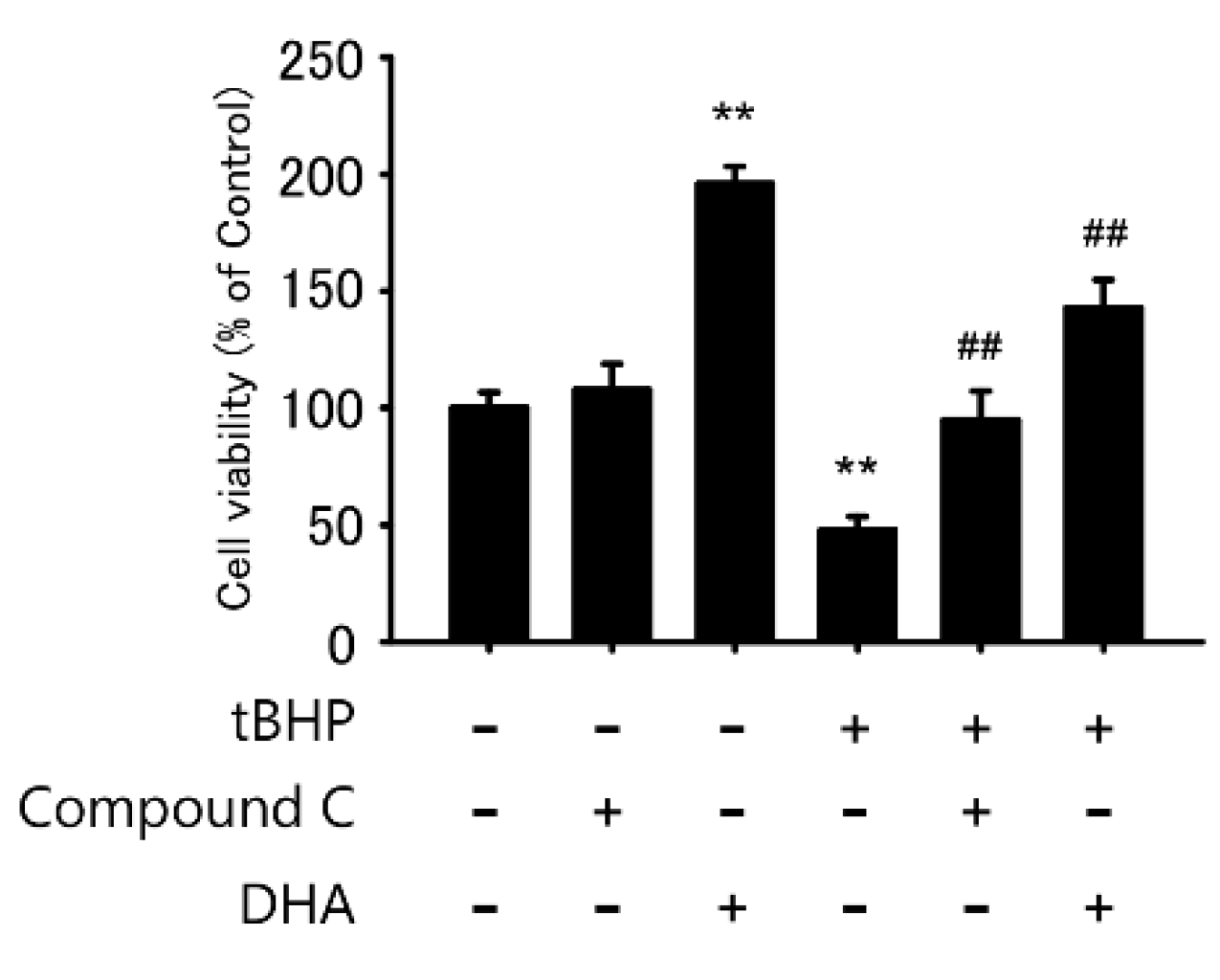
2.5. AMPK Inhibitor Protects tBHP-Induced Cytotoxicity in the IFRS1 Cell Line
2.6. DHA Suppresses tBHP-Induced Autophagosomes in the IFRS1 Cell Line
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Cell Survival Assay
4.4. Measurement of Intracellular Reactive Oxygen Species (ROS)
4.5. Western Blot Analysis
4.6. Autophagy Detection with DAPRed and DALGreen Staining
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Negre-Salvayre, A.; Salvayre, R.; Augé, N.; Pamplona, R.; Portero-Otín, M. Hyperglycemia and Glycation in Diabetic Complications. Antioxid. Redox Signal. 2009, 11, 3071–3109. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, B.C.; Cheng, H.T.; Stables, C.L.; Smith, A.L.; Feldman, E.L. Diabetic Neuropathy: Clinical Manifestations and Current Treatments. Lancet Neurol. 2012, 11, 521–534. [Google Scholar] [CrossRef]
- Yagihashi, S.; Mizukami, H.; Sugimoto, K. Mechanism of Diabetic Neuropathy: Where Are We Now and Where to Go? J. Diabetes Investig. 2011, 2, 18–32. [Google Scholar] [CrossRef] [PubMed]
- Román-Pintos, L.M.; Villegas-Rivera, G.; Rodríguez-Carrizalez, A.D.; Miranda-Díaz, A.G.; Cardona-Muñoz, E.G. Diabetic Polyneuropathy in Type 2 Diabetes Mellitus: Inflammation, Oxidative Stress, and Mitochondrial Function. J. Diabetes Res. 2016, 2016, 3425617. [Google Scholar] [CrossRef]
- Kato, A.; Tatsumi, Y.; Yako, H.; Sango, K.; Himeno, T.; Kondo, M.; Kato, Y.; Kamiya, H.; Nakamura, J.; Kato, K. Recurrent Short-Term Hypoglycemia and Hyperglycemia Induce Apoptosis and Oxidative Stress via the ER Stress Response in Immortalized Adult Mouse Schwann (IMS32) Cells. Neurosci. Res. 2019, 147, 26–32. [Google Scholar] [CrossRef]
- Dhanya, R.; Kartha, C.C. Quercetin Improves Oxidative Stress-Induced Pancreatic Beta Cell Alterations via MTOR-Signaling. Mol. Cell. Biochem. 2021, 476, 3879–3887. [Google Scholar] [CrossRef]
- Du, L.J.; Pang, B.; Tan, Y.M.; Yang, Y.N.; Zhang, M.Z.; Pang, Q.; Sun, M.; Ni, Q. Banxia Xiexin Decoction Ameliorates T-BHP-Induced Apoptosis in Pancreatic Beta Cells by Activating the PI3K/AKT/FOXO1 Signaling Pathway. J. Diabetes Res. 2020, 2020, 3695689. [Google Scholar] [CrossRef]
- Wang, L.; Liu, Z.; Chen, Z.; Huang, C.; Liu, X.; Chen, C.; Liu, X.; Huang, J.; Liu, L.; Lin, D. Metabonomic Analysis of the Therapeutic Effect of Exendin-4 for the Treatment of TBHP-Induced Injury in Mouse Glomerulus Mesangial Cells. Free. Radic. Res. 2018, 52, 544–555. [Google Scholar] [CrossRef]
- Tatsumi, Y.; Kato, A.; Sango, K.; Himeno, T.; Kondo, M.; Kato, Y.; Kamiya, H.; Nakamura, J.; Kato, K. Omega-3 Polyunsaturated Fatty Acids Exert Anti-oxidant Effects through the Nuclear Factor (Erythroid-derived 2)-related Factor 2 Pathway in Immortalized Mouse Schwann Cells. J. Diabetes Investig. 2019, 10, 602–612. [Google Scholar] [CrossRef]
- Sango, K.; Yanagisawa, H.; Takaku, S.; Kawakami, E.; Watabe, K. Immortalized Adult Rodent Schwann Cells as in Vitro Models to Study Diabetic Neuropathy. Exp. Diabetes Res. 2011, 2011, 374943. [Google Scholar] [CrossRef]
- Niimi, N.; Yako, H.; Tsukamoto, M.; Takaku, S.; Yamauchi, J.; Kawakami, E.; Yanagisawa, H.; Watabe, K.; Utsunomiya, K.; Sango, K. Involvement of Oxidative Stress and Impaired Lysosomal Degradation in Amiodarone-Induced Schwannopathy. Eur. J. Neurosci. 2016, 44, 1723–1733. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.S.; Hashimoto, M.; Gamoh, S.; Masumura, S. Antioxidative Effects of Docosahexaenoic Acid in the Cerebrum versus Cerebellum and Brainstem of Aged Hypercholesterolemic Rats. J. Neurochem. 1999, 72, 1133–1138. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Omega-3 Fatty Acids and Inflammatory Processes. Nutrients 2010, 2, 355–374. [Google Scholar] [CrossRef] [PubMed]
- Lavie, C.J.; Milani, R.v.; Mehra, M.R.; Ventura, H.O. Omega-3 Polyunsaturated Fatty Acids and Cardiovascular Diseases. J. Am. Coll. Cardiol. 2009, 54, 585–594. [Google Scholar] [CrossRef]
- Shimazawa, M.; Nakajima, Y.; Mashima, Y.; Hara, H. Docosahexaenoic Acid (DHA) Has Neuroprotective Effects against Oxidative Stress in Retinal Ganglion Cells. Brain Res. 2009, 1251, 269–275. [Google Scholar] [CrossRef]
- Johansson, I.; Monsen, V.T.; Pettersen, K.; Mildenberger, J.; Misund, K.; Kaarniranta, K.; Schønberg, S.; Bjørkøy, G. The Marine N-3 PUFA DHA Evokes Cytoprotection against Oxidative Stress and Protein Misfolding by Inducing Autophagy and NFE2L2 in Human Retinal Pigment Epithelial Cells. Autophagy 2015, 11, 1636–1651. [Google Scholar] [CrossRef]
- Pacheco, F.J.; Almaguel, F.G.; Evans, W.; Rios-Colon, L.; Filippov, V.; Leoh, L.S.; Rook-Arena, E.; Mediavilla-Varela, M.; de Leon, M.; Casiano, C.A. Docosahexanoic Acid Antagonizes TNF-α-Induced Necroptosis by Attenuating Oxidative Stress, Ceramide Production, Lysosomal Dysfunction, and Autophagic Features. Inflamm. Res. 2014, 63, 859–871. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy Fights Disease through Cellular Self-Digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive Oxygen Species Are Essential for Autophagy and Specifically Regulate the Activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef]
- Watada, H.; Fujitani, Y. Minireview: Autophagy in Pancreatic β-Cells and Its Implication in Diabetes. Mol. Endocrinol. 2015, 29, 338–348. [Google Scholar] [CrossRef]
- Han, D.; Yang, B.; Olson, L.K.; Greenstein, A.; Baek, S.H.; Claycombe, K.J.; Goudreau, J.L.; Yu, S.W.; Kim, E.K. Activation of Autophagy through Modulation of 5′-AMP-Activated Protein Kinase Protects Pancreatic β-Cells from High Glucose. Biochem. J. 2010, 425, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Cheon, H.; Jeong, Y.T.; Quan, W.; Kim, K.H.; Cho, J.M.; Lim, Y.M.; Oh, S.H.; Jin, S.M.; Kim, J.H.; et al. Amyloidogenic Peptide Oligomer Accumulation in Autophagy-Deficient β Cells Induces Diabetes. J. Clin. Investig. 2014, 124, 3311–3324. [Google Scholar] [CrossRef] [PubMed]
- Rivera, J.F.; Costes, S.; Gurlo, T.; Glabe, C.G.; Butler, P.C. Autophagy Defends Pancreatic β Cells from Human Islet Amyloid Polypeptide-Induced Toxicity. J. Clin. Investig. 2014, 124, 3489–3500. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Endoplasmic Reticulum Stress and the Inflammatory Basis of Metabolic Disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef]
- Ma, T.; Zhu, J.; Chen, X.; Zha, D.; Singhal, P.C.; Ding, G. High Glucose Induces Autophagy in Podocytes. Exp. Cell Res. 2013, 319, 779–789. [Google Scholar] [CrossRef]
- Dong, C.; Zheng, H.; Huang, S.; You, N.; Xu, J.; Ye, X.; Zhu, Q.; Feng, Y.; You, Q.; Miao, H.; et al. Heme Oxygenase-1 Enhances Autophagy in Podocytes as a Protective Mechanism against High Glucose-Induced Apoptosis. Exp. Cell Res. 2015, 337, 146–159. [Google Scholar] [CrossRef]
- Lenoir, O.; Jasiek, M.; Hénique, C.; Guyonnet, L.; Hartleben, B.; Bork, T.; Chipont, A.; Flosseau, K.; Bensaada, I.; Schmitt, A.; et al. Endothelial Cell and Podocyte Autophagy Synergistically Protect from Diabetes-Induced Glomerulosclerosis. Autophagy 2015, 11, 1130–1145. [Google Scholar] [CrossRef]
- Jin, Y.; Liu, S.; Ma, Q.; Xiao, D.; Chen, L. Berberine Enhances the AMPK Activation and Autophagy and Mitigates High Glucose-Induced Apoptosis of Mouse Podocytes. Eur. J. Pharmacol. 2017, 794, 106–114. [Google Scholar] [CrossRef]
- Towns, R.; Kabeya, Y.; Yoshimori, T.; Guo, C.; Shangguan, Y.; Hong, S.; Kaplan, M.; Klionsky, D.J.; Wiley, J.W. Sera from Patients with Type 2 Diabetes and Neuropathy Induce Autophagy and Colocalization with Mitochondria in SY5Y Cells. Autophagy 2005, 1, 163–170. [Google Scholar] [CrossRef]
- Iwashita, H.; Sakurai, H.T.; Nagahora, N.; Ishiyama, M.; Shioji, K.; Sasamoto, K.; Okuma, K.; Shimizu, S.; Ueno, Y. Small Fluorescent Molecules for Monitoring Autophagic Flux. FEBS Lett. 2018, 592, 559–567. [Google Scholar] [CrossRef]
- Park, E.Y.; Park, J.B. High Glucose-Induced Oxidative Stress Promotes Autophagy through Mitochondrial Damage in Rat Notochordal Cells. Int. Orthop. 2013, 37, 2507–2514. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.Y.; Yang, B.; Shi, Y.X.; Zhang, W.L.; Liu, F.; Zhao, W.; Yang, M.W. High Glucose Downregulates the Effects of Autophagy on Osteoclastogenesis via the AMPK/MTOR/ULK1 Pathway. Biochem. Biophys. Res. Commun. 2018, 503, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Ariyoshi, M.; Okawa, Y.; Kaimoto, S.; Uchihashi, M.; Fukai, K.; Iwai-Kanai, E.; Ikeda, K.; Ueyama, T.; Ogata, T.; et al. Inhibition of P53 Preserves Parkin-Mediated Mitophagy and Pancreatic -Cell Function in Diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 3116–3121. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.S.; Chung, K.W.; Won Kim, J.; Kim, J.; Komatsu, M.; Tanaka, K.; Nguyen, Y.H.; Kang, T.M.; Yoon, K.H.; Kim, J.W.; et al. Loss of Autophagy Diminishes Pancreatic β Cell Mass and Function with Resultant Hyperglycemia. Cell Metab. 2008, 8, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Quan, W.; Hur, K.Y.; Lim, Y.; Oh, S.H.; Lee, J.C.; Kim, K.H.; Kim, G.H.; Kim, S.W.; Kim, H.L.; Lee, M.K.; et al. Autophagy Deficiency in Beta Cells Leads to Compromised Unfolded Protein Response and Progression from Obesity to Diabetes in Mice. Diabetologia 2012, 55, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Masini, M.; Bugliani, M.; Lupi, R.; del Guerra, S.; Boggi, U.; Filipponi, F.; Marselli, L.; Masiello, P.; Marchetti, P. Autophagy in Human Type 2 Diabetes Pancreatic Beta Cells. Diabetologia 2009, 52, 1083–1086. [Google Scholar] [CrossRef] [PubMed]
- Xin, W.; Li, Z.; Xu, Y.; Yu, Y.; Zhou, Q.; Chen, L.; Wan, Q. Autophagy Protects Human Podocytes from High Glucose-Induced Injury by Preventing Insulin Resistance. Metab. Clin. Exp. 2016, 65, 1307–1315. [Google Scholar] [CrossRef]
- Martinez-Vicente, M.; Cuervo, A.M. Autophagy and Neurodegeneration: When the Cleaning Crew Goes on Strike. Lancet Neurol. 2007, 6, 352–361. [Google Scholar] [CrossRef]
- Sifuentes-Franco, S.; Pacheco-Moisés, F.P.; Rodríguez-Carrizalez, A.D.; Miranda-Díaz, A.G. The Role of Oxidative Stress, Mitochondrial Function, and Autophagy in Diabetic Polyneuropathy. J. Diabetes Res. 2017, 2017, 1–15. [Google Scholar] [CrossRef]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy Promotes Tumor Cell Survival and Restricts Necrosis, Inflammation, and Tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef]
- Rosenfeldt, M.T.; Ryan, K.M. The Multiple Roles of Autophagy in Cancer. Carcinogenesis 2011, 32, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-Consumption: The Interplay of Autophagy and Apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Pfister, A.S. Emerging Role of the Nucleolar Stress Response in Autophagy. Front. Cell. Neurosci. 2019, 13, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Jing, K.; Song, K.-S.; Shin, S.; Kim, N.; Jeong, S.; Oh, H.-R.; Park, J.-H.; Seo, K.-S.; Heo, J.-Y.; Han, J.; et al. Docosahexaenoic Acid Induces Autophagy through P53/AMPK/MTOR Signaling and Promotes Apoptosis in Human Cancer Cells Harboring Wild-Type P53. Autophagy 2011, 7, 1348–1358. [Google Scholar] [CrossRef]
- Tan, X.; Zou, L.; Qin, J.; Xia, D.; Zhou, Y.; Jin, G.; Jiang, Z.; Li, H. SQSTM1/P62 Is Involved in Docosahexaenoic Acid–Induced Cellular Autophagy in Glioblastoma Cell Lines. Vitr. Cell. Dev. Biol.-Anim. 2019, 55, 703–712. [Google Scholar] [CrossRef]
- Matsuzawa, Y.; Oshima, S.; Nibe, Y.; Kobayashi, M.; Maeyashiki, C.; Nemoto, Y.; Nagaishi, T.; Okamoto, R.; Tsuchiya, K.; Nakamura, T.; et al. RIPK3 Regulates P62–LC3 Complex Formation via the Caspase-8-Dependent Cleavage of P62. Biochem. Biophys. Res. Commun. 2015, 456, 298–304. [Google Scholar] [CrossRef]
- Inoki, K.; Kim, J.; Guan, K.L. AMPK and MTOR in Cellular Energy Homeostasis and Drug Targets. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 381–400. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and MTOR Regulate Autophagy through Direct Phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Zhou, L.; Cheng, Y. Alpha-Lipoic Acid Alleviated 6-OHDA-Induced Cell Damage by Inhibiting AMPK/MTOR Mediated Autophagy. Neuropharmacology 2019, 155, 98–103. [Google Scholar] [CrossRef]
- Kishida, E.; Tajiri, M.; Masuzawa, Y. Docosahexaenoic Acid Enrichment Can Reduce L929 Cell Necrosis Induced by Tumor Necrosis Factor. Biochim. Et Biophys. Acta-Mol. Cell Biol. Lipids 2006, 1761, 454–462. [Google Scholar] [CrossRef]
- Yano, M.; Kishida, E.; Iwasaki, M.; Kojo, S.; Masuzawa, Y. Docosahexaenoic Acid and Vitamin E Can Reduce Human Monocytic U937 Cell Apoptosis Induced by Tumor Necrosis Factor. J. Nutr. 2000, 130, 1095–1101. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sridhar, S.; Botbol, Y.; MacIan, F.; Cuervo, A.M. Autophagy and Disease: Always Two Sides to a Problem. J. Pathol. 2012, 226, 255–273. [Google Scholar] [CrossRef] [PubMed]
- Woodworth-Hobbs, M.E.; Hudson, M.B.; Rahnert, J.A.; Zheng, B.; Franch, H.A.; Price, S.R. Docosahexaenoic Acid Prevents Palmitate-Induced Activation of Proteolytic Systems in C2C12 Myotubes. J. Nutr. Biochem. 2014, 25, 868–874. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tatsumi, Y.; Kato, A.; Niimi, N.; Yako, H.; Himeno, T.; Kondo, M.; Tsunekawa, S.; Kato, Y.; Kamiya, H.; Nakamura, J.; et al. Docosahexaenoic Acid Suppresses Oxidative Stress-Induced Autophagy and Cell Death via the AMPK-Dependent Signaling Pathway in Immortalized Fischer Rat Schwann Cells 1. Int. J. Mol. Sci. 2022, 23, 4405. https://doi.org/10.3390/ijms23084405
Tatsumi Y, Kato A, Niimi N, Yako H, Himeno T, Kondo M, Tsunekawa S, Kato Y, Kamiya H, Nakamura J, et al. Docosahexaenoic Acid Suppresses Oxidative Stress-Induced Autophagy and Cell Death via the AMPK-Dependent Signaling Pathway in Immortalized Fischer Rat Schwann Cells 1. International Journal of Molecular Sciences. 2022; 23(8):4405. https://doi.org/10.3390/ijms23084405
Chicago/Turabian StyleTatsumi, Yasuaki, Ayako Kato, Naoko Niimi, Hideji Yako, Tatsuhito Himeno, Masaki Kondo, Shin Tsunekawa, Yoshiro Kato, Hideki Kamiya, Jiro Nakamura, and et al. 2022. "Docosahexaenoic Acid Suppresses Oxidative Stress-Induced Autophagy and Cell Death via the AMPK-Dependent Signaling Pathway in Immortalized Fischer Rat Schwann Cells 1" International Journal of Molecular Sciences 23, no. 8: 4405. https://doi.org/10.3390/ijms23084405
APA StyleTatsumi, Y., Kato, A., Niimi, N., Yako, H., Himeno, T., Kondo, M., Tsunekawa, S., Kato, Y., Kamiya, H., Nakamura, J., Higai, K., Sango, K., & Kato, K. (2022). Docosahexaenoic Acid Suppresses Oxidative Stress-Induced Autophagy and Cell Death via the AMPK-Dependent Signaling Pathway in Immortalized Fischer Rat Schwann Cells 1. International Journal of Molecular Sciences, 23(8), 4405. https://doi.org/10.3390/ijms23084405