
QSAR, Docking, and Molecular Dynamics Simulation Studies of Sigmacidins as Antimicrobials against Streptococci


Abstract
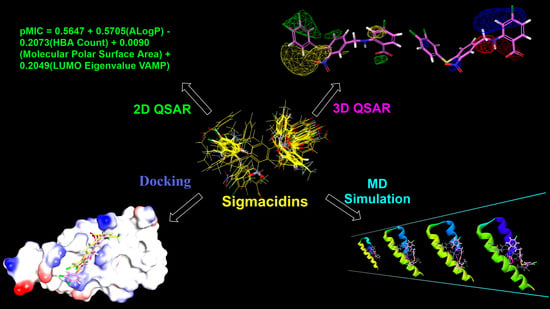
:
1. Introduction
2. Results and Discussion
2.1. Two-Dimensional QSAR
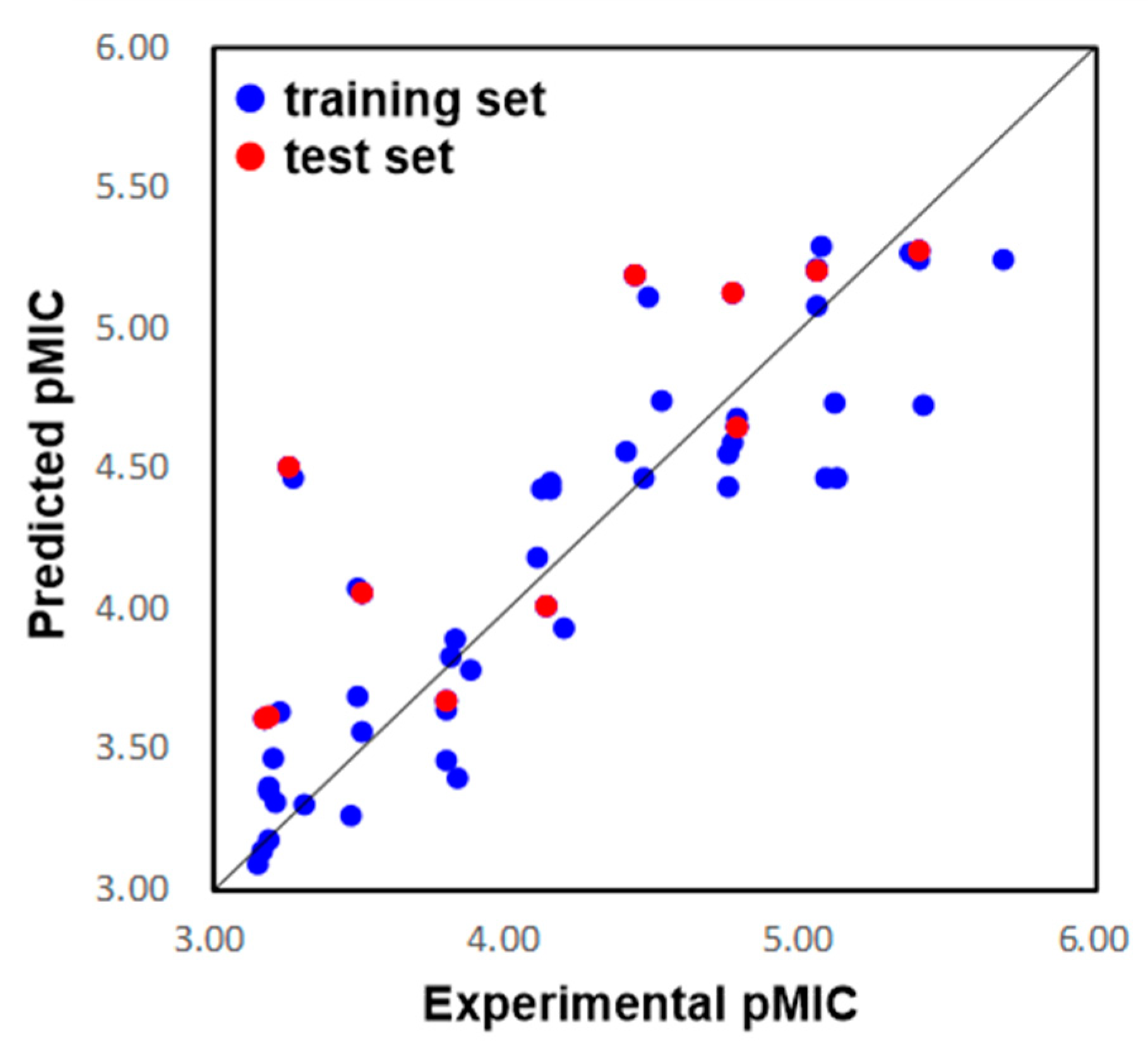
2.1.1. Two-Dimensional QSAR Study
2.1.2. Two-Dimensional QSAR Model Analysis
2.2. Three-Dimensional QSAR
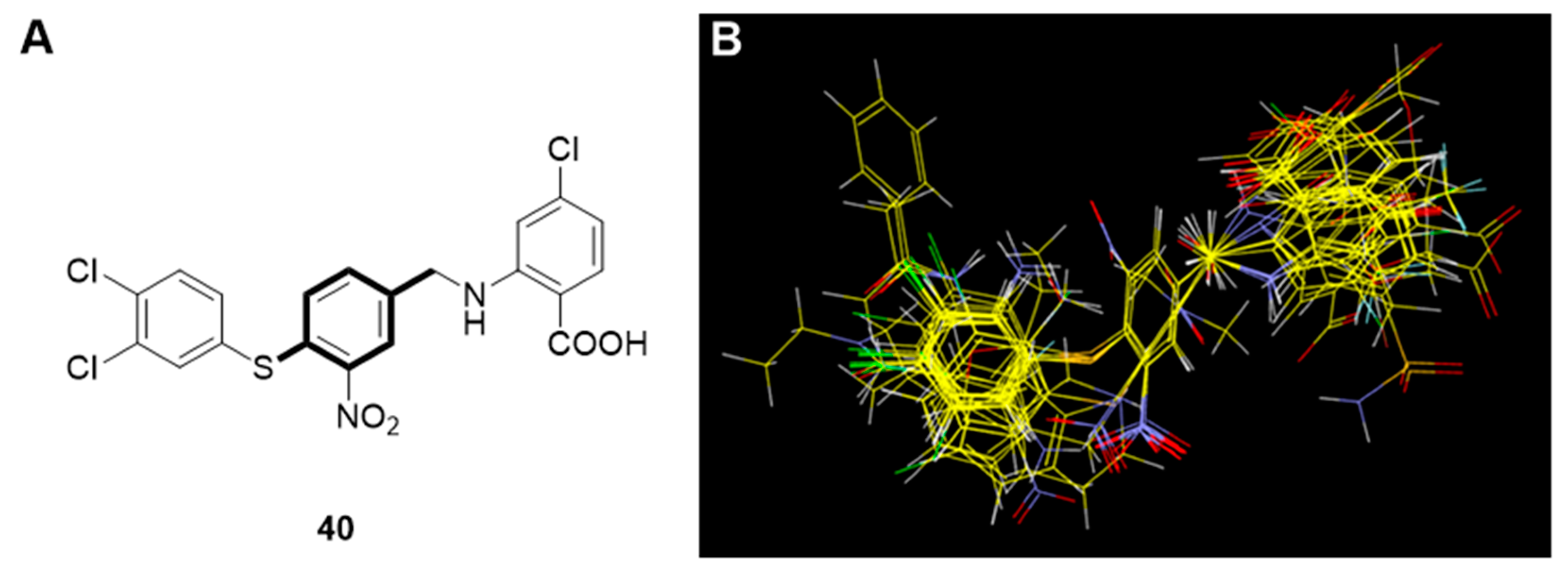
2.2.1. Molecular Alignment
2.2.2. Three-Dimensional QSAR Study
2.2.3. Interpretation of the 3D QSAR Contour Maps
2.3. Docking and MD Simulations Studies of Compound 1 and 40
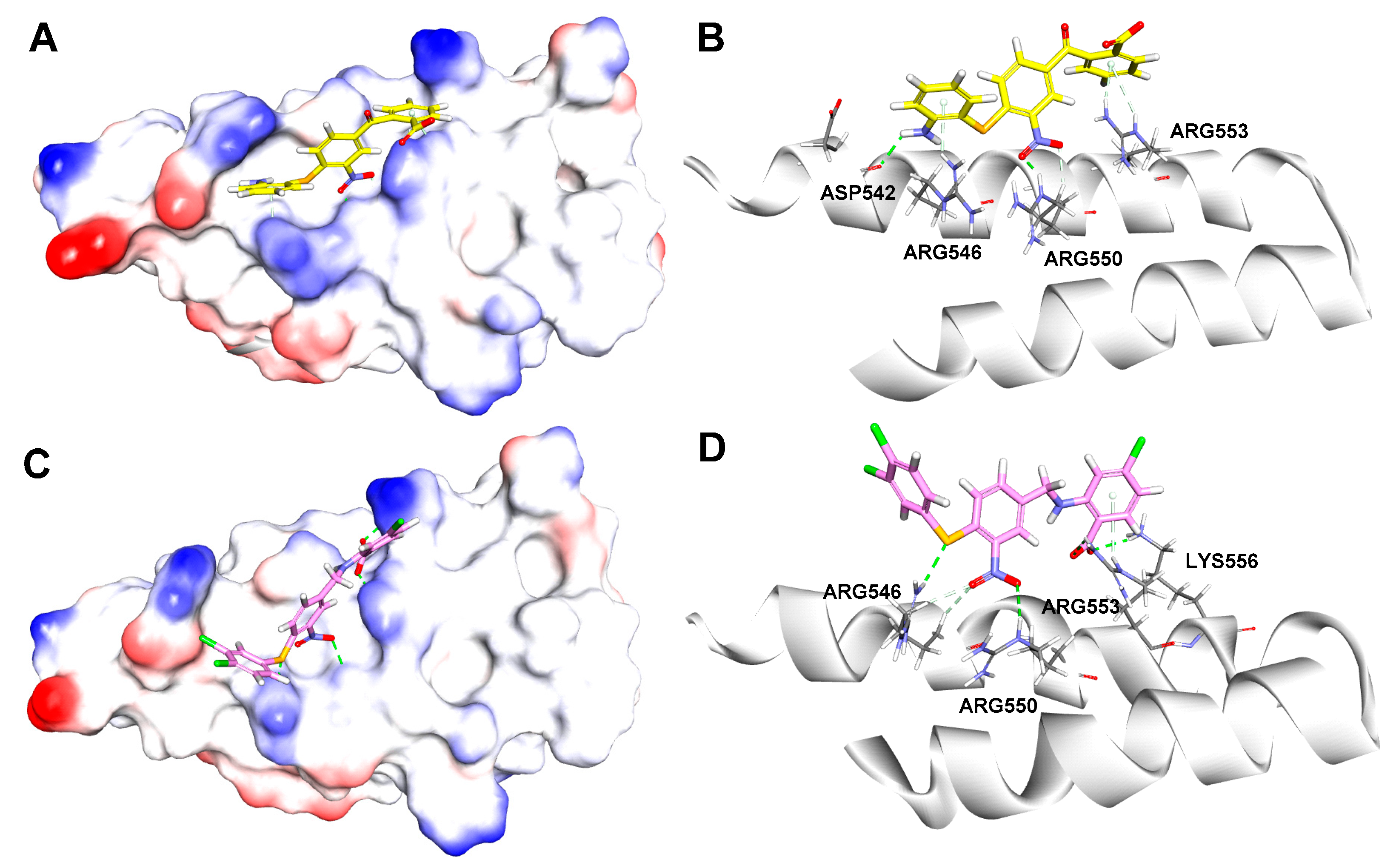
2.3.1. Docking Studies
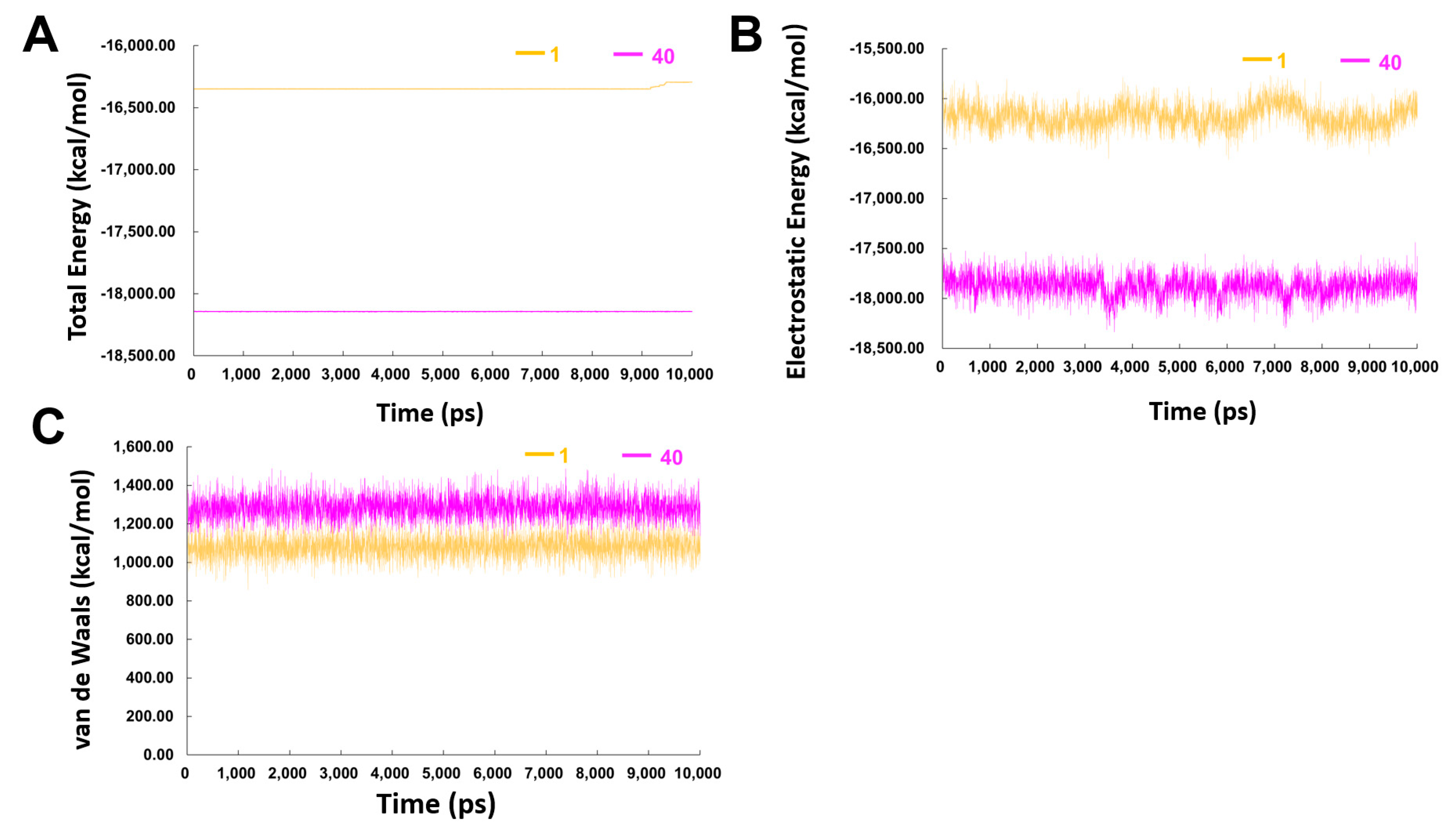
2.3.2. MD Simulation Studies
3. Materials and Methods
3.1. Dataset
3.2. Preparation of the Small Molecules
3.3. Two-Dimensional QSAR Model Construction
3.4. Three-Dimensional QSAR Model Construction
3.5. Docking and Molecular Dynamic Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leonard, A.; Gierok, P.; Methling, K.; Gómez-Mejia, A.; Hammerschmidt, S.; Lalk, M. Metabolic inventory of Streptococcus pneumoniae growing in a chemical defined environment. Int. J. Med. Microbiol. 2018, 308, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Avire, N.J.; Whiley, H.; Ross, K. A review of Streptococcus pyogenes: Public health risk factors, prevention and control. Pathogens 2021, 10, 248. [Google Scholar] [CrossRef] [PubMed]
- Raabe, V.N.; Shane, A.L. Group B Streptococcus (Streptococcus agalactiae). Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossitto, P.V.; Ruiz, L.; Kikuchi, Y.; Glenn, K.; Luiz, K.; Watts, J.L.; Cullor, J.S. Antibiotic susceptibility patterns for environmental streptococci isolated from bovine mastitis in central California dairies. J. Dairy Sci. 2002, 85, 132–138. [Google Scholar] [CrossRef]
- Heelan, J.S.; Hasenbein, M.E.; McAdam, A.J. Resistance of Group B Streptococcus to Selected Antibiotics, Including Erythromycin and Clindamycin. J. Clin. Microbiol. 2004, 42, 1263–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, M.; Saab, M.; Heider, L.; McClure, J.T.; Rodriguez-Lecompte, J.C.; Sanchez, J. Antimicrobial susceptibility patterns of environmental streptococci recovered from bovine milk samples in the Maritime Provinces of Canada. Front. Vet. Sci. 2016, 3, 79. [Google Scholar] [CrossRef] [Green Version]
- Facklam, R. What happened to the streptococci: Overview of taxonomic and nomenclature changes. Clin. Microbiol. Rev. 2002, 15, 613–630. [Google Scholar] [CrossRef] [Green Version]
- Appelbaum, P.C. Resistance among Streptococcus pneumoniae: Implications for drug selection. Clin. Infect. Dis. 2002, 34, 1613–1620. [Google Scholar] [CrossRef] [Green Version]
- WHO. Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics. 2017. Available online: https://www.who.int/medicines/publications/WHO-PPL-Short_Summary_25Feb-ET_NM_WHO.pdf (accessed on 28 February 2022).
- Kahan, R.; Worm, D.J.; De Castro, G.V.; Ng, S.; Barnard, A. Modulators of protein-protein interactions as antimicrobial agents. RSC Chem. Biol. 2021, 2, 387–409. [Google Scholar] [CrossRef]
- Ma, C.; Yang, X.; Lewis, P.J. Bacterial transcription as a target for antibacterial drug development. Microbiol. Mol. Biol. Rev. 2016, 80, 139–160. [Google Scholar] [CrossRef] [Green Version]
- Keller, A.N.; Yang, X.; Wiedermannová, J.; Delumeau, O.; Krásný, L.; Lewis, P.J. ε, a new subunit of RNA polymerase found in gram-positive bacteria. J. Bacteriol. 2014, 196, 3622–3632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Lewis, P.J. Overproduction and purification of recombinant Bacillus subtilis RNA polymerase. Protein Expr. Purif. 2008, 59, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Tsang, T.F.; Qiu, Y.; Lin, L.; Ye, J.; Ma, C.; Yang, X. Simple method for studying in vitro protein-protein interactions based on protein complementation and its application in drug screening targeting bacterial transcription. ACS Infect. Dis. 2019, 5, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Burgess, R.R.; Anthony, L. How sigma docks to RNA polymerase and what sigma does. Curr. Opin. Microbiol. 2001, 4, 126–131. [Google Scholar] [CrossRef]
- Murakami, K.S.; Darst, S.A. Bacterial RNA polymerases: The wholo story. Curr. Opin. Struct. Biol. 2003, 13, 31–39. [Google Scholar] [CrossRef]
- Vassylyev, D.G.; Sekine, S.I.; Yokoyama, S.; Yokoyama, S. Crystal structure of a bacterial RNA polymerase holoenzyme at 2.6. Å resolution. Nature 2002, 417, 712–719. [Google Scholar] [CrossRef]
- Ma, C.; Yang, X.; Lewis, P.J. Bacterial Transcription Inhibitor of RNA Polymerase Holoenzyme Formation by Structure-Based Drug Design: From In Silico Screening to Validation. ACS Infect. Dis. 2016, 2, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Chu, A.J.; Lin, L.; Yang, X.; Ma, C. First-in-class inhibitors targeting the interaction between bacterial RNA polymerase and sigma initiation factor affect the viability and toxin release of Streptococcus pneumoniae. Molecules 2019, 24, 2902. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Chu, A.J.; Harper, R.; Chan, S.T.; Shek, T.L.; Zhang, Y.; Ip, M.; Sambir, M.; Artsimovitch, I.; Zuo, Z.; et al. Discovery of Antibacterials That Inhibit Bacterial RNA Polymerase Interactions with Sigma Factors. J. Med. Chem. 2020, 63, 7695–7720. [Google Scholar] [CrossRef]
- Ye, J.; Chu, A.J.; Lin, L.; Chan, S.T.; Harper, R.; Xiao, M.; Artsimovitch, I.; Zuo, Z.; Ma, C.; Yang, X. Benzyl and benzoyl benzoic acid inhibitors of bacterial RNA polymerase-sigma factor interaction. Eur. J. Med. Chem. 2020, 208, 112671. [Google Scholar] [CrossRef]
- Lewis, R.A.; Wood, D. Modern 2D QSAR for drug discovery. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 505–522. [Google Scholar] [CrossRef]
- Lill, M.A. Multi-dimensional QSAR in drug discovery. Drug Discov. Today 2007, 12, 1013–1017. [Google Scholar] [CrossRef] [PubMed]
- Verma, J.; Khedkar, V.M.; Coutinho, E.C. 3D-QSAR in drug design—A review. Curr. Top. Med. Chem. 2010, 10, 95–115. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.; Sharma, P.; Prakash, O.; Singh, M.; Kalani, K.; Khan, F.; Bawankule, D.U.; Luqman, S.; Srivastava, S.K. QSAR and docking studies on capsazepine derivatives for immunomodulatory and anti-inflammatory activity. PLoS ONE 2014, 9, e100797. [Google Scholar] [CrossRef] [Green Version]
- Datar, P.A. 2D-QSAR study of indolylpyrimidines derivative as antibacterial against Pseudomonas aeruginosa and Staphylococcus aureus: A comparative approach. J. Comput. Med. 2014, 2014, 765457. [Google Scholar] [CrossRef] [Green Version]
- Caron, G.; Ermondi, G.; Scherrer, R.A. Lipophilicity, polarity, and hydrophobicity. In Comprehensive Medicinal Chemistry II; Elsevier: Amsterdam, The Netherlands, 2006; Volume 5, pp. 425–452. [Google Scholar]
- Graton, J.; Le Questel, J.Y.; Maxwell, P.; Popelier, P. Hydrogen-Bond Accepting Properties of New Heteroaromatic Ring Chemical Motifs: A Theoretical Study. J. Chem. Inf. Model. 2016, 56, 322–334. [Google Scholar] [CrossRef] [Green Version]
- Clark, D.E. What has polar surface area ever done for drug discovery? Future Med. Chem. 2011, 3, 469–484. [Google Scholar] [CrossRef]
- Lohray, B.B.; Gandhi, N.; Srivastava, B.K.; Lohray, V.B. 3D QSAR studies of N-4-arylacryloylpiperazin-1-yl-phenyl-oxazolidinones: A novel class of antibacterial agents. Bioorganic Med. Chem. Lett. 2006, 16, 3817–3823. [Google Scholar] [CrossRef]
- Shakil, S.; Danish Rizvi, S.M.; Greig, N.H. High throughput virtual screening and molecular dynamics simulation for identifying a putative inhibitor of bacterial CTX-M-15. Antibiotics 2021, 10, 474. [Google Scholar] [CrossRef]
- Raharjo, S.J.; Mahdi, C.; Nurdiana, N.; Kikuchi, T.; Fatchiyah, F. Binding energy calculation of patchouli alcohol isomer cyclooxygenase complexes suggested as COX-1/COX-2 selective inhibitor. Adv. Bioinform. 2014, 2014, 850628. [Google Scholar] [CrossRef]
- MacroModel, Version 10.2; Schrödinger, LLC: New York, NY, USA, 2013.
- Meduru, H.; Wang, Y.T.; Tsai, J.J.P.; Chen, Y.C. Finding a potential dipeptidyl peptidase-4 (DPP-4) inhibitor for type-2 diabetes treatment based on molecular docking, pharmacophore generation, and molecular dynamics simulation. Int. J. Mol. Sci. 2016, 17, 920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Ma, C.; Lewis, P. A vector system that allows simple generation of mutant Escherichia coli RNA polymerase. Plasmid 2014, 75, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Ma, C.; Lewis, P.J. Identification of inhibitors of bacterial RNA polymerase. Methods 2015, 86, 45–50. [Google Scholar] [CrossRef] [PubMed]
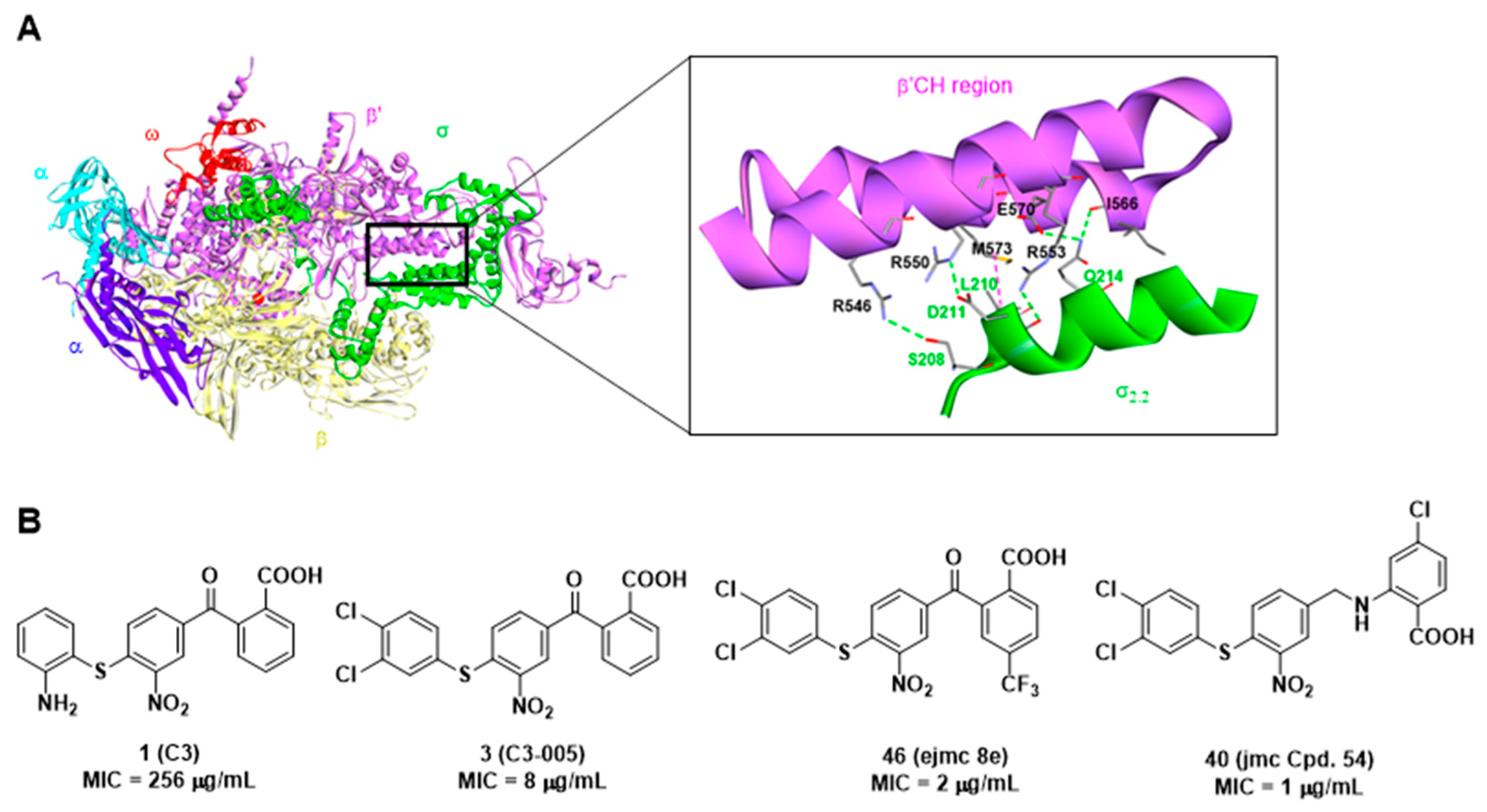





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd. | MIC (μg/mL) | MW | MIC (M) | pMIC | 2D QSAR | 3D QSAR | ||
|---|---|---|---|---|---|---|---|---|
| Predicted pMIC | Δ b | Predicted pMIC | Δ b | |||||
| 1 a | 256 | 394.40 | 6.49 × 10−4 | 3.188 | 3.503 | −0.315 | 3.616 | −0.428 |
| 2 a | 128 | 409.41 | 3.13 × 10−4 | 3.505 | 3.579 | −0.074 | 4.060 | −0.555 |
| 3 | 8 | 448.28 | 1.78 × 10−5 | 4.748 | 4.406 | 0.342 | 4.552 | 0.196 |
| 4 | 256 | 362.34 | 7.07 × 10−4 | 3.151 | 3.295 | −0.144 | 3.089 | 0.062 |
| 5 | 256 | 354.36 | 7.22 × 10−4 | 3.141 | 2.899 | 0.242 | 2.994 | 0.148 |
| 6 | 128 | 409.42 | 3.13 × 10−4 | 3.505 | 3.119 | 0.386 | 3.564 | −0.059 |
| 7 a | 64 | 395.43 | 1.62 × 10−4 | 3.791 | 3.529 | 0.262 | 3.674 | 0.117 |
| 8 | 64 | 394.40 | 1.62 × 10−4 | 3.790 | 3.508 | 0.282 | 3.642 | 0.148 |
| 9 | 128 | 394.40 | 3.25 × 10−4 | 3.489 | 3.516 | −0.027 | 3.692 | −0.203 |
| 10 | 256 | 523.52 | 4.89 × 10−4 | 3.311 | 3.942 | −0.631 | 3.303 | 0.008 |
| 11 | 256 | 376.36 | 6.80 × 10−4 | 3.167 | 3.224 | −0.057 | 3.140 | 0.027 |
| 12 | 256 | 394.35 | 6.49 × 10−4 | 3.188 | 3.328 | −0.14 | 3.181 | 0.007 |
| 13 | 128 | 376.36 | 3.40 × 10−4 | 3.468 | 3.575 | −0.107 | 3.261 | 0.207 |
| 14 | 8 | 449.31 | 1.78 × 10−5 | 4.749 | 4.541 | 0.208 | 4.437 | 0.313 |
| 15 a | 256 | 463.29 | 5.53 × 10−4 | 3.258 | 3.996 | −0.738 | 4.507 | −1.249 |
| 16 | 256 | 418.44 | 6.12 × 10−4 | 3.213 | 4.095 | −0.882 | 3.311 | −0.098 |
| 17 | 128 | 397.38 | 3.22 × 10−4 | 3.492 | 3.802 | −0.31 | 4.072 | −0.580 |
| 18 a | 32 | 436.48 | 7.33 × 10−5 | 4.135 | 4.154 | −0.019 | 4.012 | 0.123 |
| 19 | 256 | 409.42 | 6.25 × 10−4 | 3.204 | 3.069 | 0.135 | 3.467 | −0.263 |
| 20 | 64 | 409.42 | 1.56 × 10−4 | 3.806 | 3.229 | 0.577 | 3.827 | −0.021 |
| 21 | 32 | 407.44 | 7.85 × 10−5 | 4.105 | 4.247 | −0.142 | 4.181 | −0.076 |
| 22 a | 256 | 383.38 | 6.68 × 10−4 | 3.175 | 3.173 | 0.002 | 3.612 | −0.437 |
| 23 | 32 | 448.28 | 7.14 × 10−5 | 4.146 | 4.418 | −0.272 | 4.426 | −0.280 |
| 24 | 32 | 498.51 | 6.42 × 10−5 | 4.193 | 4.182 | 0.011 | 3.936 | 0.256 |
| 25 | 64 | 428.85 | 1.49 × 10−4 | 3.826 | 3.860 | −0.034 | 3.894 | −0.068 |
| 26 | 64 | 431.23 | 1.48 × 10−4 | 3.829 | 4.013 | −0.184 | 3.401 | 0.427 |
| 27 | 64 | 395.43 | 1.62 × 10−4 | 3.791 | 3.660 | 0.131 | 3.462 | 0.329 |
| 28 | 256 | 395.43 | 6.47 × 10−4 | 3.189 | 3.581 | −0.392 | 3.366 | −0.177 |
| 29 | 256 | 436.44 | 5.87 × 10−4 | 3.232 | 3.228 | 0.004 | 3.632 | −0.400 |
| 30 | 4 | 483.75 | 8.27 × 10−6 | 5.083 | 4.915 | 0.168 | 4.469 | 0.614 |
| 31 | 16 | 479.33 | 3.34 × 10−5 | 4.477 | 4.297 | 0.180 | 5.112 | −0.635 |
| 32 | 16 | 534.41 | 2.99 × 10−5 | 4.524 | 4.359 | 0.165 | 4.744 | −0.221 |
| 33 | 64 | 484.38 | 1.32 × 10−4 | 3.879 | 4.246 | −0.367 | 3.787 | 0.092 |
| 34 | 16 | 463.33 | 3.45 × 10−5 | 4.462 | 4.776 | −0.314 | 4.466 | −0.004 |
| 35 | 256 | 396.78 | 6.45 × 10−4 | 3.190 | 3.656 | −0.466 | 3.353 | −0.163 |
| 36 | 4 | 528.20 | 7.57 × 10−6 | 5.121 | 4.962 | 0.159 | 4.467 | 0.654 |
| 37 | 256 | 483.75 | 5.29 × 10−4 | 3.276 | 4.912 | −1.636 | 4.470 | −1.194 |
| 38 a | 4 | 449.31 | 8.90 × 10−6 | 5.050 | 4.500 | 0.550 | 5.204 | −0.154 |
| 39 | 16 | 403.28 | 3.97 × 10−5 | 4.401 | 4.082 | 0.319 | 4.561 | −0.160 |
| 40 | 1 | 483.75 | 2.07 × 10−6 | 5.685 | 4.868 | 0.817 | 5.246 | 0.439 |
| 41 a | 16 | 434.29 | 3.68 × 10−5 | 4.434 | 4.798 | −0.364 | 5.189 | −0.755 |
| 42 | 32 | 418.29 | 7.65 × 10−5 | 4.116 | 3.891 | 0.225 | 4.428 | −0.311 |
| 43 | 32 | 446.35 | 7.17 × 10−5 | 4.145 | 4.213 | −0.068 | 4.449 | −0.304 |
| 44 a | 8 | 482.72 | 1.66 × 10−5 | 4.781 | 4.773 | 0.008 | 4.648 | 0.133 |
| 45 | 8 | 482.72 | 1.66 × 10−5 | 4.781 | 4.766 | 0.015 | 4.653 | 0.128 |
| 46 | 2 | 516.27 | 3.87 × 10−6 | 5.412 | 4.925 | 0.487 | 4.725 | 0.687 |
| 47 | 4 | 468.74 | 8.53 × 10−6 | 5.069 | 5.159 | −0.090 | 5.289 | −0.221 |
| 48 | 4 | 448.32 | 8.92 × 10−6 | 5.050 | 5.073 | −0.023 | 5.081 | −0.031 |
| 49 | 8 | 478.30 | 1.67 × 10−5 | 4.777 | 4.278 | 0.499 | 4.681 | 0.096 |
| 50 | 8 | 466.27 | 1.72 × 10−5 | 4.766 | 4.512 | 0.254 | 4.592 | 0.174 |
| 51 | 4 | 516.27 | 7.75 × 10−6 | 5.111 | 4.914 | 0.197 | 4.737 | 0.374 |
| 52 a | 2 | 502.29 | 3.98 × 10−6 | 5.400 | 5.271 | 0.129 | 5.273 | 0.126 |
| 53 a | 8 | 464.32 | 1.72 × 10−5 | 4.764 | 4.611 | 0.153 | 5.126 | −0.362 |
| 54 | 2 | 468.74 | 4.27 × 10−6 | 5.370 | 5.169 | 0.201 | 5.271 | 0.099 |
| 55 | 4 | 452.28 | 8.84 × 10−6 | 5.053 | 4.908 | 0.145 | 5.218 | −0.164 |
| 56 | 2 | 502.29 | 3.98 × 10−6 | 5.400 | 5.251 | 0.149 | 5.243 | 0.157 |
| Property | AlogP | HBA Count | LUMO Eigenvalue VAMP | Molecular Polar Surface Area |
|---|---|---|---|---|
| AlogP | 1.000 | −0.367 | −0.264 | −0.490 |
| HBA Count | 1.000 | −0.114 | 0.405 | |
| LUMO Eigenvalue VAMP | 1.000 | −0.350 | ||
| Molecular Polar Surface Area | 1.000 |
| R | r2 | r2 (Adjusted) | r2 (Prediction) | RMS Residual Error | q2 (Cross-Validation) | RMS Residual Error (Cross-Validation) |
|---|---|---|---|---|---|---|
| 0.856 | 0.732 | 0.705 | 0.613 | 0.399 | 0.562 | 0.526 |
| R | r2 | r2 (Adjusted) | r2 (Prediction) | RMS Residual Error | q2 (Cross-Validation) | RMS Residual Error (Cross-Validation) |
|---|---|---|---|---|---|---|
| 0.882 | 0.777 | 0.753 | 0.721 | 0.375 | 0.690 | 0.447 |
| SD | r2 | r2 CV | r2 Scramble | Stability | F | P | RMSE | Q2 | Pearson-r |
|---|---|---|---|---|---|---|---|---|---|
| 0.356 | 0.805 | 0.568 | 0.482 | 0.883 | 56.4 | 1.31×10−14 | 0.52 | 0.528 | 0.835 |
| Steric | Electrostatic | Hydrophobic | H-Bond Acceptor | H-Bond Donor |
|---|---|---|---|---|
| 36.1 | 9.4 | 29.8 | 9.8 | 14.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, J.; Yang, X.; Ma, C. QSAR, Docking, and Molecular Dynamics Simulation Studies of Sigmacidins as Antimicrobials against Streptococci. Int. J. Mol. Sci. 2022, 23, 4085. https://doi.org/10.3390/ijms23084085
Ye J, Yang X, Ma C. QSAR, Docking, and Molecular Dynamics Simulation Studies of Sigmacidins as Antimicrobials against Streptococci. International Journal of Molecular Sciences. 2022; 23(8):4085. https://doi.org/10.3390/ijms23084085
Chicago/Turabian StyleYe, Jiqing, Xiao Yang, and Cong Ma. 2022. "QSAR, Docking, and Molecular Dynamics Simulation Studies of Sigmacidins as Antimicrobials against Streptococci" International Journal of Molecular Sciences 23, no. 8: 4085. https://doi.org/10.3390/ijms23084085
APA StyleYe, J., Yang, X., & Ma, C. (2022). QSAR, Docking, and Molecular Dynamics Simulation Studies of Sigmacidins as Antimicrobials against Streptococci. International Journal of Molecular Sciences, 23(8), 4085. https://doi.org/10.3390/ijms23084085