1. Introduction
The detailed visualization of tissue samples is the basis to understanding, detecting, and characterizing complex pathologies, and represents an indispensable step in modern disease diagnosis. Classical histopathology, which is the current gold standard routinely used in pathology laboratories, focuses on the analysis of representative two-dimensional (2D) physical sections of samples. Besides the intrinsic limitations of the physical sectioning process, such as tissue loss or artifacts including tissue distortion, the classical 2D histopathology process is severely limited in accuracy, integrity, and spatial information, as a thin sliced specimen section is not an adequate representation of an entire volumetric specimen [
1]. To visualize and subsequently interpret the precise morphological and molecular aspects of tissue samples, three-dimensional (3D) histopathology is required, as the specimen can be visualized in its entirety, which ultimately results in increased diagnostic and predictive accuracy of the tissue analysis [
2].
Standard immunofluorescence stainings are routinely based on mammalian IgG antibodies, which are used to detect distinguished biomarkers in tissue specimens [
3]. However, structural and chemical properties of conventional antibodies, such as their relatively large size, poor tissue penetration, heterogeneous distribution, and strong background signal, oftentimes prevent an efficient staining of the entire sample [
4]. As an alternative staining reagent, nanobodies (Nbs) have been established within the scientific community, as they can produce superior immunostaining results and feature distinct technological benefits concerning generation, production, and versatility [
5]. In contrast to intricate multi-chained IgG antibodies, Nbs are single-domain antibodies (sdAbs) that are composed of the antigen-binding variable domain (VHH) of heavy-chain only antibodies (HCAb), a unique antibody class found in the Camelidae species [
6]. With molecular weights varying around 15 kDa, they are the smallest naturally occurring antigen-binding molecules described, as of today [
7]. Moreover, Nbs feature several advantageous properties for immunostainings, including high solubility [
8], high resistance to chemical and proteolytic degradation [
9], a long shelf-life [
10], as well as faster tissue penetration rates compared to standard IgG antibodies [
11,
12]. Due to their high affinity [
4], specificity, and small size [
13], they can also reliably bind to epitopes, otherwise inaccessible, for conventional antibodies [
14,
15]. Furthermore, owing to its small size, the Nb genetic sequence can comfortably be integrated into conventional plasmid-based vectors and thus be expressed in high-yield bacterial production cultures in a time- and cost-efficient manner [
7]. In addition to the animal-free production procedure, plasmid expression vectors also allow for versatile genetic modifications of the expression construct, e.g., by addition or replacement of small protein tags, regulatory protein domains, and fusion protein constructs [
5].
Endothelial cell-selective adhesion molecule (ESAM) is a transmembrane glycoprotein expressed by endothelial cells, activated platelets, and megakaryocytes, where it facilitates cell–cell interactions at cell junctions [
16,
17,
18] or regulates thrombus formation [
19]. Due to its high expression in blood endothelial cells, ESAM serves as a distinctive blood vessel marker used for immunostainings in the context of vascular research [
20,
21]. However, although several commercial ESAM antibodies are commonly used in 2D immunofluorescence stainings, there is no commercial ESAM-staining reagent available for high-quality whole-mount 3D imaging.
To overcome the present limitations of 2D microscopy as well as the lack of 3D imaging tools visualizing ESAM-positive blood vascular networks, we generated and methodologically validated innovative Nbs targeting the human ESAM protein. These novel agents generate detailed 3D representations of blood vessels in volumetric human specimens, enabling spatial morphological analysis in academic research and potential applications in clinical histopathological diagnostics.
3. Discussion
Thorough understanding of complex pathologies and spatial biological structures necessitates high-quality visualization of histological samples in their 3D morphological context. However, the current gold standard in histopathology still mainly relies on stainings of representative 2D physical sections due to technical limitations for 3D visualization of entire specimens, as well as the absence of suitable labeling reagents for whole-mount sample stainings. Aggravated by the usage of IgG antibodies, which are oftentimes not suitable for 3D imaging due to their relatively large size and consequently limited tissue penetration, the current 2D histopathological approach does not provide sufficient spatial information for appropriate diagnosis.
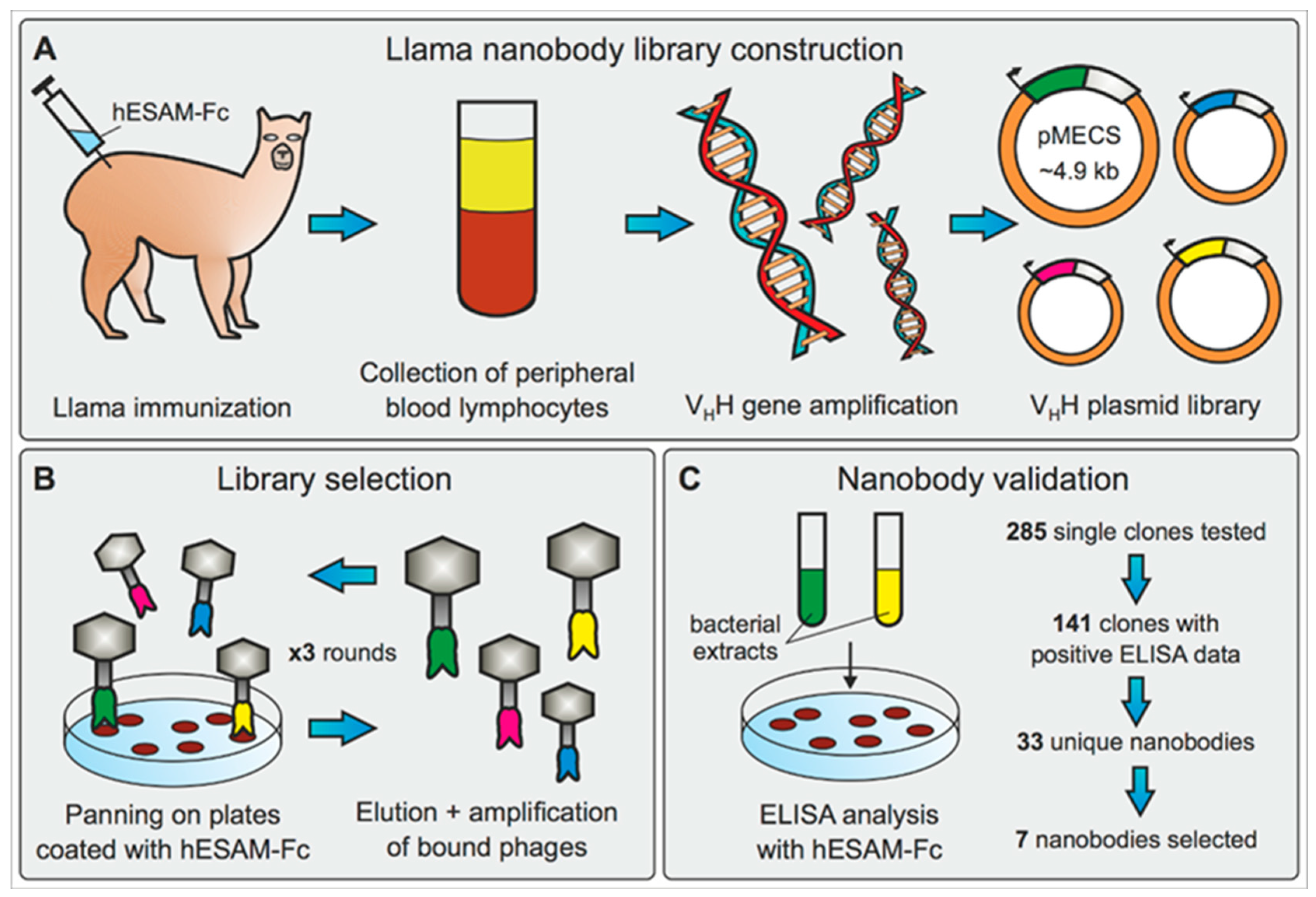
In this study, we therefore aimed to generate and experimentally validate a novel single-domain antibody (nanobody, Nb) directed against the human endothelial cell-selective adhesion molecule (hESAM), which can be used to specifically label blood endothelial cells in whole-mount 3D imaging approaches. To this end, we immunized llamas with a recombinant hESAM-Fc protein, composed of the hESAM extracellular domain (ECD) linked to the Fc domain of human IgG1, which ultimately resulted in the identification of 33 unique Nb clones. Although limited in sequence variety, as represented by only six different sequences for the complementary determining region (CDR) 3, the identified Nb clones demonstrated robust antigen-specific binding affinities in ELISA screenings. Low diversity of detected CDR3 sequences as well as the general moderate number of unique Nb clones may be caused by high glycosylation of the hESAM ECD [
25,
26], which might sterically hinder potential epitope recognition [
27].
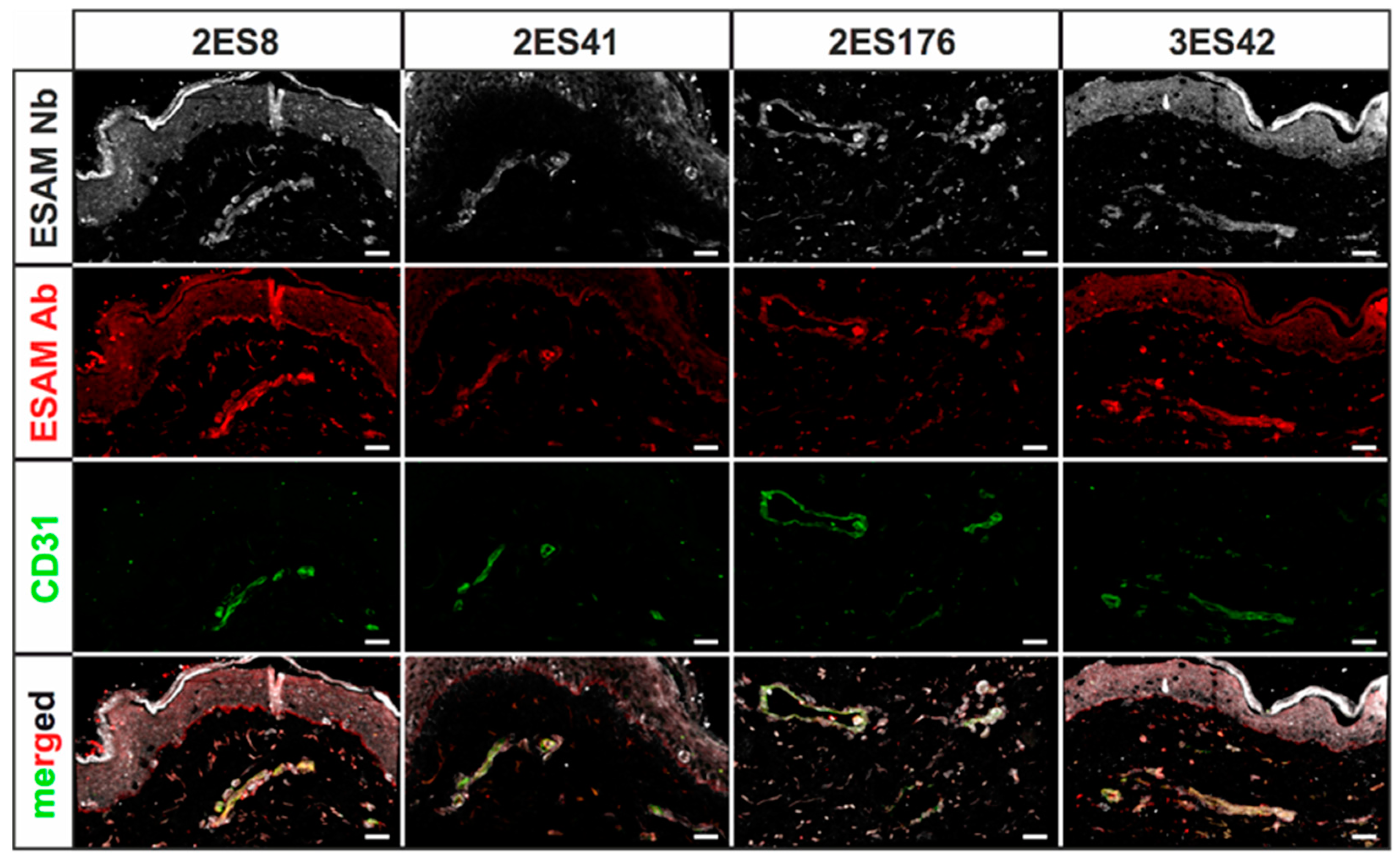
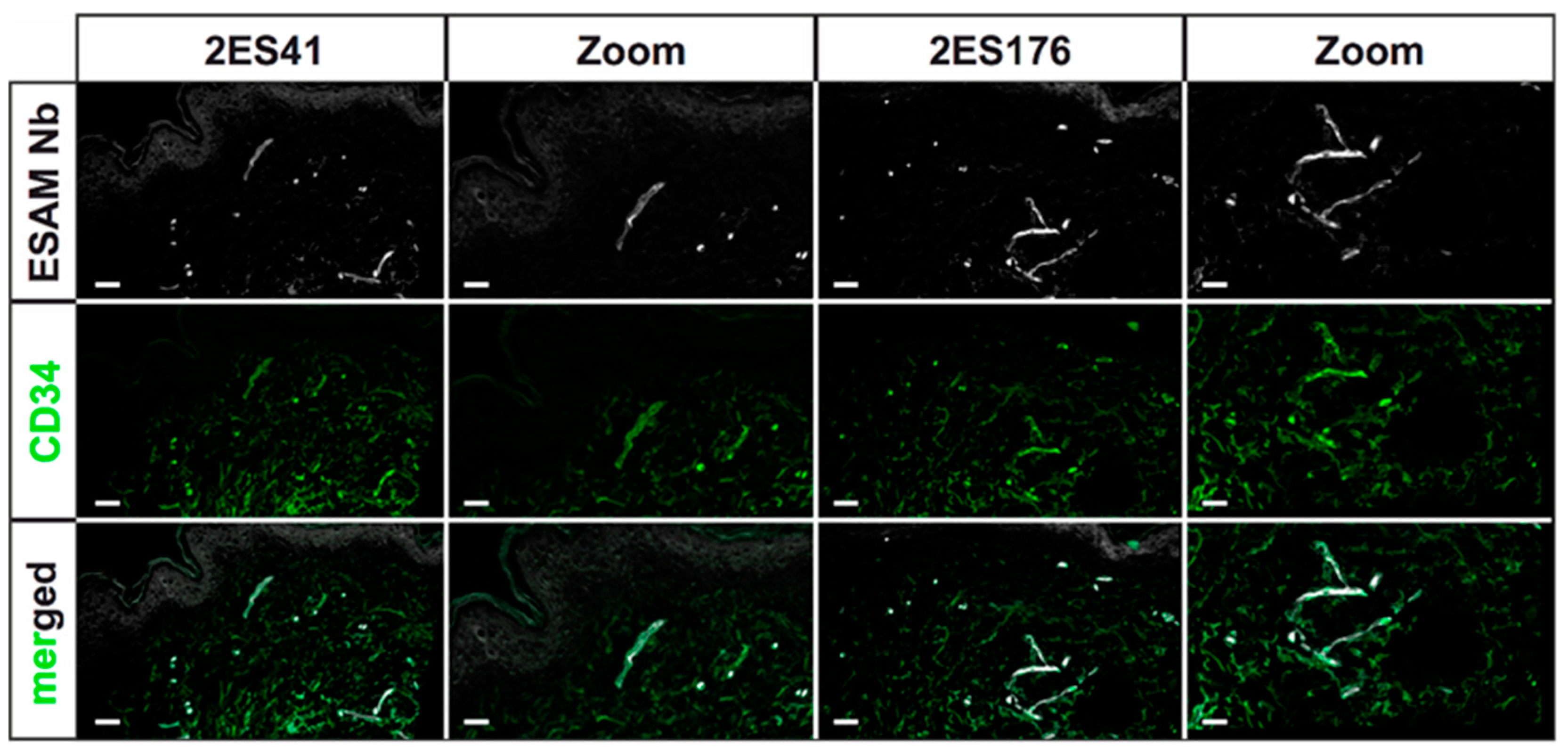
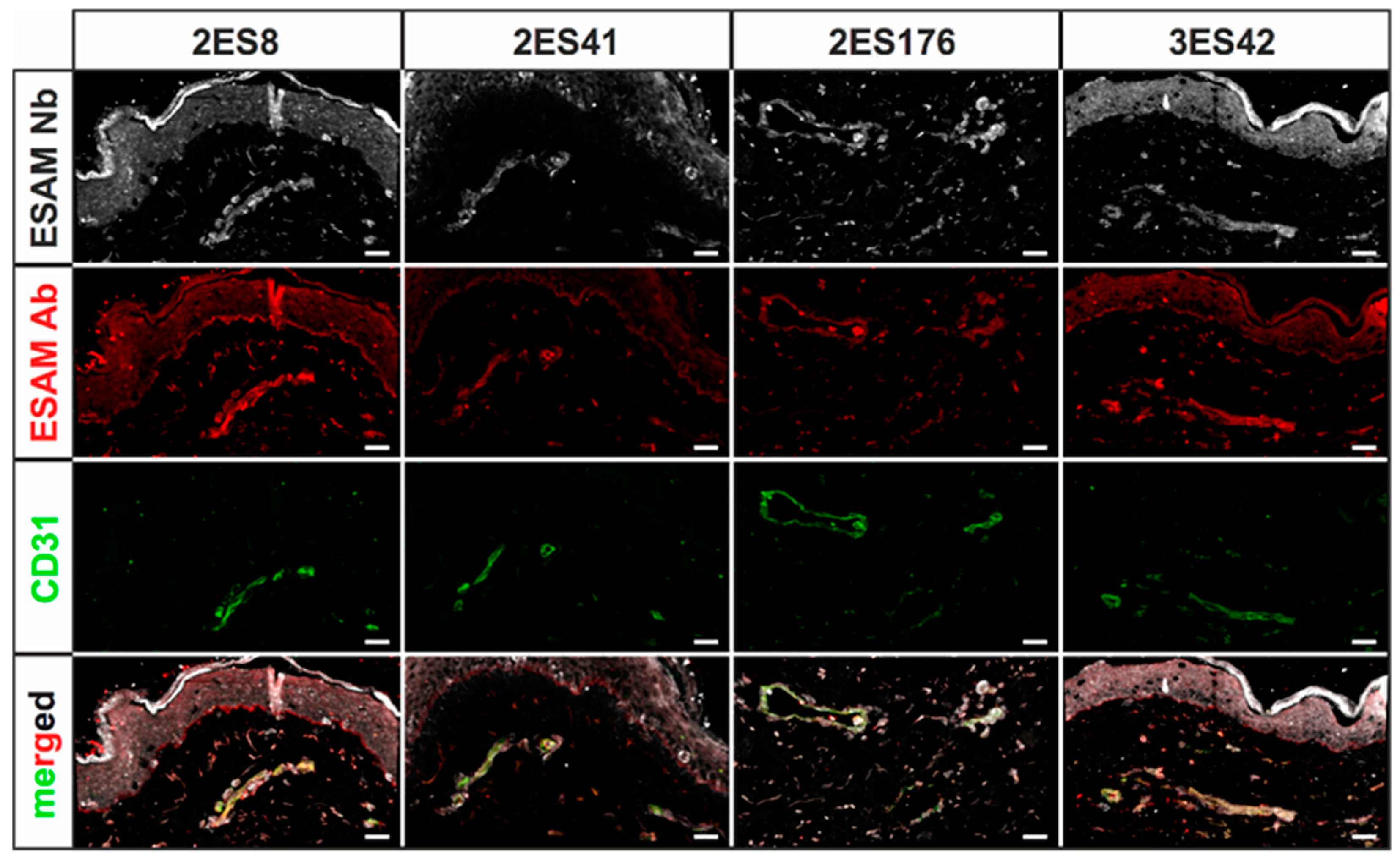
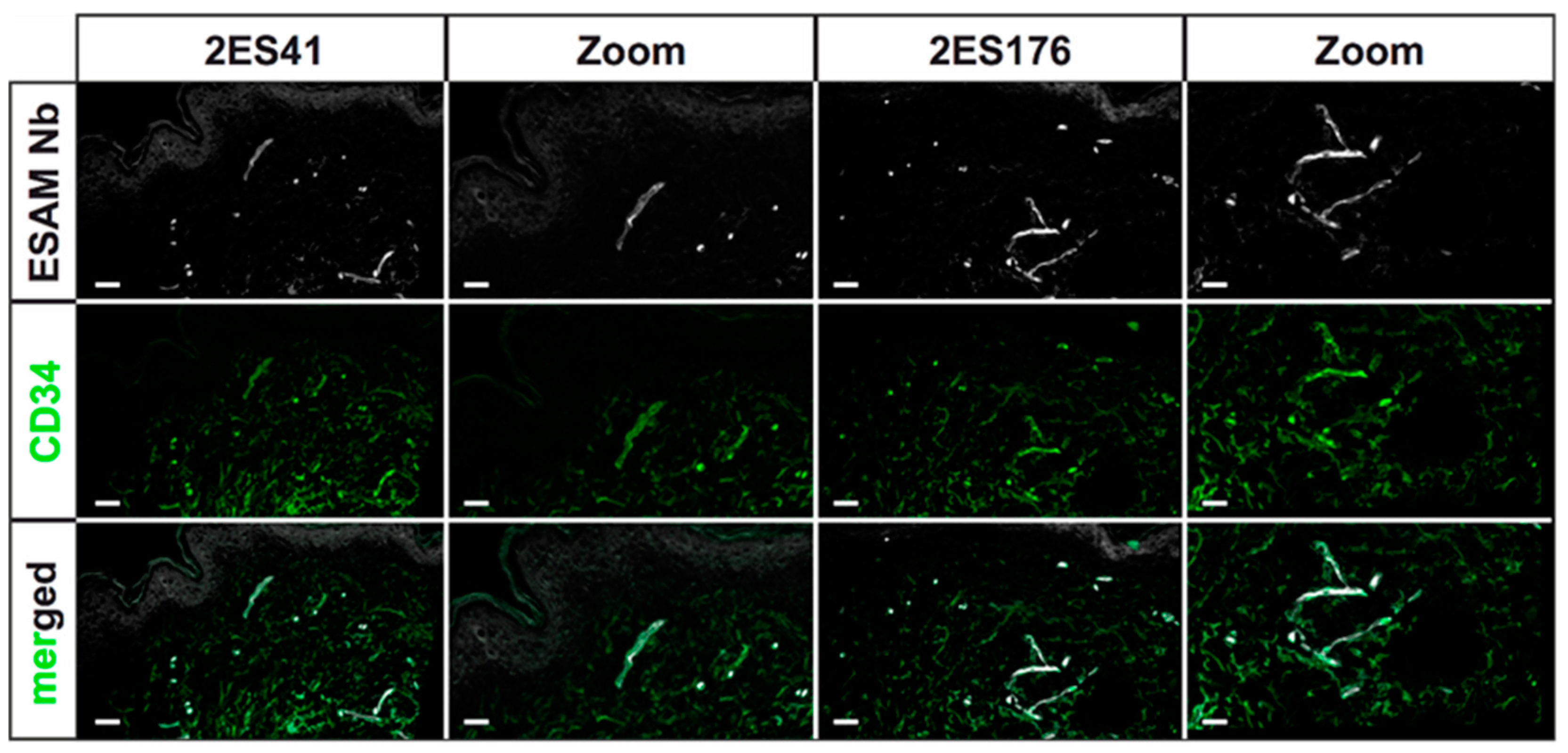
Human skin 2D stainings of selected Nbs reflected heterogenous results, as some Nb clones depicted strong unspecific background signals and only faint antigen-specific labeling. This finding was not unexpected, as antigen-specific ELISA screenings do not reflect the vast plethora of potential biological antigens, which may additionally be bound by the Nbs, and ultimately underlines the importance of experimental validation of every single Nb clone. Of interest, a single hESAM-Nb also exhibited moderate staining performance in 2D immunofluorescence stainings of murine embryonic tissue, highlighting the broad variety in target recognition among Nb clones with otherwise close sequence homology. Unexpectedly, however, Nb clones with sufficient staining quality in human samples also exhibited additional unspecific labeling of the epidermis, which, although also shown by the commercially available hESAM antibody control, prevents high-quality imaging. This phenomenon was bypassed through direct-labeling of selected Nbs with fluorescent dyes using NHS ester chemistry, additionally reducing incubation times and unspecific background signals. Yet, NHS ester-mediated crosslinking of fluorophores and lysine amino acids may elicit conformational changes within the inherent Nb structure, possibly resulting in altered binding affinities of specific as well as unspecific targets. Notably, we also observed changes in Nb binding specificity, since undesirable binding events were strongly enhanced in some Nb clones. Consequently, due to its unpredictable nature, NHS ester chemistry was not applicable to every Nb, so other labeling strategies may be more favorable, such as sortase- or maleimide-mediated protein labeling approaches [
28,
29,
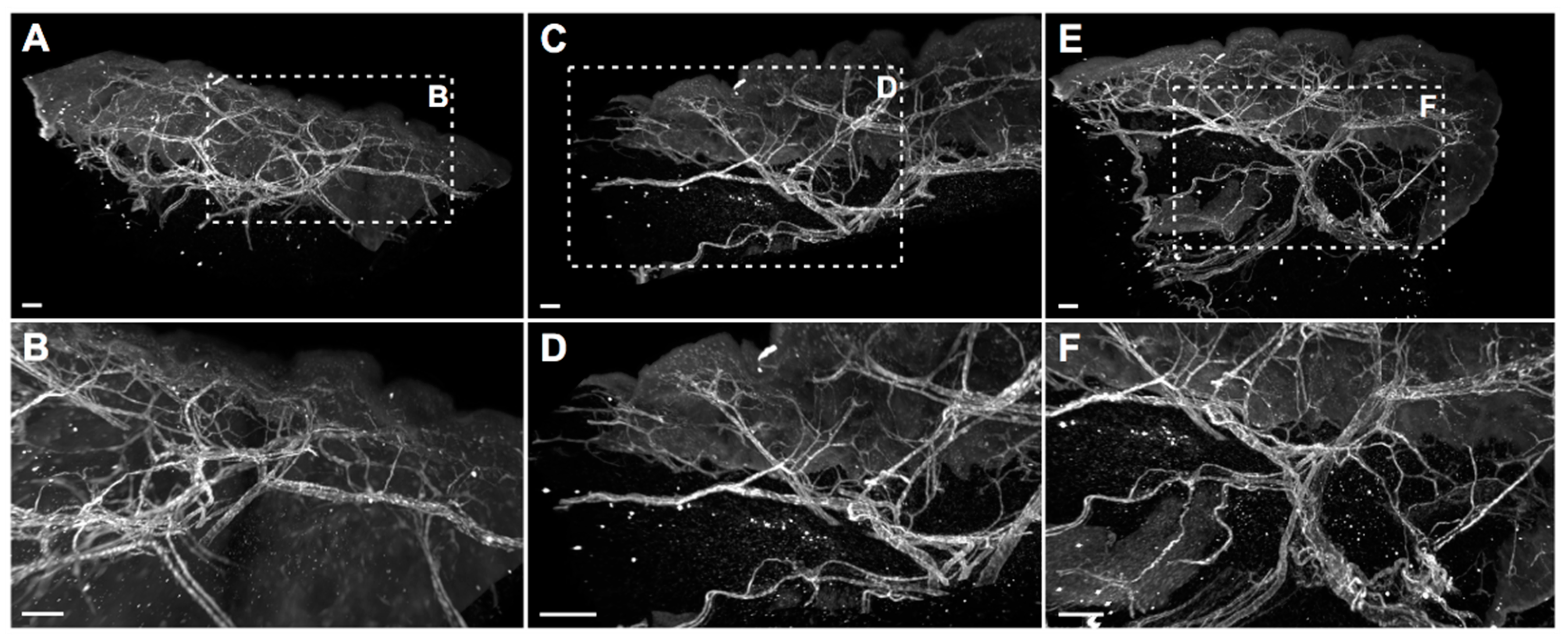
30]. Nonetheless, for other clones, imaging quality was considerably improved, which was demonstrated in high-quality 2D and whole-mount 3D immunofluorescence stainings. In 3D stainings of human skin biopsies, the hESAM-Nbs allowed unprecedented visualization of ESAM-positive target structures, displaying the delicate blood vascular network of the entire tissue sample.
In conclusion, in this study, we generated and experimentally validated hESAM-Nbs, which not only featured similar 2D staining properties to a commercially available IgG antibody, but also unparalleled 3D imaging quality. Previously impossible by using commercial ESAM antibodies, for the first time, the hESAM-Nbs enabled unrestricted and detailed spatial representations of ESAM-expressing blood endothelial cells in 3D imaging approaches. Owing to the animal-free, high-yield, and low-cost production, the hESAM-Nbs represent a novel and cost-efficient imaging tool for 3D histopathological analysis in academic research and potentially clinical diagnostics.
4. Materials and Methods
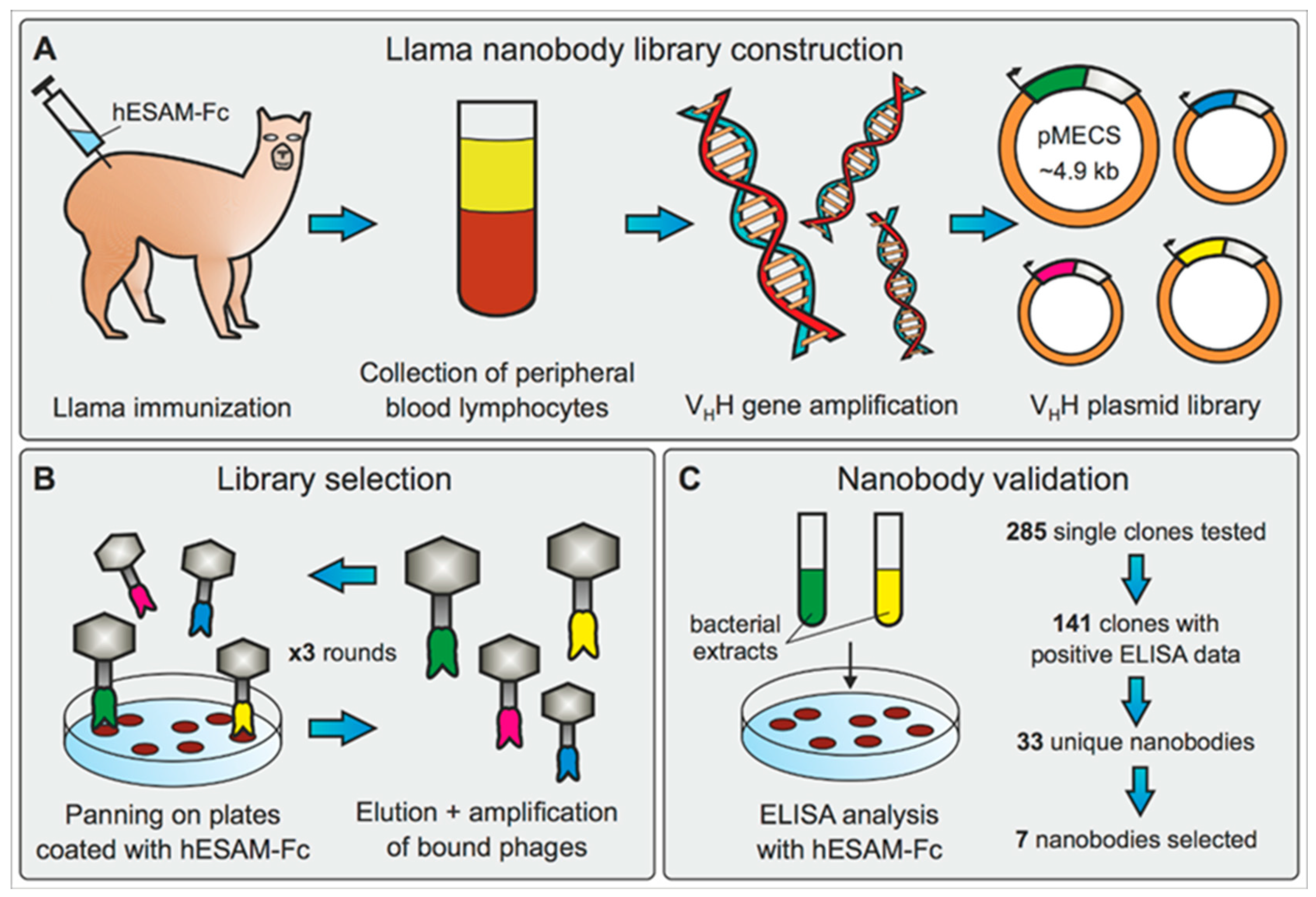
4.1. Llama Immunization and Construction of Nanobody Library
For the generation of Nbs targeting the hESAM protein, llamas (
Lama glama) were immunized with the extracellular part of the hESAM protein (amino acids 1-247) fused to the Fc domain of human IgG1 (hESAM-Fc,
Supplementary Figure S1A). The recombinant hESAM-Fc protein was expressed in CHO cells and purified from the CHO cell culture supernatant using Protein G Sepharose
® columns (ab193260, Abcam, Cambridge, UK), followed by dialysis against PBS. The immunoadjuvant used during immunizations was Adjuvant P (3111, GERBU Biotechnik GmbH, Heidelberg, Germany). In detail, two llamas were subcutaneously injected with 100 µg carrier-free, recombinant hESAM-Fc protein on days 0, 10, 16, 23, 30, and 37, followed by collection of 100 mL anti-coagulated blood on day 42 for isolation of peripheral blood lymphocytes (PBLs). For each llama, separate VHH gene libraries were constructed by using total RNA from PBLs as a template for first strand cDNA synthesis with oligo(dT) primers. Using the PBL cDNA, VHH encoding sequences were amplified via a multi-step PCR, digested with SapI (R0569, New England Biolabs, Ipswich, MA, USA), and cloned into the SapI restriction site of the pMECS phagemid vector (Prof. Serge Muyldermans, Laboratory of Cellular and Molecular Immunology, Vrije Universiteit, Brussel, Belgium). After ligation, electrocompetent
E. coli TG1 cells (60502, Lucigen, Middleton, WI, USA) were transformed with the recombinant phagemid vectors, resulting in two VHH libraries of about 10
8 independent transformants each. A detailed description of the workflow has been published elsewhere [
22].
4.2. Biopanning and Screening for hESAM-Specific Nanobodies
Phage enrichment and biopanning was performed as previously described [
22], with the following specifications: For the selection of hESAM-specific Nbs, three consecutive panning rounds were performed on 96-well Microtiter™ microplates (2205, Thermo Fisher Scientific, Waltham, MA, USA) coated with different concentrations of the recombinant hESAM-Fc protein (1st round: 100 µg/mL, 2nd + 3rd round: 50 µg/mL). The coating buffer was 100 mM NaHCO
3, with pH 8.2. To reduce unspecific human IgG1-Fc binding events, the solution was supplemented with 1 µM recombinant human IgG Fc protein (110-HG, R&D Systems, Minneapolis, MN, USA). Enrichment of antigen-specific phages was about 10
2-fold and 2 × 10
3-fold after the 2nd and 3rd round, respectively. After each round of biopanning,
E. coli TG1 cells were transfected with the phage output and used to amplify phages for the next round of biopanning and/or subsequent ELISA analysis. After enrichment, 285 clones were randomly selected and subjected to ELISA analysis. In detail, crude periplasmic extracts of bacterial clones were incubated on 96-well Microtiter™ microplates coated with 1 µg/mL hESAM immunogen in blocking buffer (‘hESAM’), 2 µg/mL hIgG1-Fc in blocking buffer (‘Fc’), or blocking buffer only (‘control’). The blocking buffer was 100 mM NaHCO
3, with pH 8.2. ELISA measurements were performed as described elsewhere [
22].
For expression of Nbs linked to the 6xHis protein tag only, Nb sequences were recloned from the pMECS phagemid vector into pHEN6c expression vector [
23]. To this end, Nb sequences were PCR amplified using generic framework primers with the following sequences:
Forward Primer1: 5′-GATGTGCAGCTGCAGGAGTCTGGGGGAGG-3′ (all clones expect ‘3ES42’);
Forward Primer2: 5′-GATGTGCAGCTGCAGGAGTCTGGAGGAGG-3′ (‘3ES42’);
Reverse Primer: 5′-CTAGTGCGGCCGCTGAGGAGACGGTGACCTGGGT-3′ (all clones).
After purification (QIAquick PCR Purification Kit, 28104, Qiagen, Hilden, Germany), obtained PCR products were digested with PstI-HF (R3140, New England Biolabs, Ipswich, MA, USA) and BstEII-HF (R3162, New England Biolabs, Ipswich, MA, USA) restriction enzymes for 20 min at 37 °C. Simultaneously, empty pHEN6c plasmids were subjected to PstI-HF/BstEII-HF restriction digestion, additionally supplemented with 5 units of FastAP™ alkaline phosphatase (EF0651, Thermo Fisher Scientific, Waltham, MA, USA). FastAP™ activity was heat-inactivated by incubation for 5 min at 80 °C, following the incubation for 20 min at 37 °C. After purification (QIAquick PCR Purification Kit, 28104, Qiagen, Hilden, Germany), restricted Nb sequences and pHEN6c plasmids were subjected to T4 DNA igase-mediated ligation reactions at 16 °C for 16 h, containing 2.5 units of T4 DNA ligase (M0202, New England Biolabs, Ipswich, MA, USA). Subsequently, ligation products were transformed into WK6 E. coli cells (C303006, Thermo Fisher Scientific, Waltham, MA, USA), which were analyzed by Sanger DNA sequencing for the correct integration of the respective Nb sequences, using primers with the following sequences:
4.3. Nanobody Expression and Purification
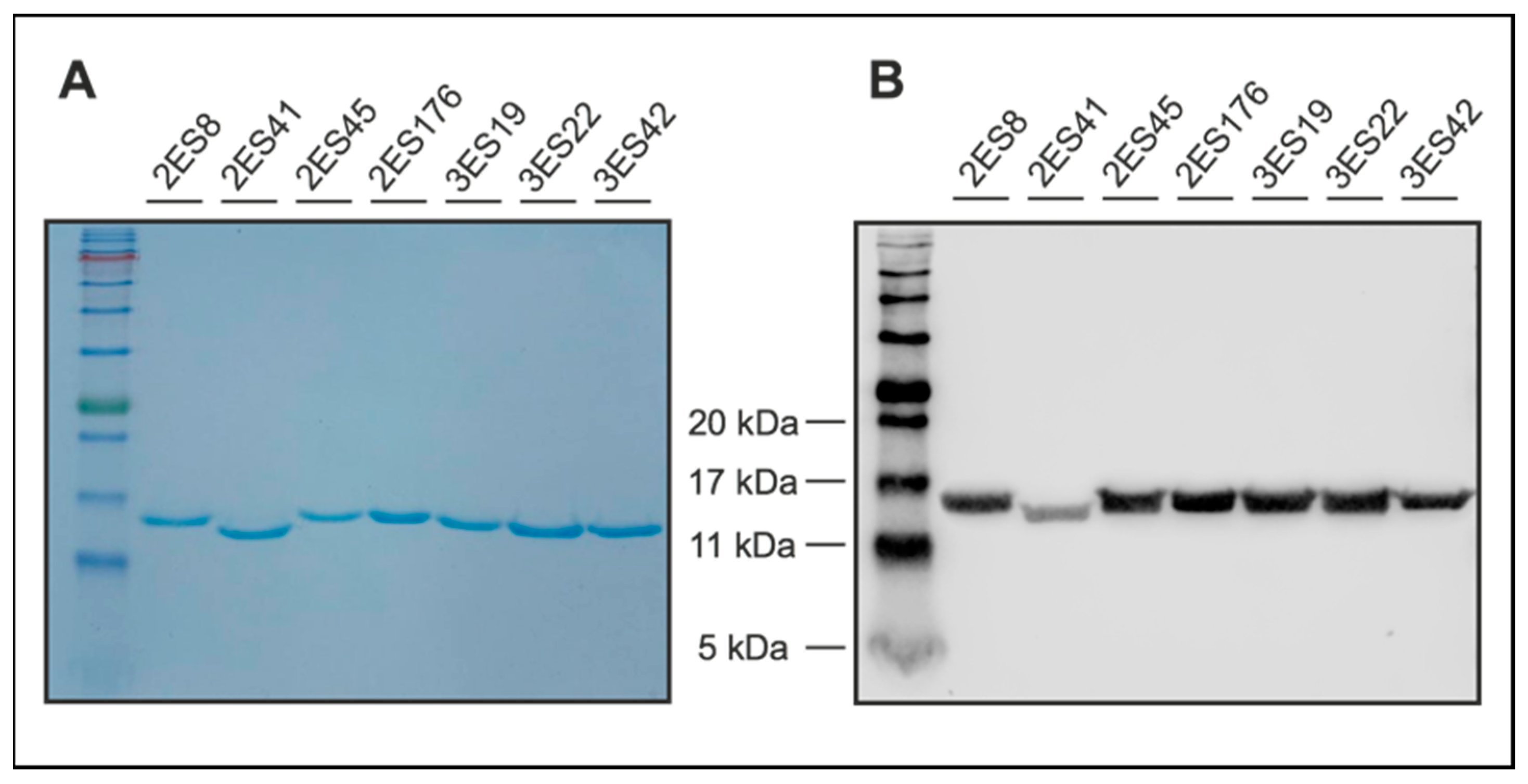
For production of Nbs, WK6 E. coli harboring the pHEN6c-Nb plasmid of interest were grown in 1 L of ‘Terrific Broth’ medium (2.3 g/L KH2PO4, 16.4 g/L K2HPO4-3H2O, 12 g/L tryptone, 24 g/L yeast extract, 0.4% (v/v) glycerol) supplemented with 100 µg/mL ampicillin, 2 mM MgCl2, and 0.1% (w/v) glucose. Bacterial cultures were incubated at 37 °C with constant shaking until an OD600 of 0.6–0.9 was reached. Then, Nb expression was induced by adding isopropyl ß-D-1-thiogalactopyranoside (IPTG) to a final concentration of 1 mM, and cultures were incubated at 28 °C with constant shaking for 16 h. For extraction of Nbs, bacterial cells were centrifuged (8000× g, 8 min, RT), resuspended in 12 mL TES buffer (0.2 M Tris [pH 8.0], 0.5 mM EDTA, 0.5 M sucrose), and shaken for 1 h on ice. Subsequently, 18 mL TES/4 buffer (0.05 M Tris [pH 8.0], 0.125 mM EDTA, 0.125 M sucrose) was added to the cell solution, followed by shaking for 1 h on ice and centrifugation (8000× g, 30 min, 4 °C). The supernatants containing the periplasmic proteins were collected and 6xHis-tagged Nbs were purified using HIS-Select® nickel affinity gel (P6611, Sigma-Aldrich, Darmstadt, Germany) according to the manufacturer’s instructions. Briefly, 2 mL HIS-Select® solution was equilibrated in 48 mL of phosphate buffered saline (PBS, pH 7.4), mixed and centrifuged (2000× g, 2 min), followed by removal of the supernatant. The equilibration procedure was repeated for two additional times, after which bacterial periplasmic extracts were added to the equilibrated HIS-Select® resin and incubated for 1 h at RT with gentle shaking. After incubation, solutions were loaded on PD-10 columns (17-0435-01, GE healthcare, Chicago, IL, USA) and washed three times with 50 mL PBS. Nbs were eluted by using 3 × 1 mL 0.5 M imidazole in PBS (I2399, Sigma-Aldrich, Darmstadt, Germany). To remove imidazole, eluted Nb solutions were subjected to overnight dialysis against PBS (3 kDa MWCO, 66382, Thermo Fisher Scientific, Waltham, MA, USA) with two changes of the dialysis buffer.
If Nb concentrations did not suffice for NHS labeling, Nb eluates were concentrated 4-fold using Pierce™ protein concentrator columns (3 kDa MWCO, 88512, Thermo Fisher Scientific, Waltham, MA, USA).
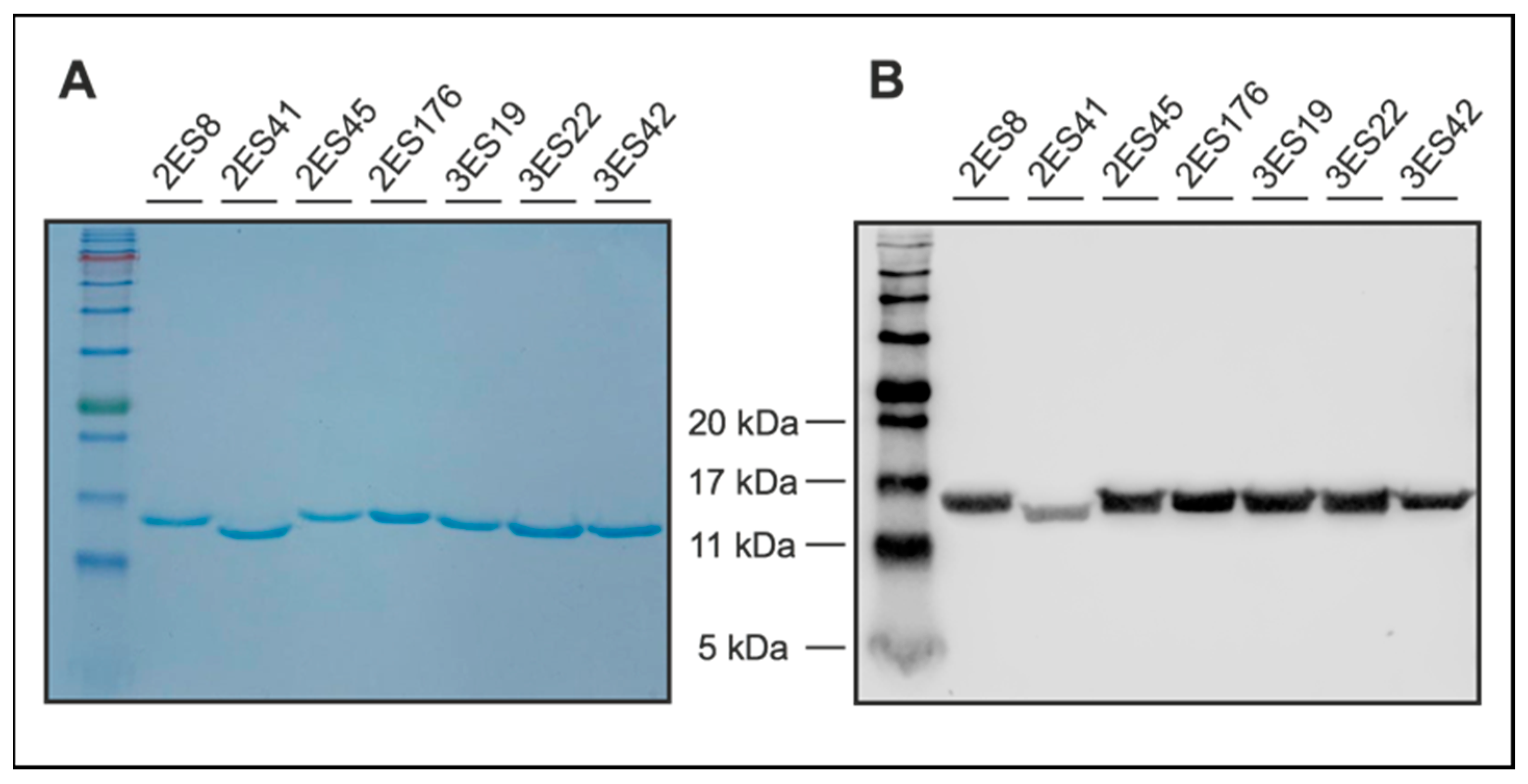
4.4. Coomassie-Blue Stained SDS-PAGE and Western Blotting
To evaluate sample purity, Nbs were separated via sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), according to standard procedures. To this end, 5 µg of Nbs were dissolved in sample denaturation buffer (0.1 M DTT, 25 mM Tris-HCl, 14% (w/v) glycerol, 0.8% (v/v) SDS, and 0.04% (w/v) OrangeG in ddH2O) and denatured for 5 min at 96 °C. In addition to the samples, a prestained protein ladder was loaded onto the gels (ab116029, Abcam, Cambridge, UK). Following electrophoresis, proteins were either directly visualized using Coomassie staining solution (0.1% (w/v), Coomassie Brilliant Blue R-250 (1610400, Bio-Rad Laboratories Inc., Hercules, CA, USA), 50% (v/v) methanol, and 10% (v/v) glacial acetic acid in ddH2O, or blotted on a nitrocellulose membrane (1620112, Bio-Rad Laboratories Inc., Hercules, CA, USA). Coomassie Blue-stained gels were de-stained after 1 h using Coomassie destaining solution (50% ddH2O, 40% methanol, 10% acetic acid (v/v/v)). Proteins transferred on nitrocellulose membranes were detected by anti-His primary antibodies (12698, Cell Signaling Technology, Danvers, MA, USA) in combination with anti-rabbit secondary antibodies (926-32211, LI-COR Biosciences, Lincoln, NE, USA) and visualized using an Odyssey® Fc Imaging System (LI-COR Biosciences, Lincoln, NE, USA).
4.5. Immunofluorescence Stainings
After fixation in 4% PFA in PBS, specimens were washed in PBS, embedded and snap-frozen in Tissue-Tek™ O.C.T. (12351753, Thermo Fisher Scientific, Waltham, MA, USA). Subsequently, 5 µm cryosections were fixed in ice-cold methanol for 15 min at −20 °C, 3× washed in PBS and blocked in blocking solution (5% chicken serum, 0.3% Triton™ X-100 in PBS) for 1 h at RT. Stainings were performed overnight at 4 °C using 10 µg/mL Nbs in dilution buffer (1% BSA, 1% chicken serum, 0.3% Triton™ X-100 in PBS). After 3× washing in PBS-T (0.1% (v/v) Tween® 20 in PBS) for 10 min, the slides were incubated overnight at 4 °C using anti-His antibody (12698, Cell Signaling Technology, Danvers, MA, USA) as well as other primary antibodies for control stainings. The slides were then washed 3× in PBS-T, and finally incubated in directly-labeled anti-rabbit tertiary antibodies (A31573, Invitrogen, Waltham, MA, USA) as well as other Alexa Fluor™ dye-conjugated secondary antibodies (Invitrogen, Waltham, MA, USA) for 1 h at RT. Following 3× washing steps in PBS-T, the sections were stained in Hoechst 33342 (62249, Thermo Fisher Scientific, Waltham, MA, USA) for 1 h at RT and subsequently washed 3x in PBS-T. After samples were mounted in fluorescence mounting medium (S3023, Agilent Technologies, Santa Clara, CA, USA), representative images were captured using a LSM 780 confocal microscope (Zeiss, Oberkochen, Germany) with 20× magnification. The following antibodies were used according to the manufacturer’s instructions: rabbit monoclonal anti-His antibody (12698, Cell Signaling Technologies, Danvers, MA, USA), mouse monoclonal anti-human CD31 antibody (131M, Cell Marque Corporation, Rocklin, CA, USA), mouse monoclonal anti-human CD34 antibody (134M, Cell Marque Corporation, Rocklin, CA, USA), goat polyclonal anti-human ESAM antibody (AF2688, R&D Systems, Minneapolis, MN, USA), rat IgG2a anti-mouse PECAM-1 antibody (102502, BioLegend, San Diego, CA, USA), donkey polyclonal anti-rabbit IgG Alexa Fluor™ 647 antibody (A31573, Invitrogen, Waltham, MA, USA), donkey polyclonal anti-mouse IgG Alexa Fluor™ 488 antibody (A21202, Invitrogen, Waltham, MA, USA), donkey polyclonal anti-goat IgG Alexa Fluor™ 568 antibody (A11057, Invitrogen, Waltham, MA, USA), and donkey polyclonal anti-rat IgG Alexa Fluor™ 488 antibody (A21208, Invitrogen, Waltham, MA, USA).
4.6. Direct Labeling of Nanobodies
Direct coupling of hESAM Nbs with Alexa Fluor™ 647 NHS-Ester dyes (A20006, Invitrogen, Waltham, MA, USA) was performed with 1 mg of Nbs dissolved in 0.1 M NaHCO3 buffer (pH 8.0) and 1 mg of amine-reactive compound dissolved in DMSO. According to the manufacturer’s instructions, the reaction was incubated in the dark for 1 h at RT with continuous stirring. Finally, reaction products were isolated using a NAP™-10 gel filtration column (prepacked with Sephadex™ G-25, 17-0854-01, Thermo Fisher Scientific, Waltham, MA, USA) and eluted in PBS. For preservation purposes, sodium azide was added to the coupled Nb solution, with a final concentration of 0.03%.
4.7. Whole-Mount Immunofluorescence Stainings
Human skin biopsies were fixated for 4 h in 4% PFA in PBS at 4 °C, followed by permeabilization (0.5% Triton™ X-100 in PBS) at 4 °C and blocking in PermBlock solution (1% BSA, 0.5% Tween® 20 in PBS) at 4 °C. Immunofluorescence stainings were performed at 4 °C with permanent shaking using directly-labeled Nbs at a final concentration of 10 µg/mL in dilution buffer (1% BSA, 1% chicken serum, 0.3% Triton™ X-100 in PBS). After 3x washing with PBS-T, stained samples were embedded in 1% low-melting point agarose and subjected to dehydration in increasing methanol concentrations (50%, 70%, 95%, >99.0%, and >99.0% (v/v) methanol in ddH2O). Finally, samples were optically cleared twice in a benzyl alcohol:benzyl benzoate solution (BABB, ratio 1:2) before being imaged on a LaVision Ultramicroscope II (LaVision BioTec, Bielefeld, Germany) at various magnifications and a step size of 2 µm. Digital 3D reconstruction of light sheet image stacks was performed using the Imaris Microscopy Image Analysis Software (Oxford Instruments, Abingdon, UK).
4.8. Homology Comparison of hESAM Amino Acid Sequences
For protein homology comparison, amino acid sequences of the human (ENST00000278927.10) as well as the murine (ENSMUST00000002011.14) ESAM proteins were exported from the ensemble genome browser (Ensembl release 105) and subjected to pairwise sequence alignment using the EMBOSS needle algorithm [
31]. The detailed localization of the respective extracellular domains (ECDs) were obtained from the Universal Protein Resource (UniProt) database [
26].


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}