
Glucose Increases STAT3 Activation, Promoting Sustained XRCC1 Expression and Increasing DNA Repair


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
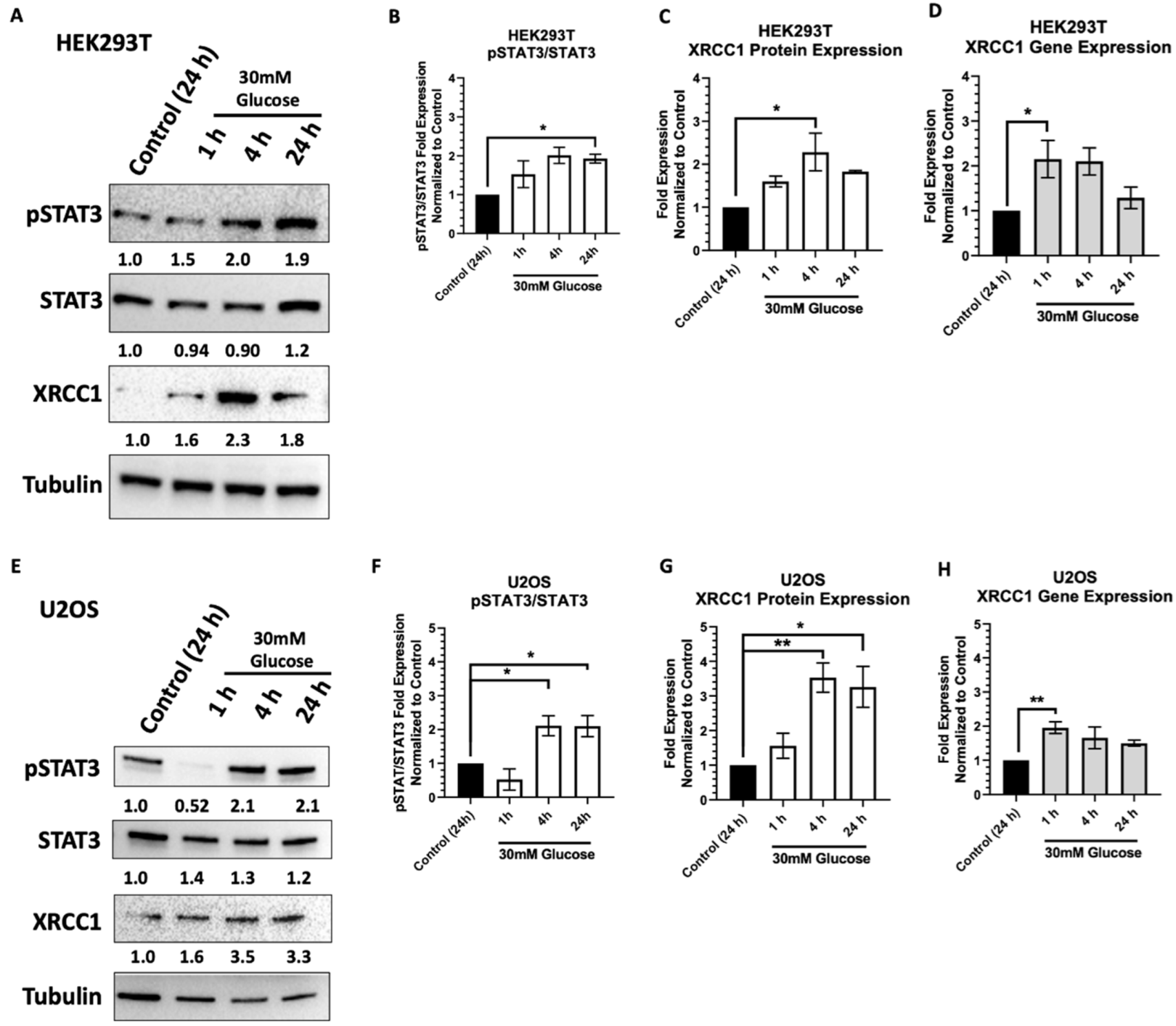
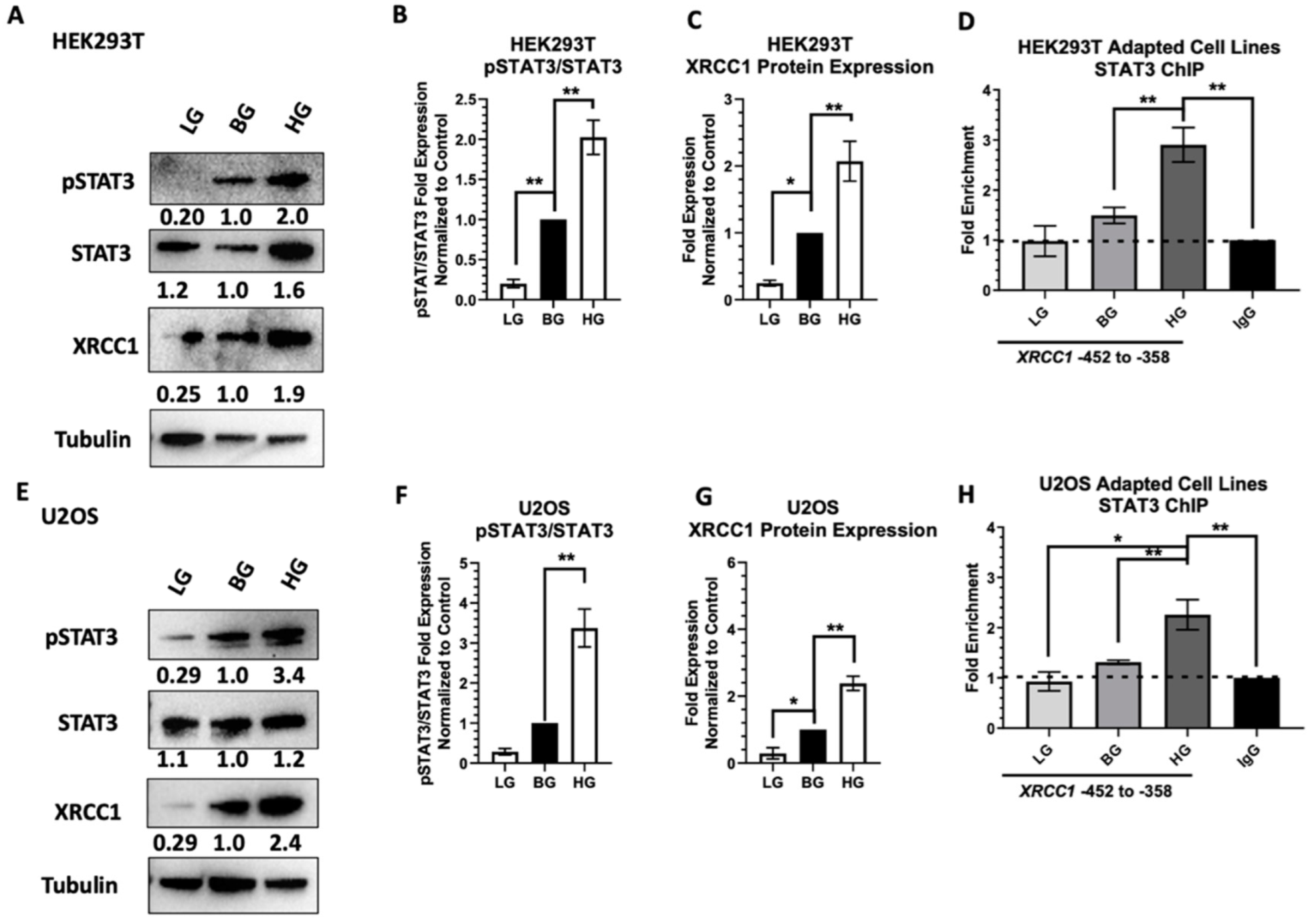
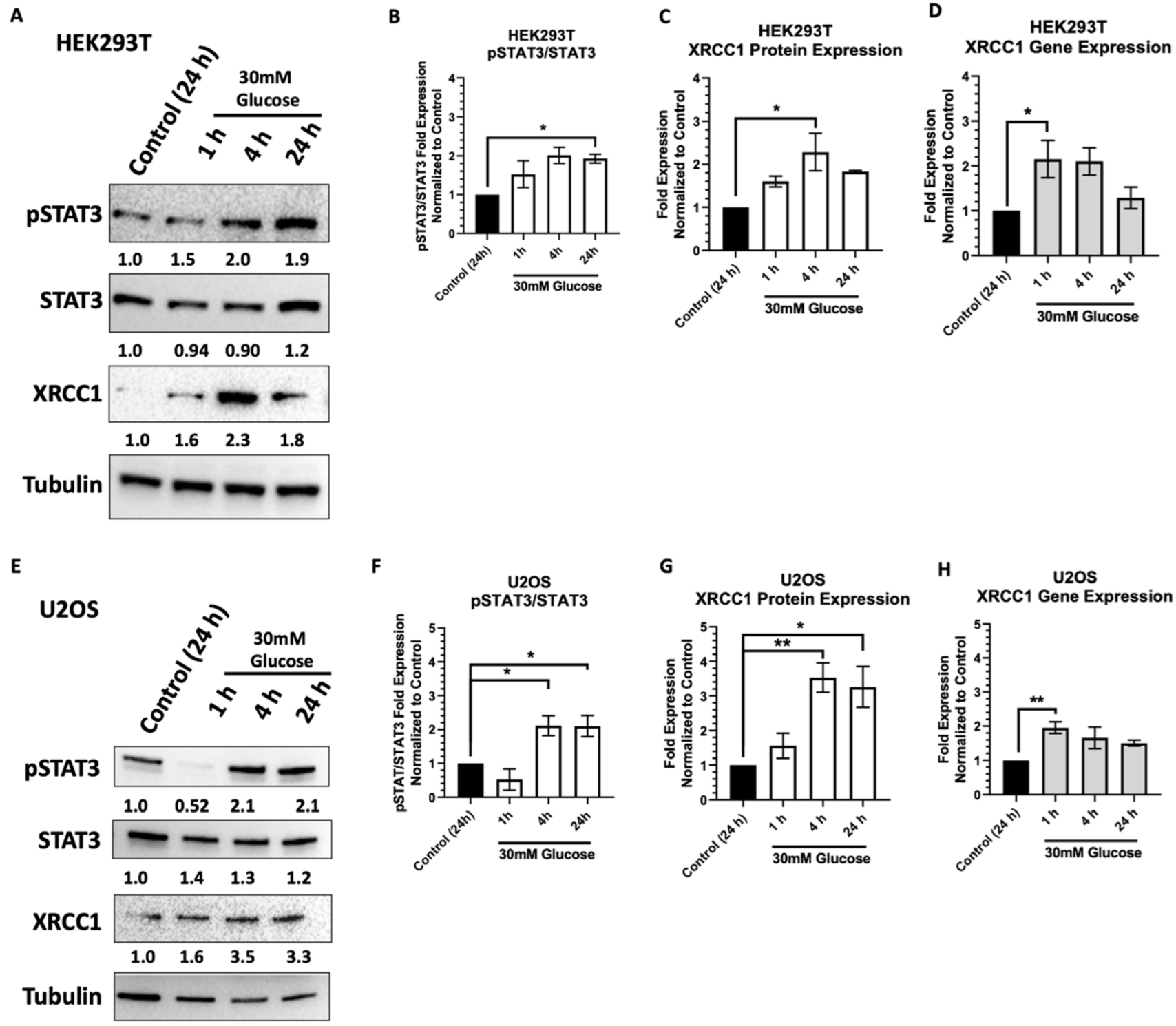
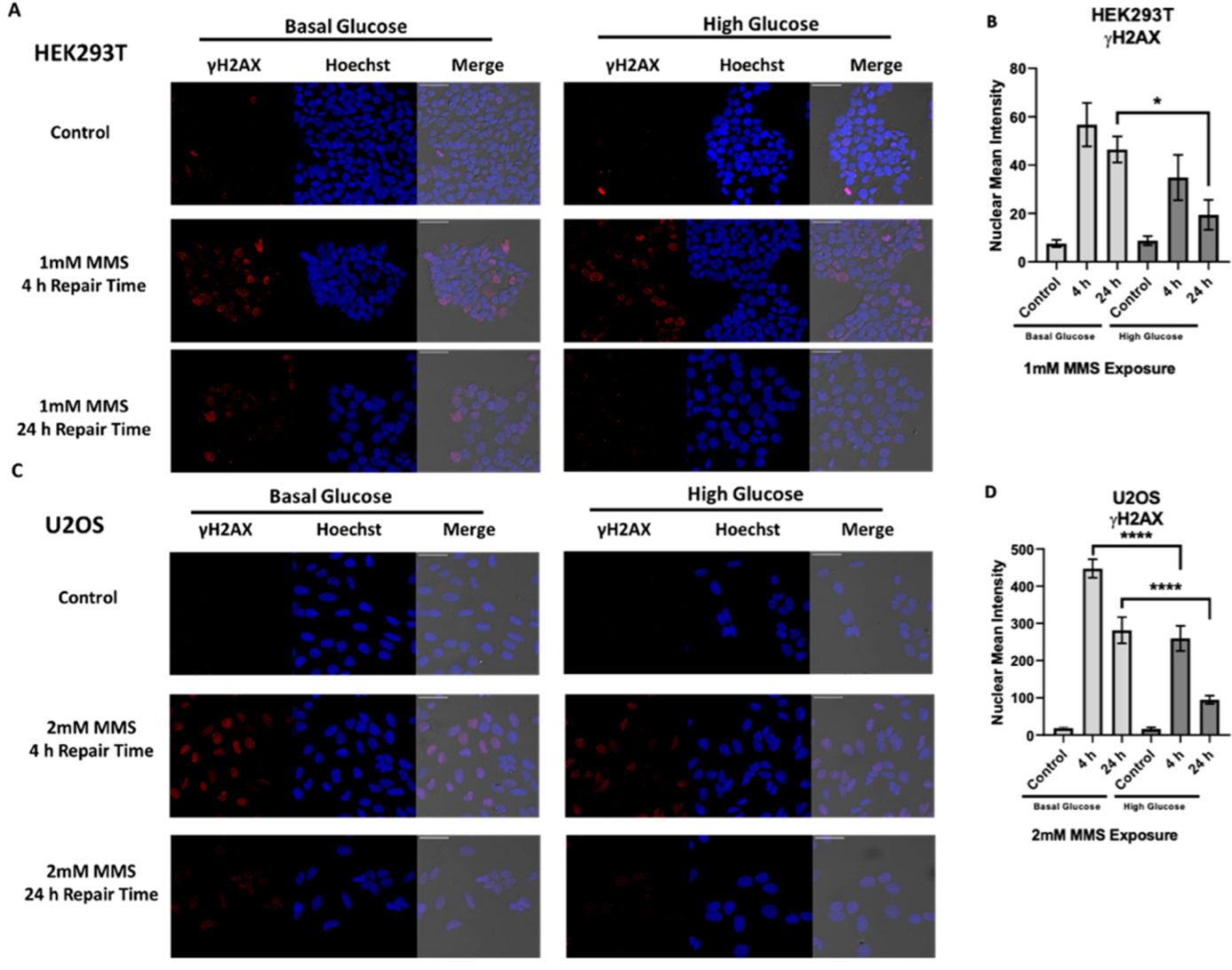
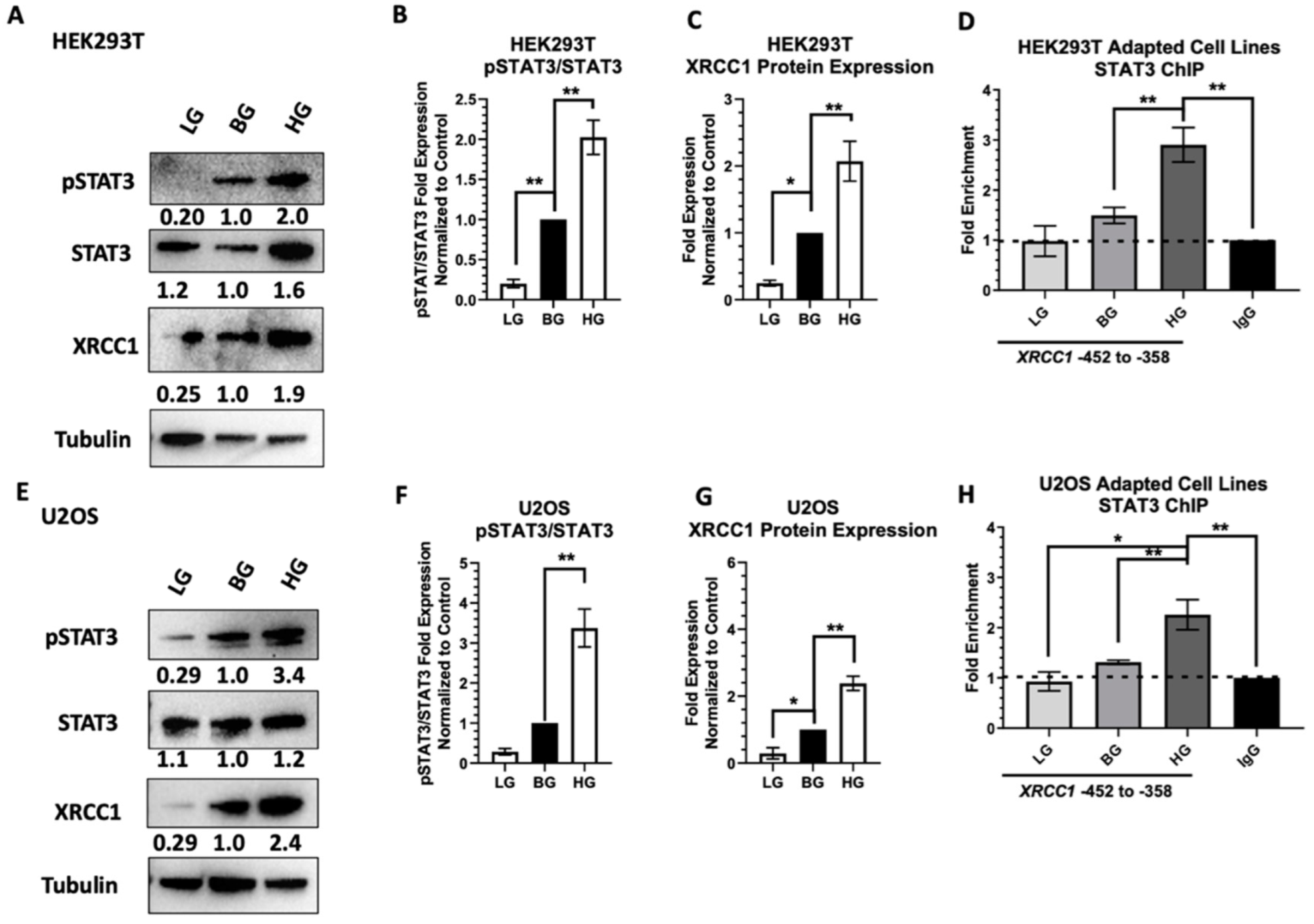
2.1. Acute High Glucose Stimulates Activation of STAT3 and XRCC1 Expression
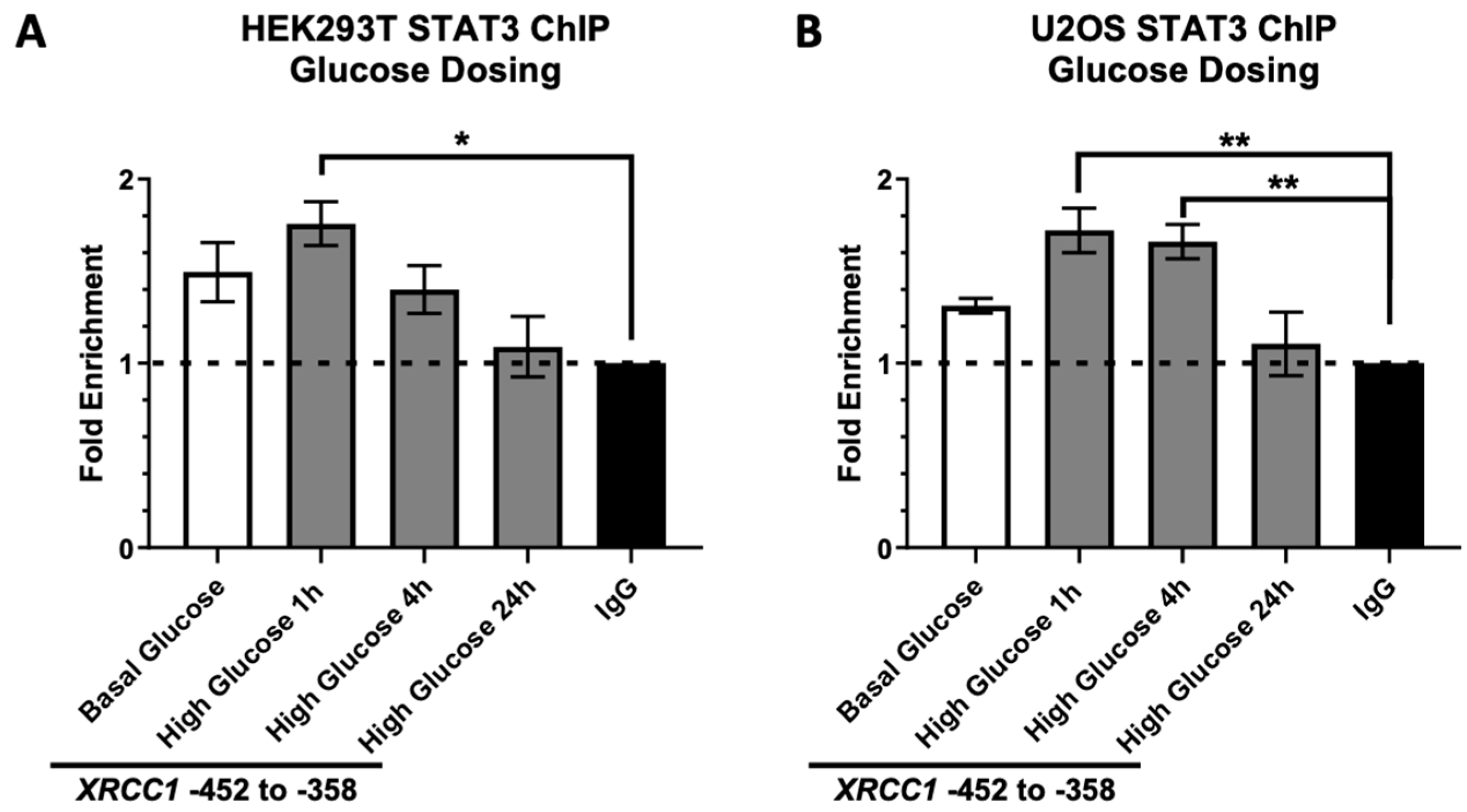
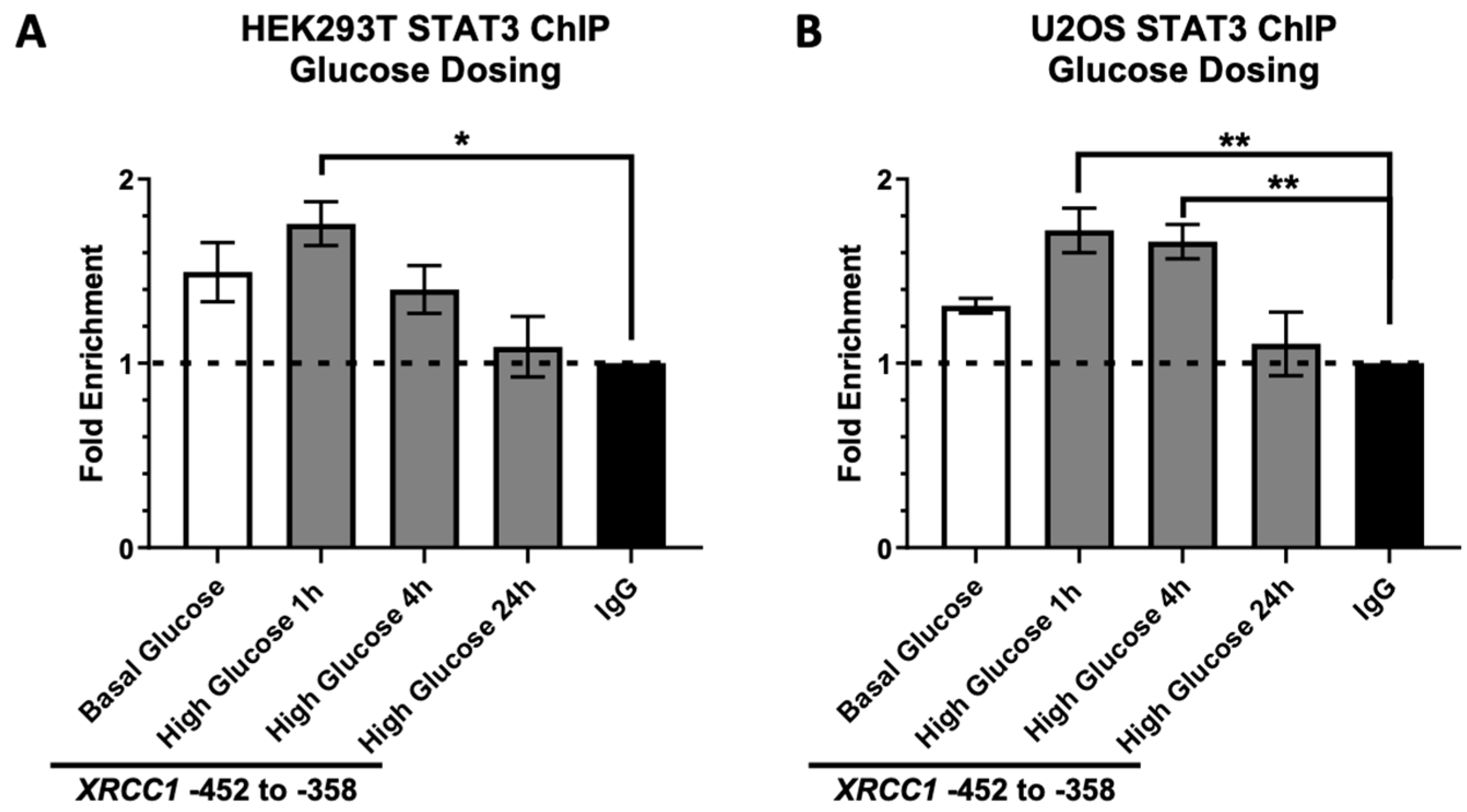
2.2. STAT3 Occupancy of the XRCC1 Promoter Increases following Acute High Glucose Exposure
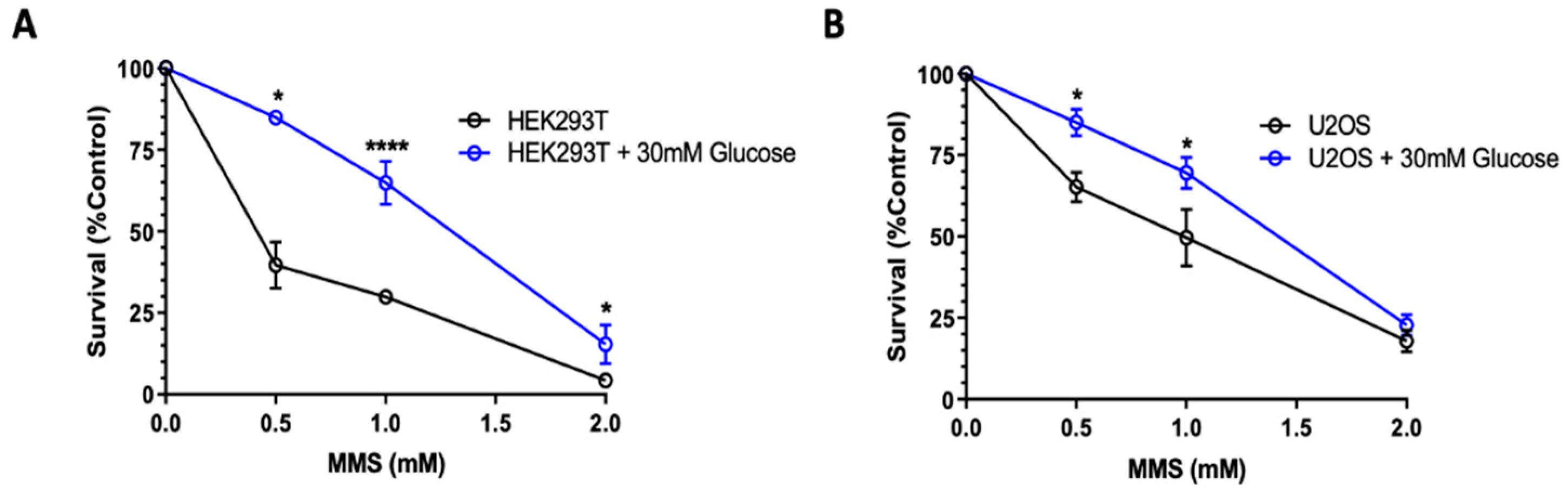
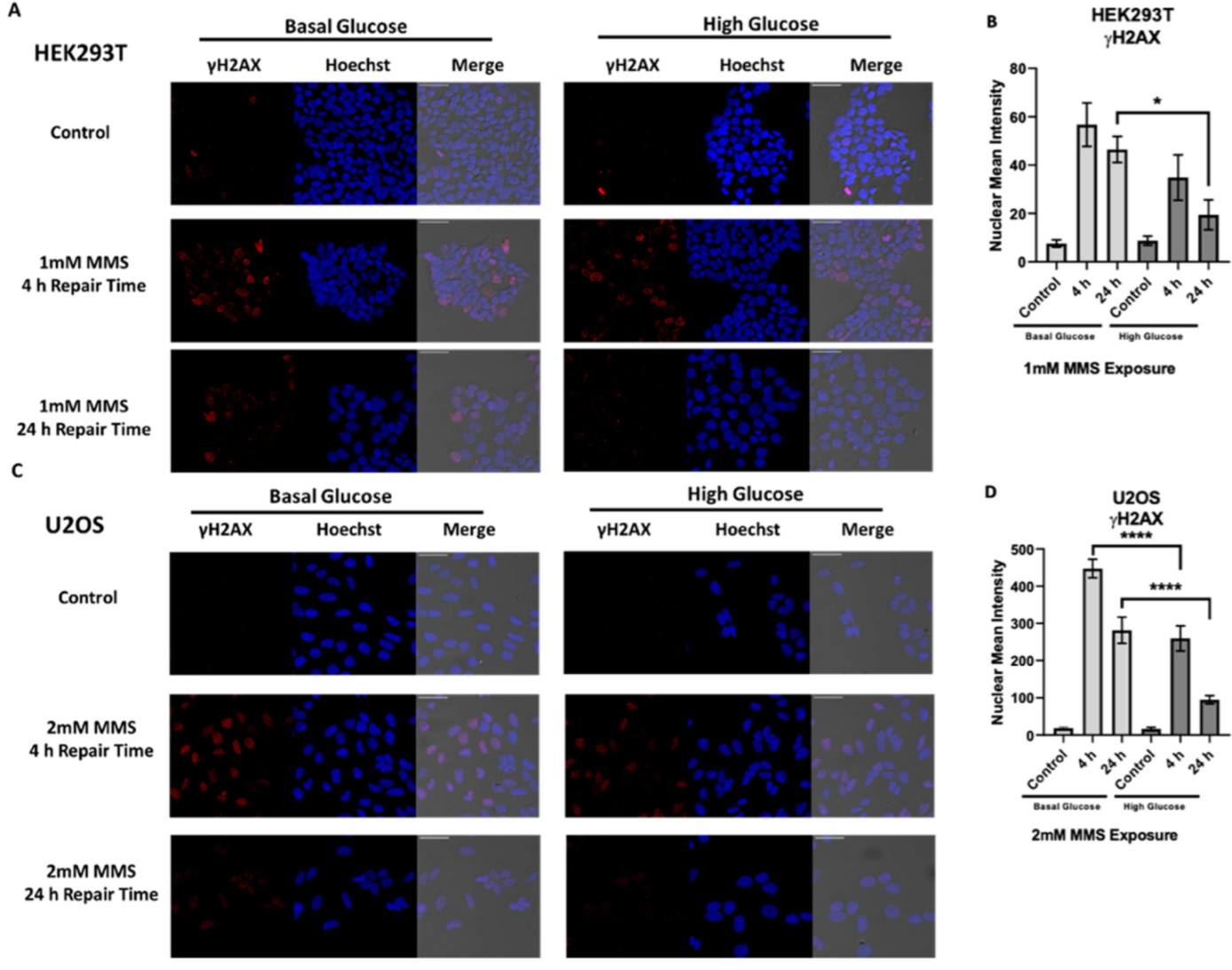
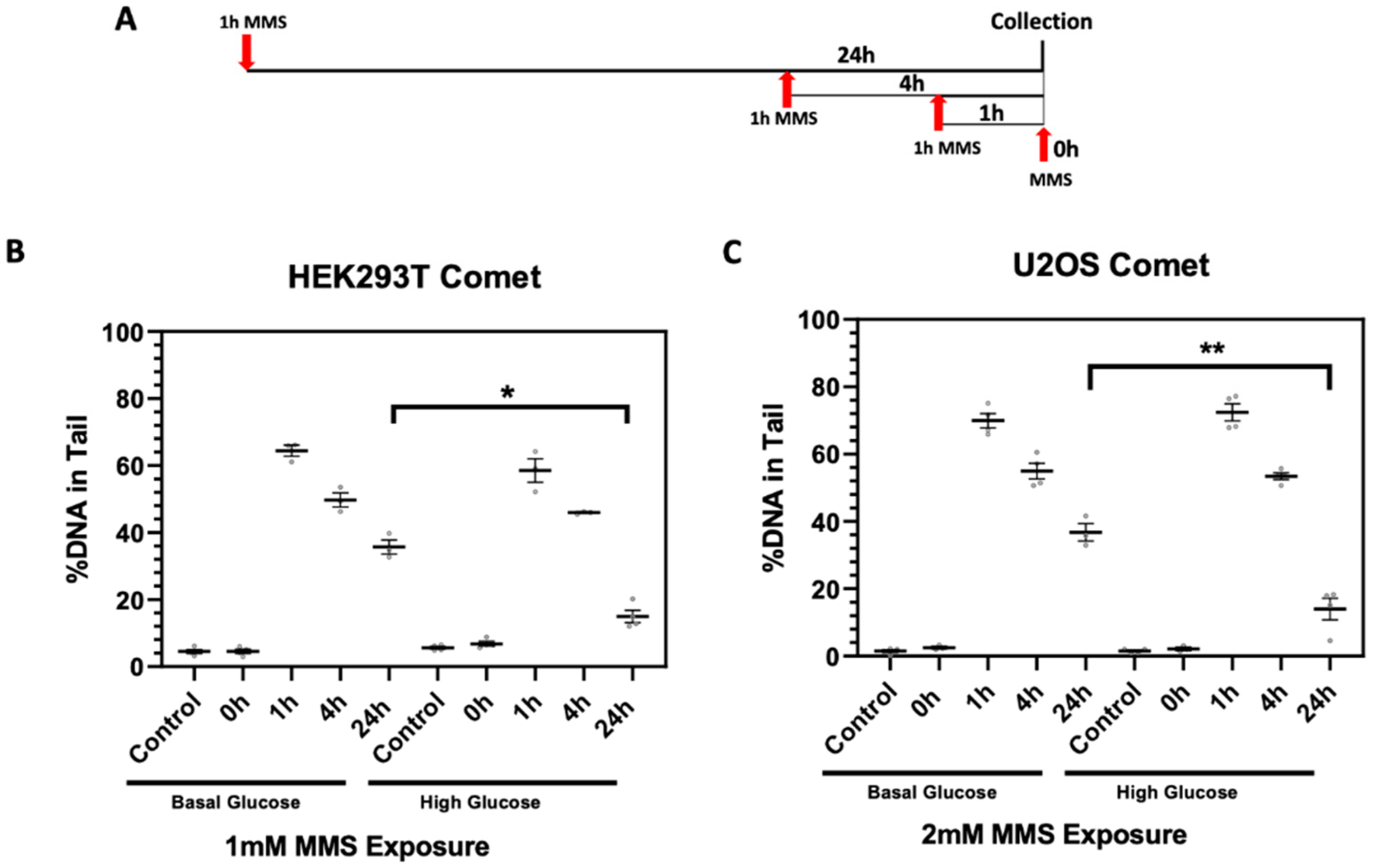
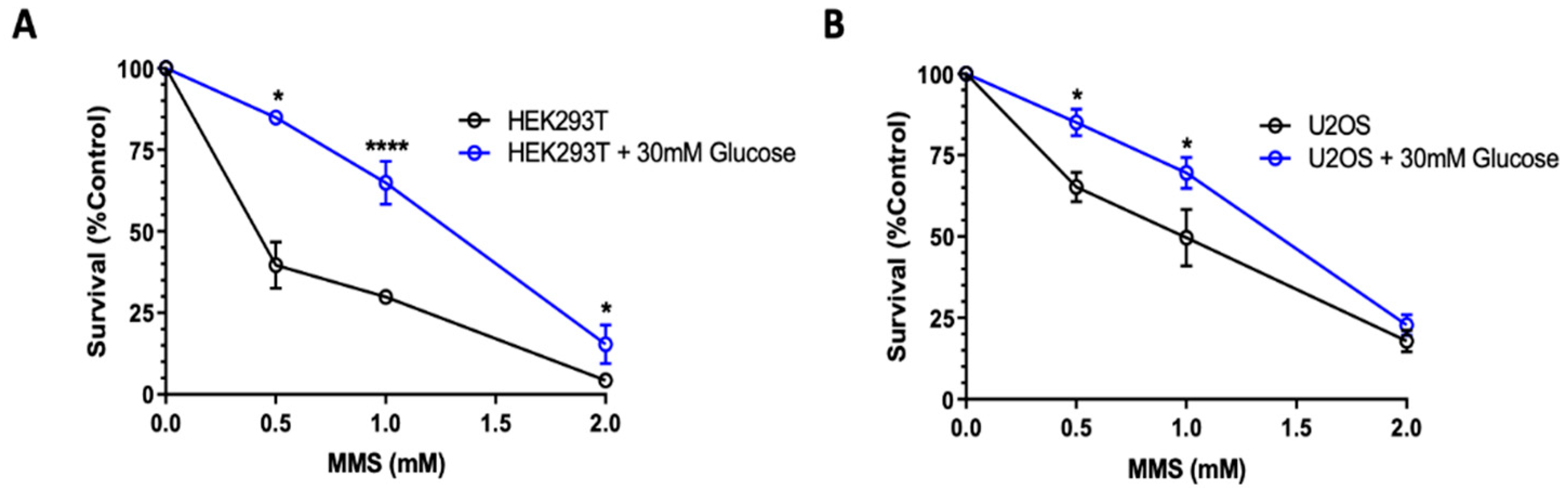
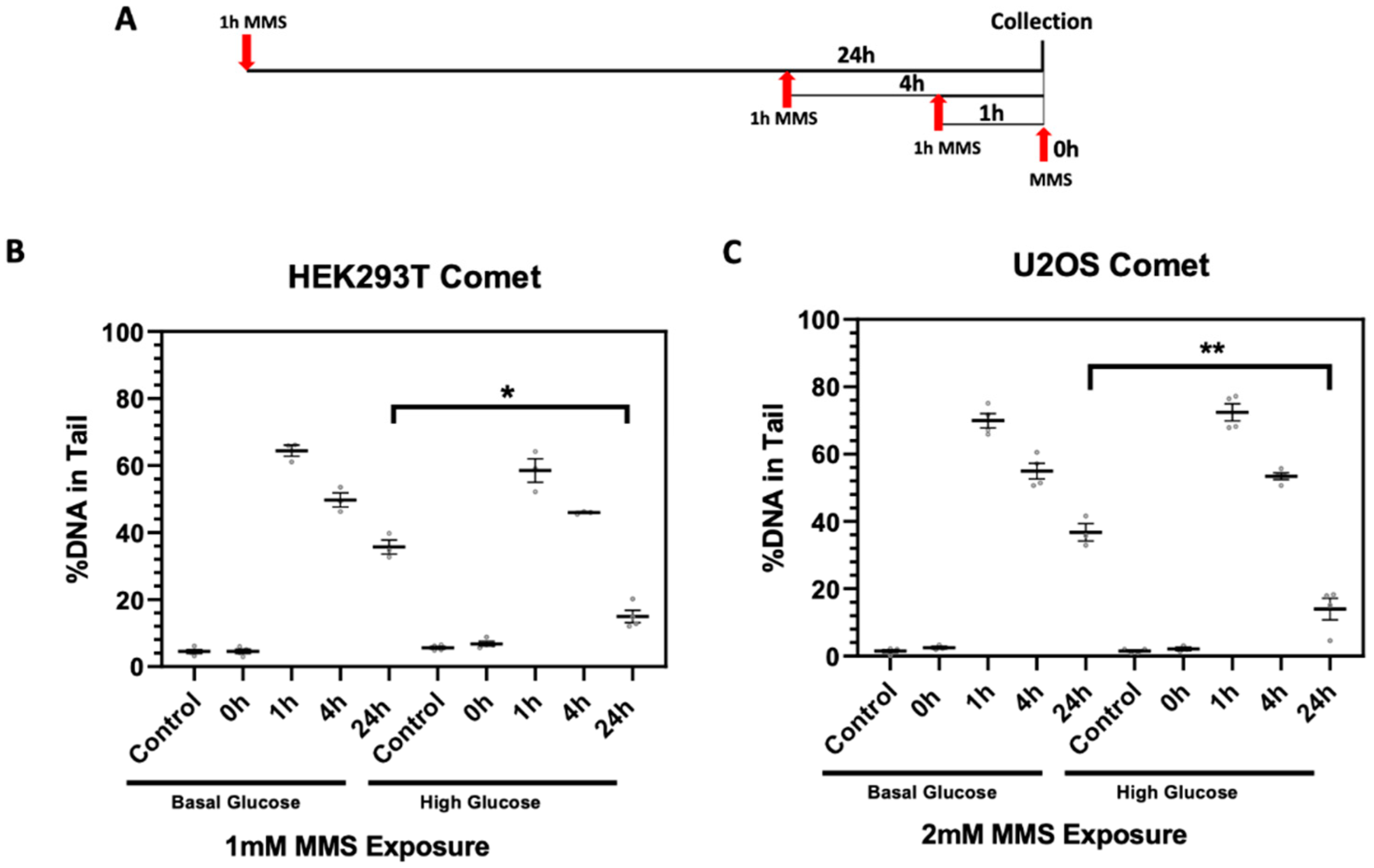
2.3. High Glucose-Induced XRCC1 Promotes Cell Survival through Increased DNA Repair
2.4. Adaptive Changes in Glucose Concentration Alter STAT3 Activation, XRCC1 Expression, and STAT3 Occupancy of the XRCC1 Promoter
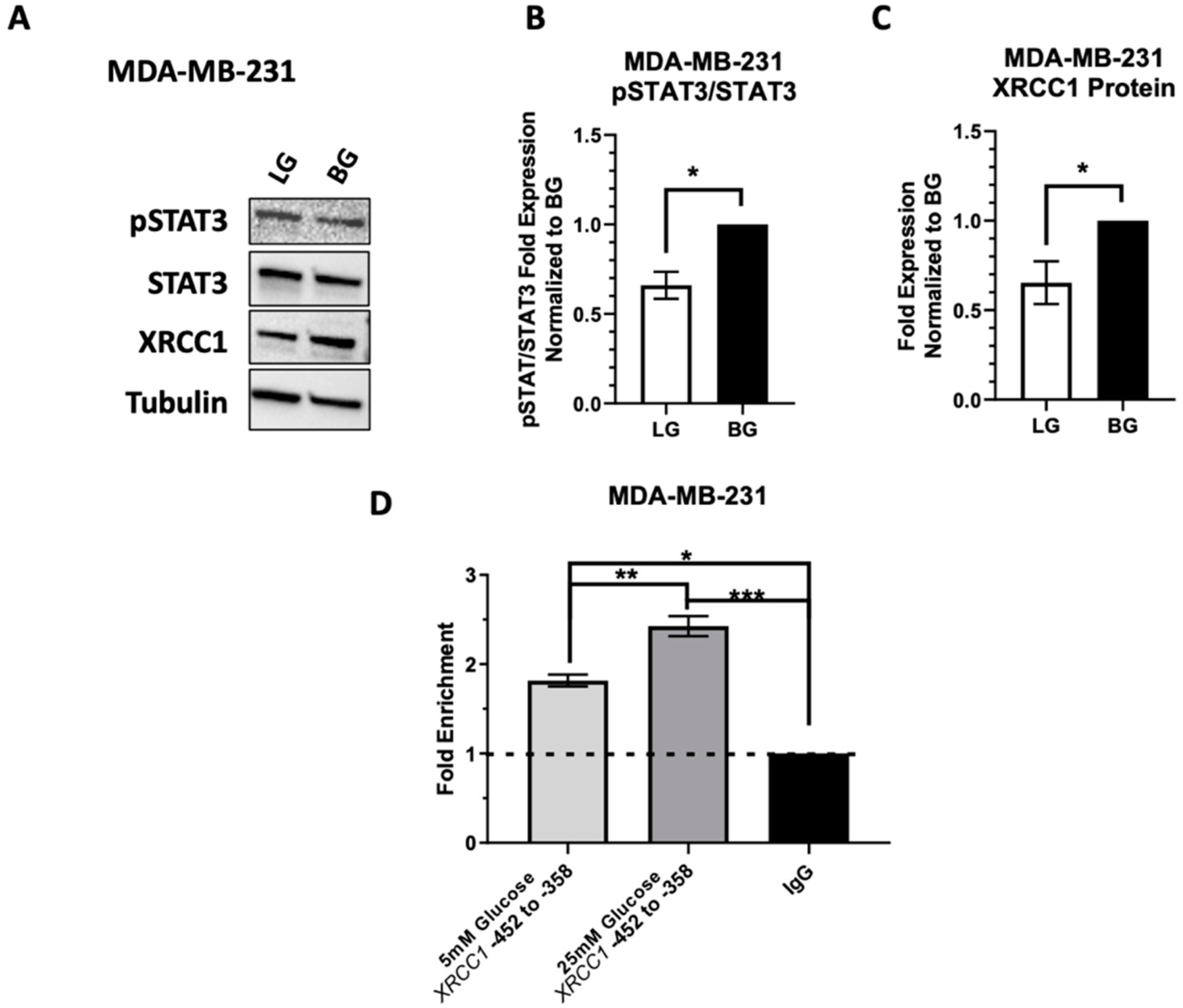
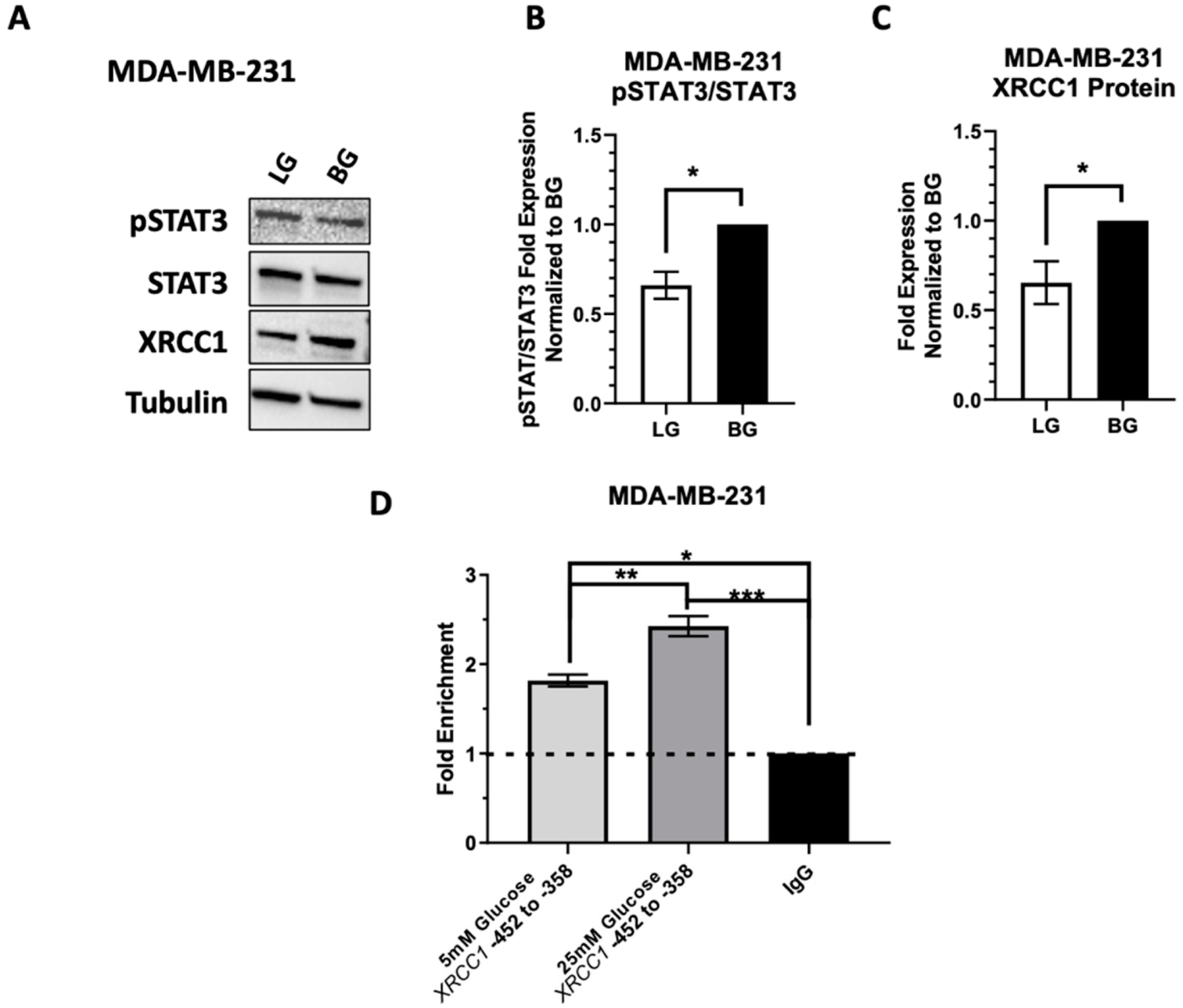
2.5. Low Glucose Adaptation of the TNBC Cell Line MDA-MB-231 Only Partially Reduces STAT3 Occupancy of the XRCC1 Promoter
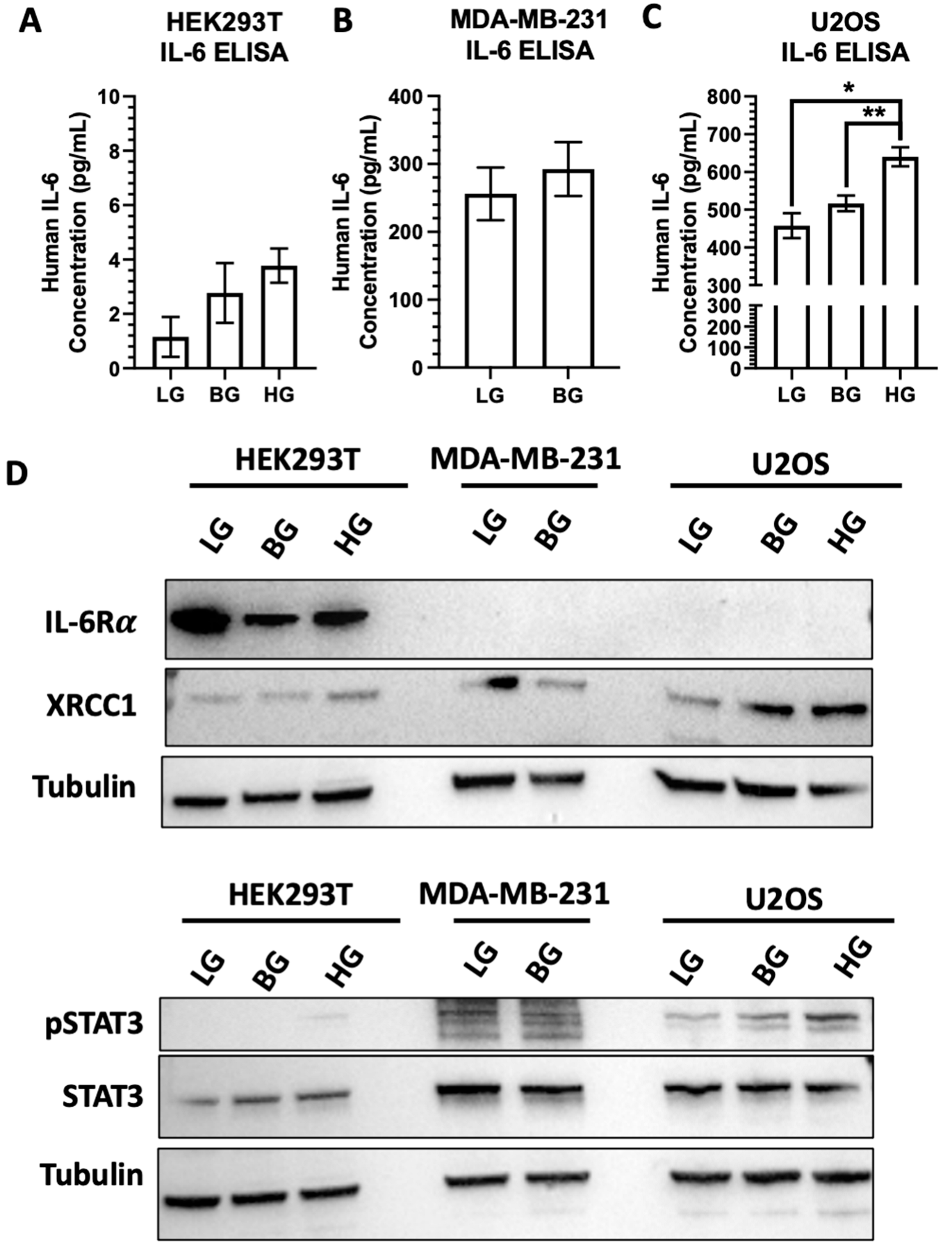
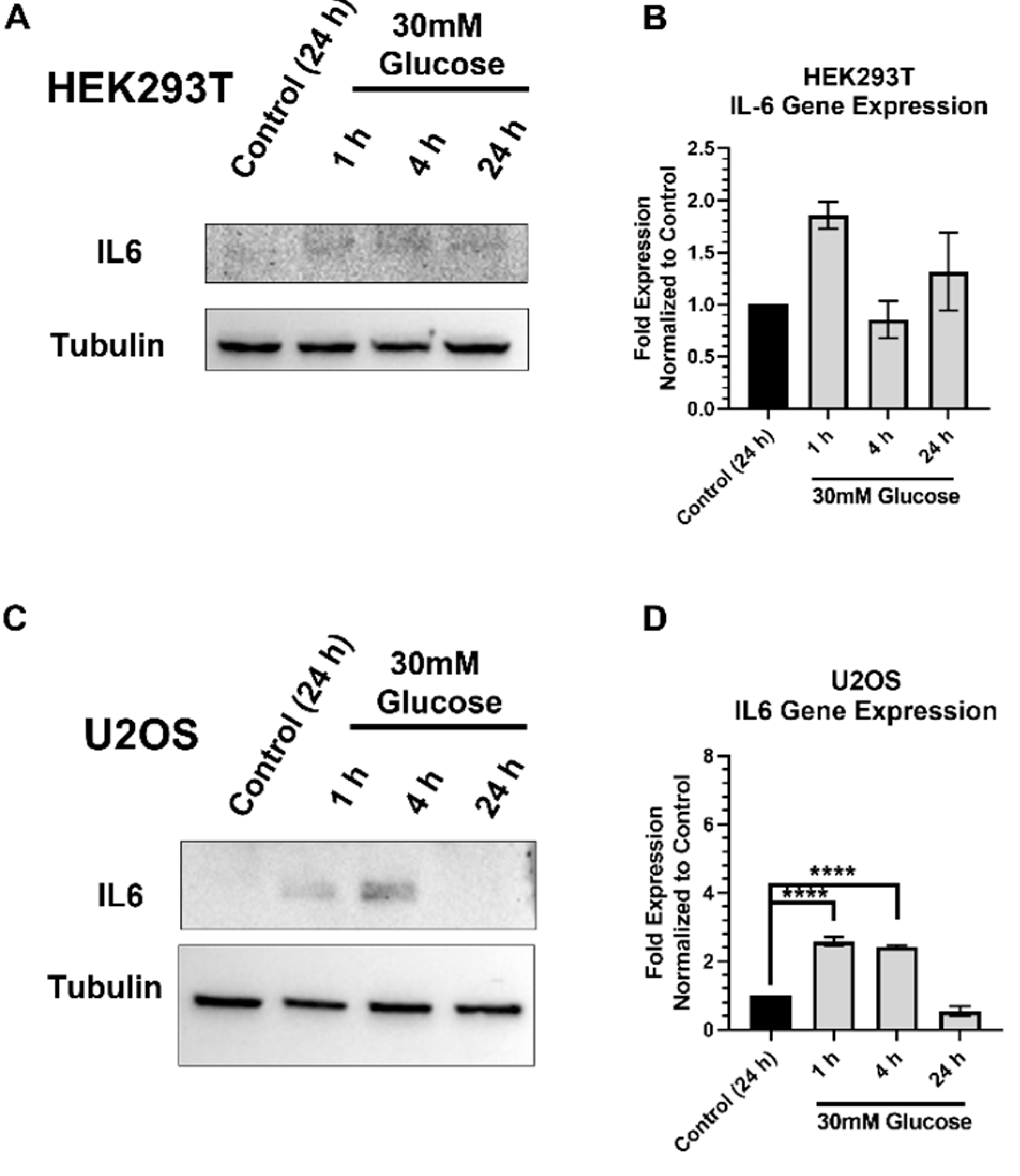
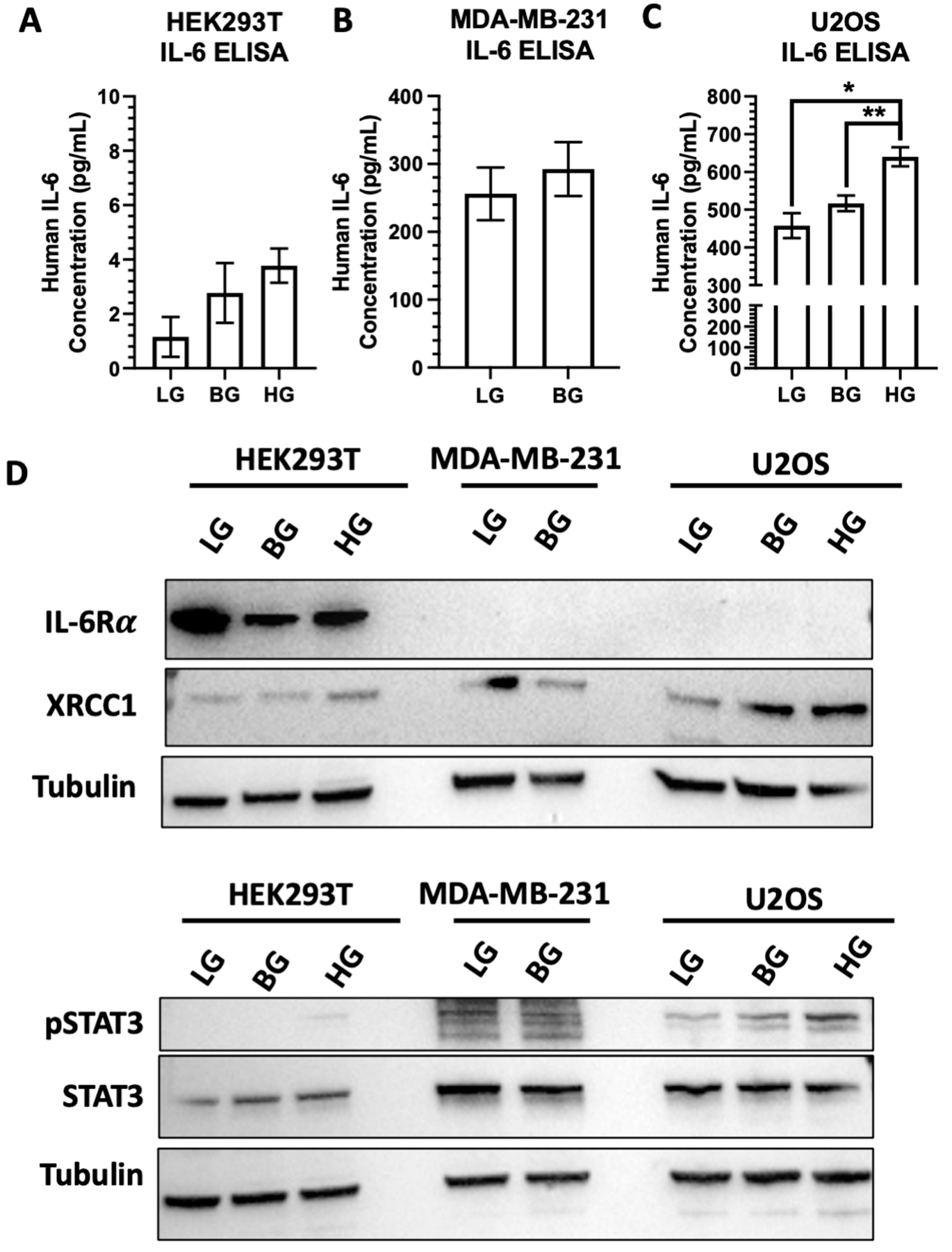
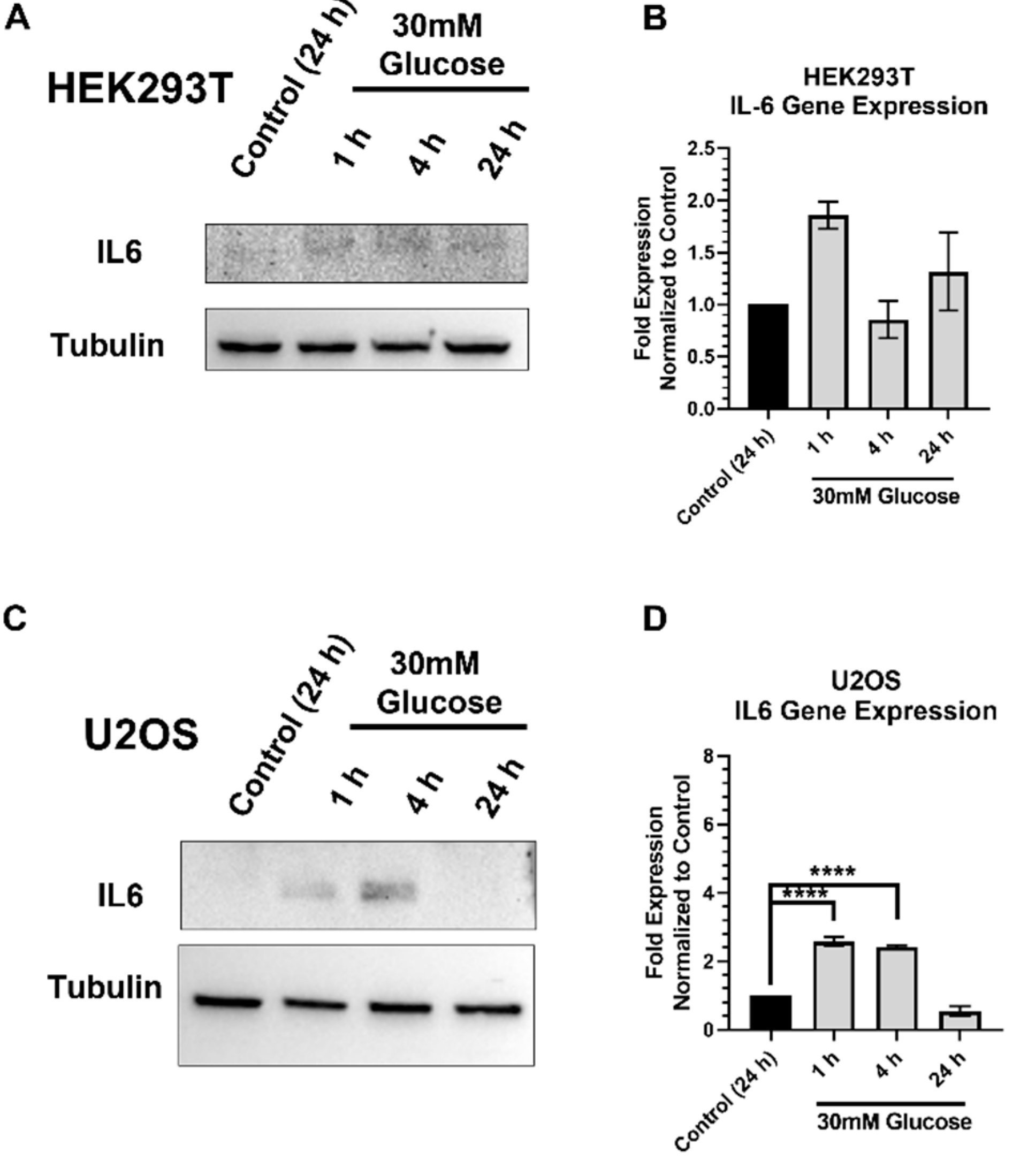
2.6. IL-6 Expression and Stimulation Increases pSTAT3 and XRCC1 in HEK293T and U2OS
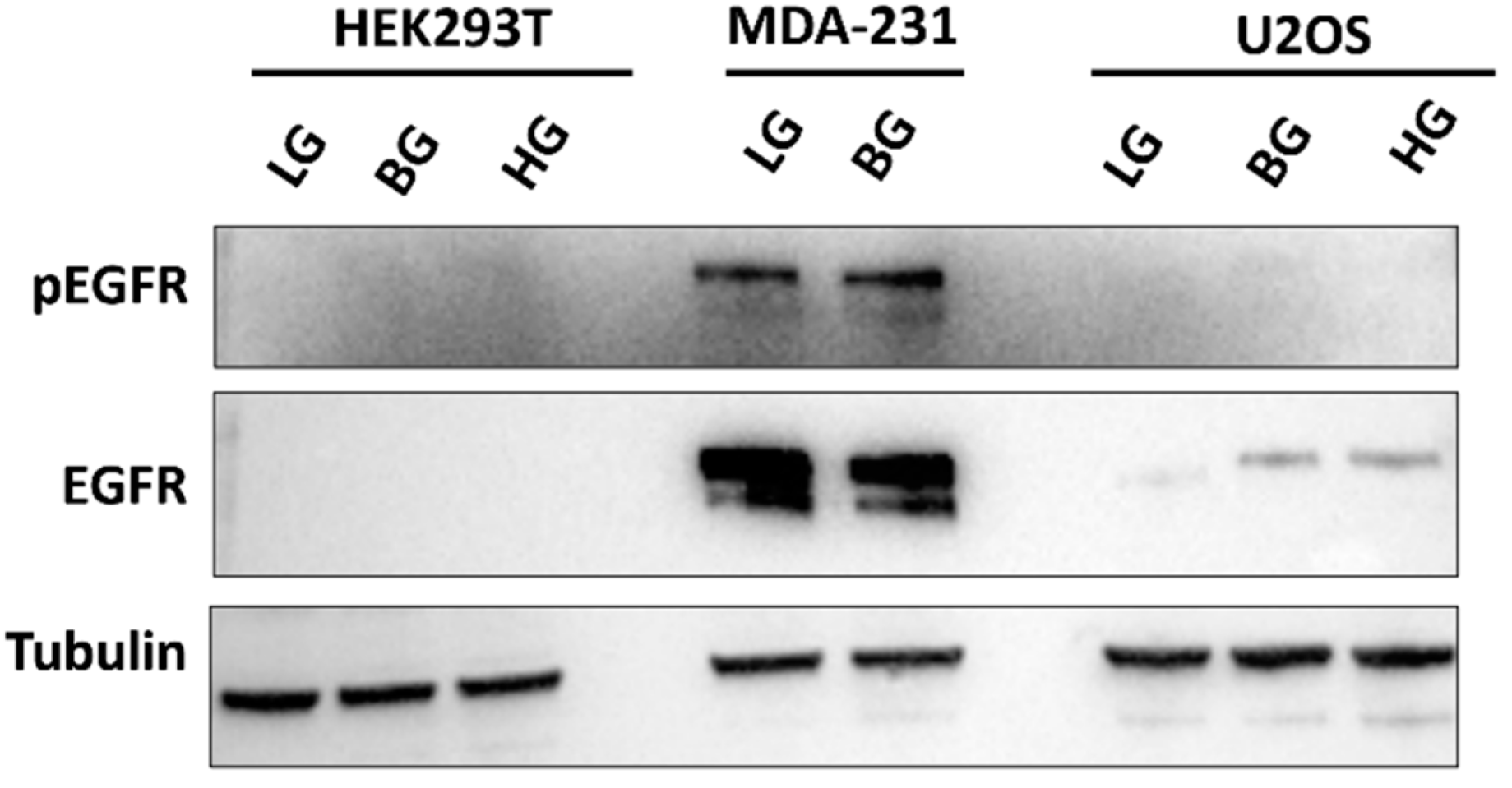
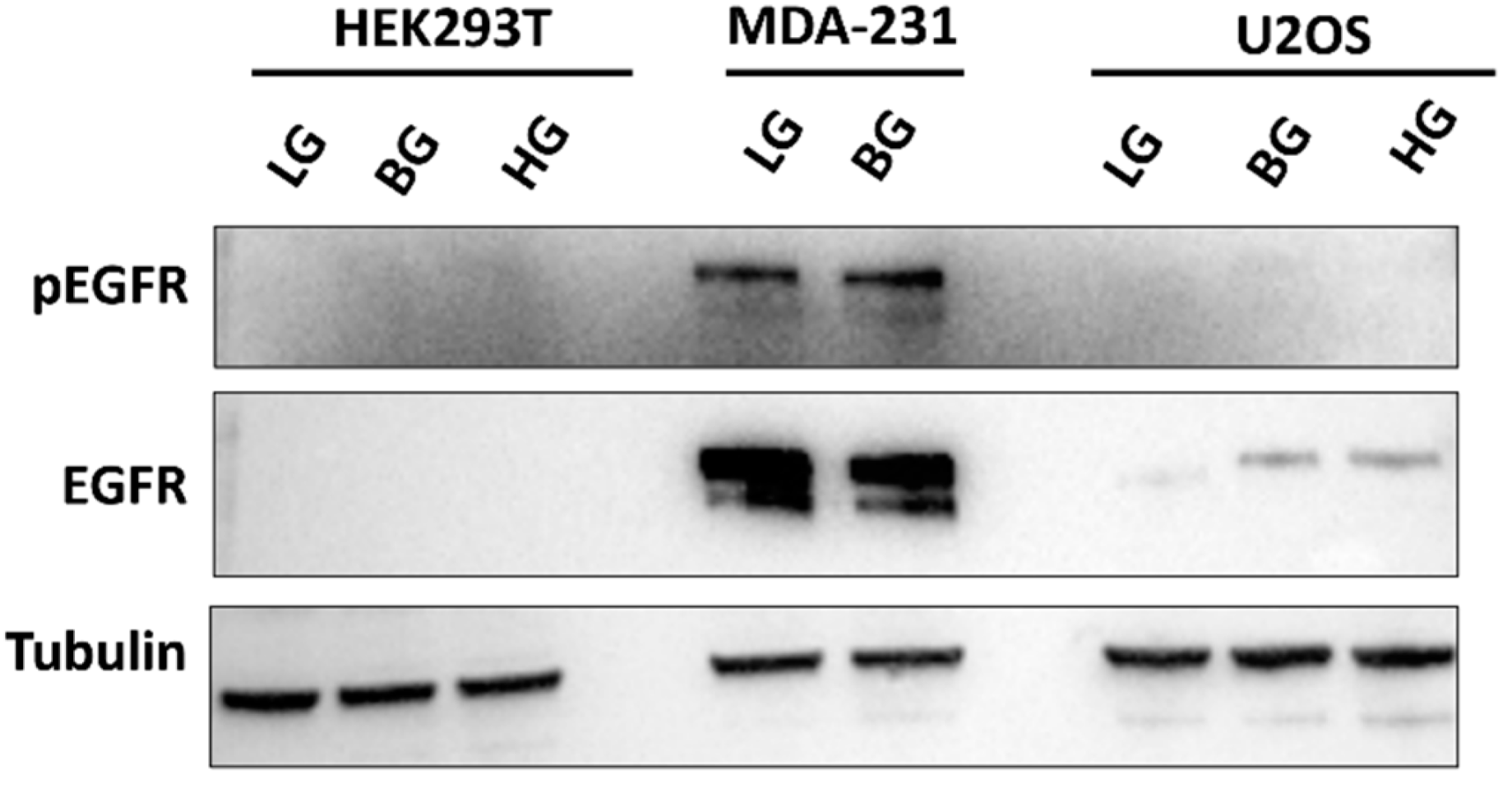
2.7. High EGFR Expression in MDA-MB-231 Mitigates Glucose-Driven Changes in pSTAT3
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Immunoblot
4.3. Gene Expression Analysis
4.4. Chromatin Immunoprecipitation
4.5. Cell Growth Inhibition
4.6. Immunofluorescence
4.7. Single Cell Gel Electrophoresis (Comet Assay)
4.8. Cell Adaptation
4.9. IL-6 ELISA Quantification
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, K.J.; Mann, E.; Wright, G.; Piett, C.G.; Nagel, Z.D.; Gassman, N.R. Exploiting DNA repair defects in triple negative breast cancer to improve cell killing. Ther. Adv. Med. Oncol. 2020, 12, 1758835920958354. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.J.; Piett, C.G.; Andrews, J.F.; Mann, E.; Nagel, Z.D.; Gassman, N.R. Defective base excision repair in the response to DNA damaging agents in triple negative breast cancer. PLoS ONE 2019, 14, e0223725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, G.; Sonavane, M.; Gassman, N.R. Activated STAT3 Is a Novel Regulator of the XRCC1 Promoter and Selectively Increases XRCC1 Protein Levels in Triple Negative Breast Cancer. Int. J. Mol. Sci. 2021, 22, 5475. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.S. Base excision repair: A critical player in many games. DNA Repair 2014, 19, 14–26. [Google Scholar] [CrossRef] [Green Version]
- London, R.E. The structural basis of XRCC1-mediated DNA repair. DNA Repair 2015, 30, 90–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audebert, M.; Salles, B.; Calsou, P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J. Biol. Chem. 2004, 279, 55117–55126. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Chen, Q.; Wang, Q.; Sun, Y.; Wang, S.; Li, A.; Xu, S.; Røe, O.D.; Wang, M.; Zhang, R.; et al. JWA reverses cisplatin resistance via the CK2-XRCC1 pathway in human gastric cancer cells. Cell Death Dis. 2014, 5, e1551. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Wang, S.; Chen, Q.; Zhang, Y.; Ni, P.; Wu, X.; Zhang, J.; Qiang, F.; Li, A.; Røe, O.D.; et al. TXNL1-XRCC1 pathway regulates cisplatin-induced cell death and contributes to resistance in human gastric cancer. Cell Death Dis. 2014, 5, e1055. [Google Scholar] [CrossRef]
- Abdel-Fatah, T.; Sultana, R.; Abbotts, R.; Hawkes, C.; Seedhouse, C.; Chan, S.; Madhusudan, S. Clinicopathological and functional significance of XRCC1 expression in ovarian cancer. Int. J. Cancer 2013, 132, 2778–2786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, J.; Xi, C.; Dai, Y.; Gong, H.; Zhang, T.M. Altered expression of base excision repair genes in response to high glucose-induced oxidative stress in HepG2 hepatocytes. Med. Sci. Monit. 2012, 18, Br281. [Google Scholar] [CrossRef] [Green Version]
- Sultana, R.; Abdel-Fatah, T.; Abbotts, R.; Hawkes, C.; Albarakati, N.; Seedhouse, C.; Ball, G.; Chan, S.; Rakha, E.A.; Ellis, I.O.; et al. Targeting XRCC1 deficiency in breast cancer for personalized therapy. Cancer Res. 2013, 73, 1621–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Miao, X.; Zhang, Y.; Li, D.; Zou, Q.; Yuan, Y.; Liu, R.; Yang, Z. XRCC1 Is a Promising Predictive Biomarker and Facilitates Chemo-Resistance in Gallbladder Cancer. Front. Mol. Biosci. 2020, 7, 70. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S.C.; Grou, C.P.; Legrand, A.J.; Chen, X.; Soderstrom, K.; Poletto, M.; Dianov, G.L. Sp1 phosphorylation by ATM downregulates BER and promotes cell elimination in response to persistent DNA damage. Nucleic Acids Res. 2018, 46, 1834–1846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Yu, Z.; Zhu, Z.; Lopez, C.D. E2F1 regulates the base excision repair gene XRCC1 and promotes DNA repair. J. Biol. Chem. 2008, 283, 15381–15389. [Google Scholar] [CrossRef] [Green Version]
- Mármol, I.; Sánchez-de-Diego, C.; Pradilla Dieste, A.; Cerrada, E.; Rodriguez Yoldi, M.J. Colorectal Carcinoma: A General Overview and Future Perspectives in Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 197. [Google Scholar] [CrossRef] [Green Version]
- Kang, C.; LeRoith, D.; Gallagher, E.J. Diabetes, Obesity, and Breast Cancer. Endocrinology 2018, 159, 3801–3812. [Google Scholar] [CrossRef] [Green Version]
- Kolb, R.; Sutterwala, F.S.; Zhang, W. Obesity and cancer: Inflammation bridges the two. Curr. Opin. Pharmacol. 2016, 29, 77–89. [Google Scholar] [CrossRef] [Green Version]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramteke, P.; Deb, A.; Shepal, V.; Bhat, M.K. Hyperglycemia Associated Metabolic and Molecular Alterations in Cancer Risk, Progression, Treatment, and Mortality. Cancers 2019, 11, 1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, J.; Zheng, Q.; Meng, X. Hyperglycemia and Chemoresistance in Breast Cancer: From Cellular Mechanisms to Treatment Response. Front. Oncol. 2021, 11, 628359. [Google Scholar] [CrossRef]
- Zhong, A.; Chang, M.; Yu, T.; Gau, R.; Riley, D.J.; Chen, Y.; Chen, P.L. Aberrant DNA damage response and DNA repair pathway in high glucose conditions. J. Can. Res. Updates 2018, 7, 64–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, C.; Li, X.; Wang, Y.; Zhang, Y.; Cai, M.; Zhu, B.; Mu, P.; Xia, X.; Zhao, Y.; Weng, J.; et al. Diabetes causes multiple genetic alterations and downregulates expression of DNA repair genes in the prostate. Lab. Invest. 2011, 91, 1363–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manoel-Caetano, F.S.; Xavier, D.J.; Evangelista, A.F.; Takahashi, P.; Collares, C.V.; Puthier, D.; Foss-Freitas, M.C.; Foss, M.C.; Donadi, E.A.; Passos, G.A.; et al. Gene expression profiles displayed by peripheral blood mononuclear cells from patients with type 2 diabetes mellitus focusing on biological processes implicated on the pathogenesis of the disease. Gene 2012, 511, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Bailey, S.K.; Metge, B.J.; Hanna, A.; Hinshaw, D.C.; Mota, M.; Forero-Torres, A.; Chatham, J.C.; Samant, R.S.; Shevde, L.A. O-GlcNAcylation of GLI transcription factors in hyperglycemic conditions augments Hedgehog activity. Lab. Investig. 2019, 99, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, K.; Resat, H. Constitutive activation of STAT3 in breast cancer cells: A review. Int. J. Cancer 2016, 138, 2570–2578. [Google Scholar] [CrossRef]
- Wegenka, U.M.; Buschmann, J.; Lütticken, C.; Heinrich, P.C.; Horn, F. Acute-phase response factor, a nuclear factor binding to acute-phase response elements, is rapidly activated by interleukin-6 at the posttranslational level. Mol. Cell Biol. 1993, 13, 276–288. [Google Scholar] [CrossRef]
- Saengboonmee, C.; Seubwai, W.; Pairojkul, C.; Wongkham, S. High glucose enhances progression of cholangiocarcinoma cells via STAT3 activation. Sci. Rep. 2016, 6, 18995. [Google Scholar] [CrossRef]
- Du, W.; Wang, N.; Li, F.; Jia, K.; An, J.; Liu, Y.; Wang, Y.; Zhu, L.; Zhao, S.; Hao, J. STAT3 phosphorylation mediates high glucose-impaired cell autophagy in an HDAC1-dependent and -independent manner in Schwann cells of diabetic peripheral neuropathy. Faseb. J. 2019, 33, 8008–8021. [Google Scholar] [CrossRef]
- Ishihara, M. Acute hyperglycemia in patients with acute myocardial infarction. Circ. J. 2012, 76, 563–571. [Google Scholar] [CrossRef] [Green Version]
- Esposito, K.; Nappo, F.; Marfella, R.; Giugliano, G.; Giugliano, F.; Ciotola, M.; Quagliaro, L.; Ceriello, A.; Giugliano, D. Inflammatory Cytokine Concentrations Are Acutely Increased by Hyperglycemia in Humans. Circulation 2002, 106, 2067–2072. [Google Scholar] [CrossRef] [Green Version]
- Panahi, G.; Pasalar, P.; Zare, M.; Rizzuto, R.; Meshkani, R. High glucose induces inflammatory responses in HepG2 cells via the oxidative stress-mediated activation of NF-κB, and MAPK pathways in HepG2 cells. Arch. Physiol. Biochem. 2018, 124, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Saengboonmee, C.; Phoomak, C.; Supabphol, S.; Covington, K.R.; Hampton, O.; Wongkham, C.; Gibbs, R.A.; Umezawa, K.; Seubwai, W.; Gingras, M.C.; et al. NF-κB and STAT3 co-operation enhances high glucose induced aggressiveness of cholangiocarcinoma cells. Life Sci. 2020, 262, 118548. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Wen, Z.; Darnell, J.E., Jr. Stat3: A STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994, 264, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.; Houlbrook, S.; Zhang, Q.M.; Harrison, M.; Hickson, I.D.; Dianov, G.L. Overexpression of DNA polymerase beta results in an increased rate of frameshift mutations during base excision repair. Mutagenesis 2007, 22, 183–188. [Google Scholar] [CrossRef] [Green Version]
- Frosina, G. Overexpression of enzymes that repair endogenous damage to DNA. Eur. J. Biochem. 2000, 267, 2135–2149. [Google Scholar] [CrossRef]
- Yoshizawa, K.; Jelezcova, E.; Brown, A.R.; Foley, J.F.; Nyska, A.; Cui, X.; Hofseth, L.J.; Maronpot, R.M.; Wilson, S.H.; Sepulveda, A.R.; et al. Gastrointestinal hyperplasia with altered expression of DNA polymerase beta. PLoS ONE 2009, 4, e6493. [Google Scholar] [CrossRef] [Green Version]
- Sak, S.C.; Harnden, P.; Johnston, C.F.; Paul, A.B.; Kiltie, A.E. APE1 and XRCC1 protein expression levels predict cancer-specific survival following radical radiotherapy in bladder cancer. Clin. Cancer Res. 2005, 11, 6205–6211. [Google Scholar] [CrossRef] [Green Version]
- De Summa, S.; Pinto, R.; Pilato, B.; Sambiasi, D.; Porcelli, L.; Guida, G.; Mattioli, E.; Paradiso, A.; Merla, G.; Micale, L.; et al. Expression of base excision repair key factors and miR17 in familial and sporadic breast cancer. Cell Death Dis. 2014, 5, e1076. [Google Scholar] [CrossRef] [Green Version]
- Horton, J.K.; Watson, M.; Stefanick, D.F.; Shaughnessy, D.T.; Taylor, J.A.; Wilson, S.H. XRCC1 and DNA polymerase beta in cellular protection against cytotoxic DNA single-strand breaks. Cell Res. 2008, 18, 48–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchins, A.P.; Diez, D.; Takahashi, Y.; Ahmad, S.; Jauch, R.; Tremblay, M.L.; Miranda-Saavedra, D. Distinct transcriptional regulatory modules underlie STAT3′s cell type-independent and cell type-specific functions. Nucleic Acids Res. 2013, 41, 2155–2170. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wright, G.M.; Gassman, N.R. Glucose Increases STAT3 Activation, Promoting Sustained XRCC1 Expression and Increasing DNA Repair. Int. J. Mol. Sci. 2022, 23, 4314. https://doi.org/10.3390/ijms23084314
Wright GM, Gassman NR. Glucose Increases STAT3 Activation, Promoting Sustained XRCC1 Expression and Increasing DNA Repair. International Journal of Molecular Sciences. 2022; 23(8):4314. https://doi.org/10.3390/ijms23084314
Chicago/Turabian StyleWright, Griffin M., and Natalie R. Gassman. 2022. "Glucose Increases STAT3 Activation, Promoting Sustained XRCC1 Expression and Increasing DNA Repair" International Journal of Molecular Sciences 23, no. 8: 4314. https://doi.org/10.3390/ijms23084314
APA StyleWright, G. M., & Gassman, N. R. (2022). Glucose Increases STAT3 Activation, Promoting Sustained XRCC1 Expression and Increasing DNA Repair. International Journal of Molecular Sciences, 23(8), 4314. https://doi.org/10.3390/ijms23084314