
Antiviral Effects of ABMA and DABMA against Influenza Virus In Vitro and In Vivo via Regulating the Endolysosomal Pathway and Autophagy

 ,
,  , ,
, ,  and
and
Abstract
1. Introduction
2. Results
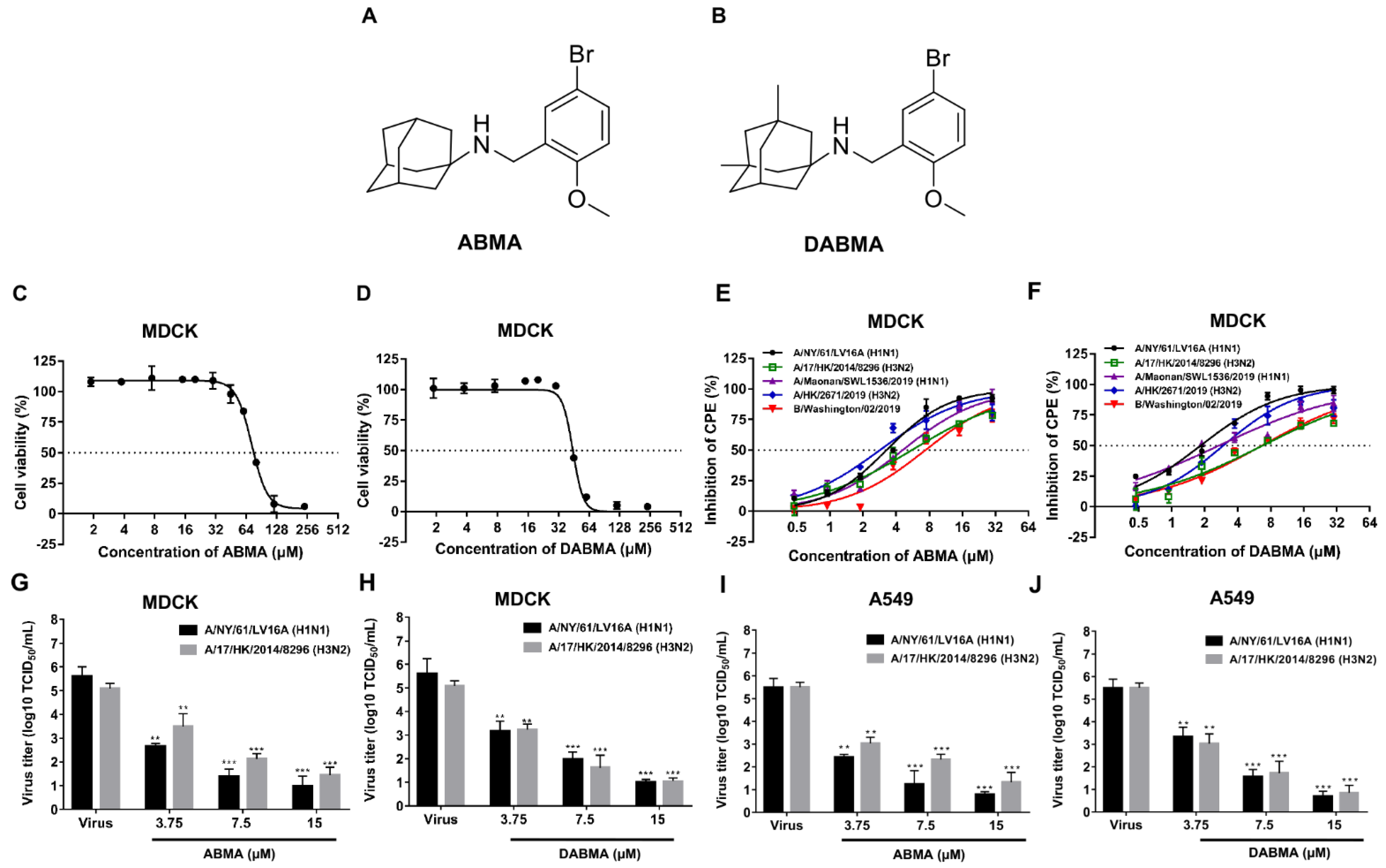
2.1. ABMA and DABMA Protect Cells against Influenza Virus Infection
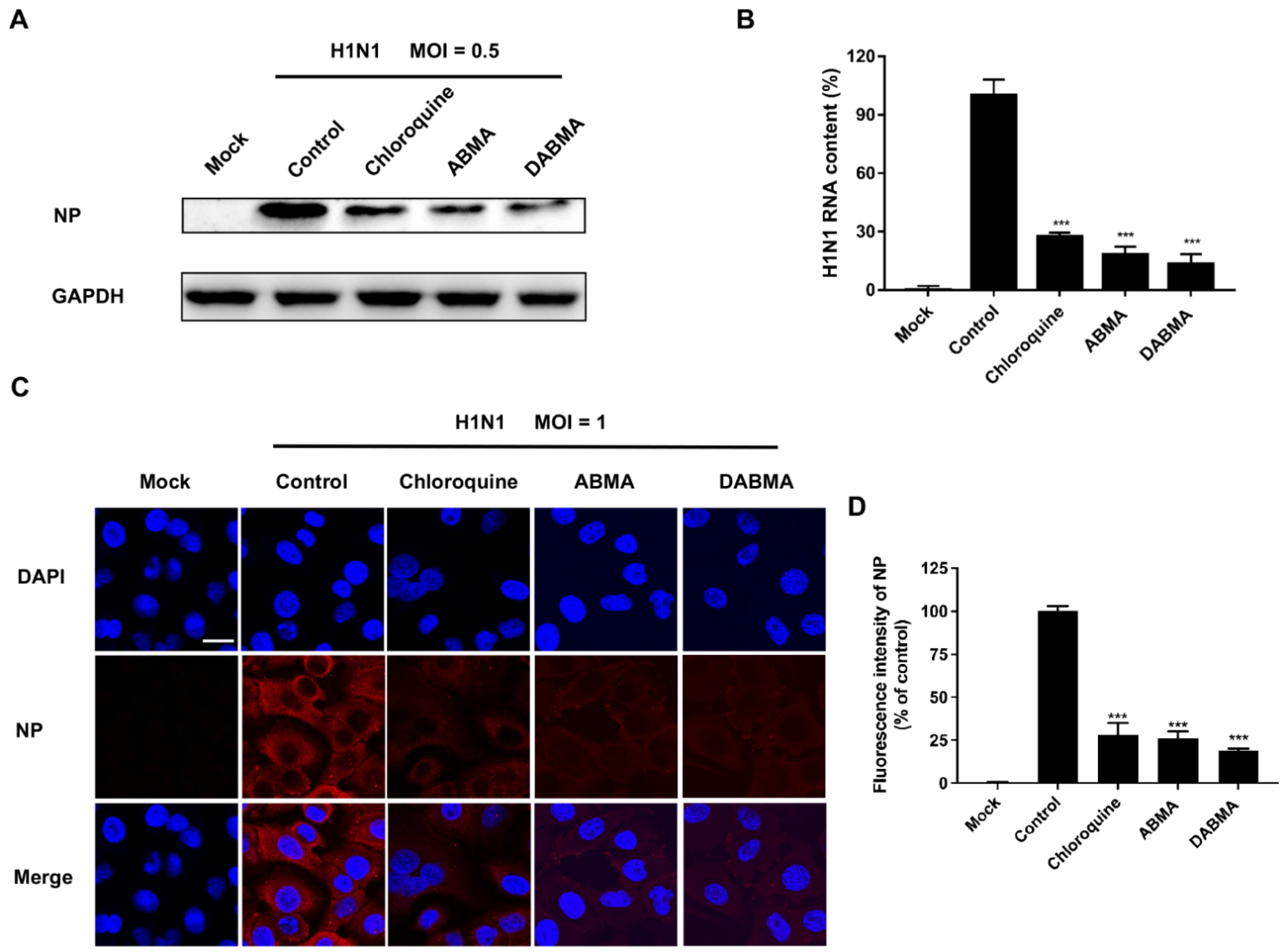
2.2. ABMA and DABMA Inhibit H1N1 Genomic RNA Replication and Protein Synthesis
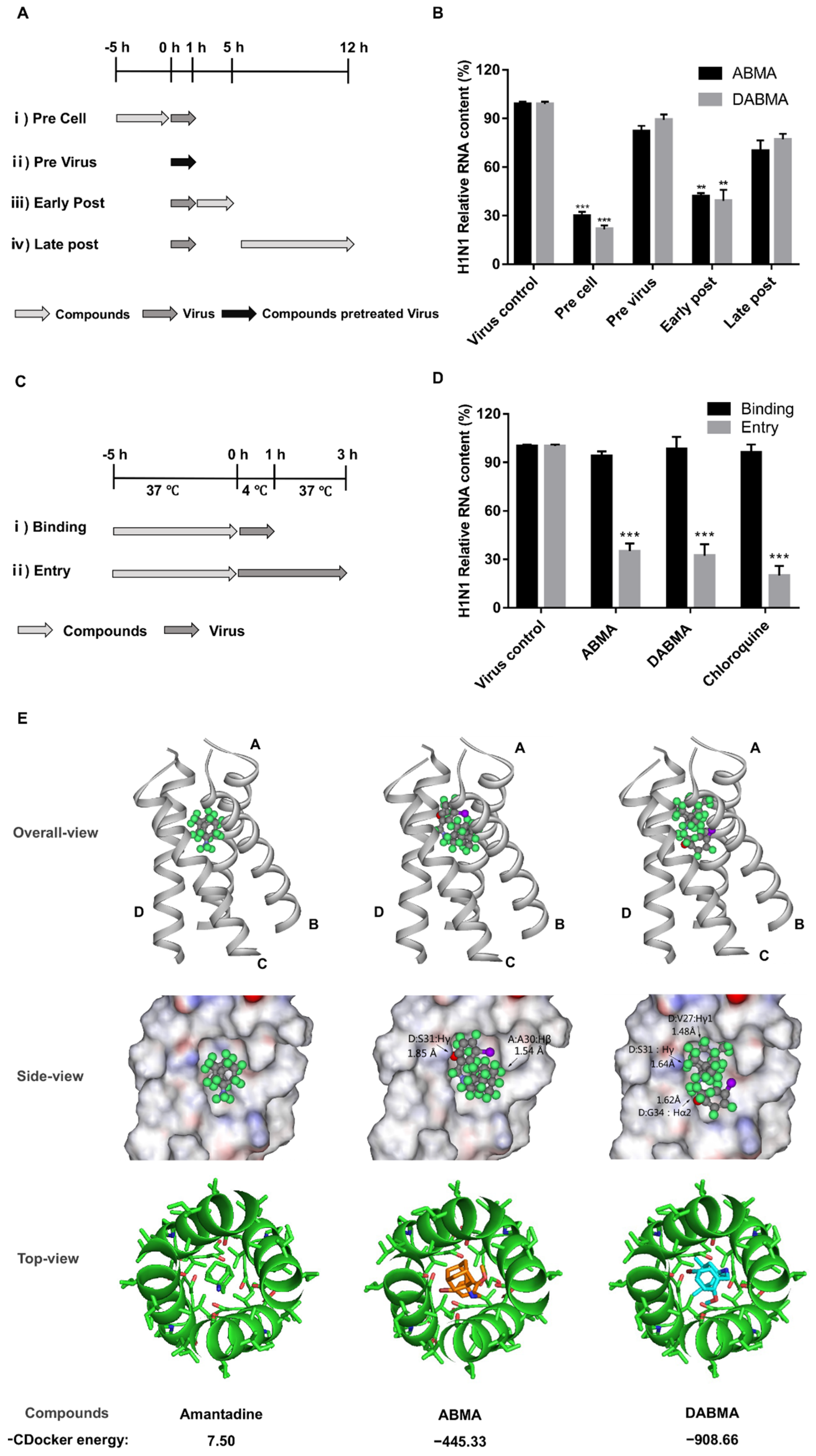
2.3. ABMA and DABMA Interfere with the Entry Stage of the H1N1 Infection Cycle
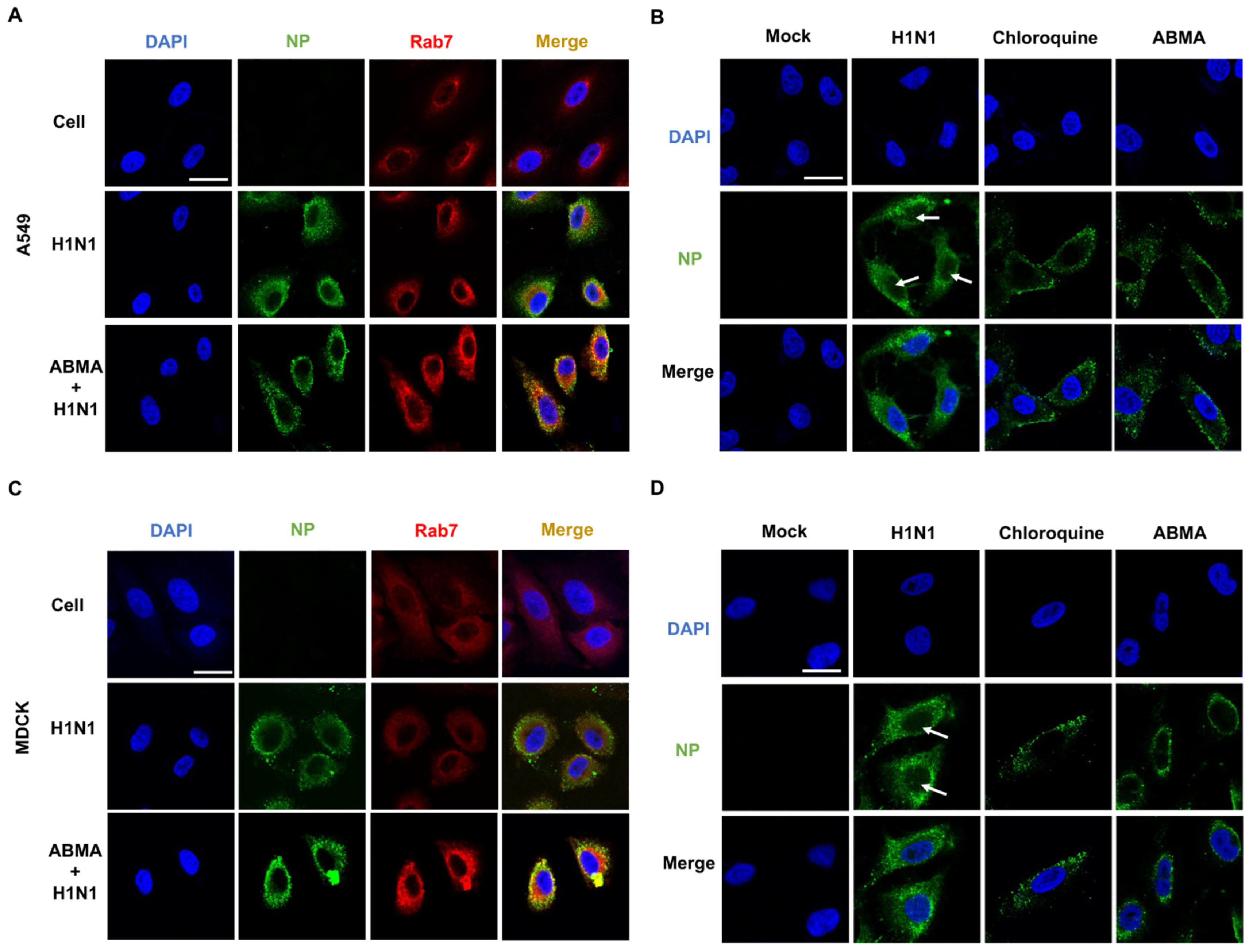
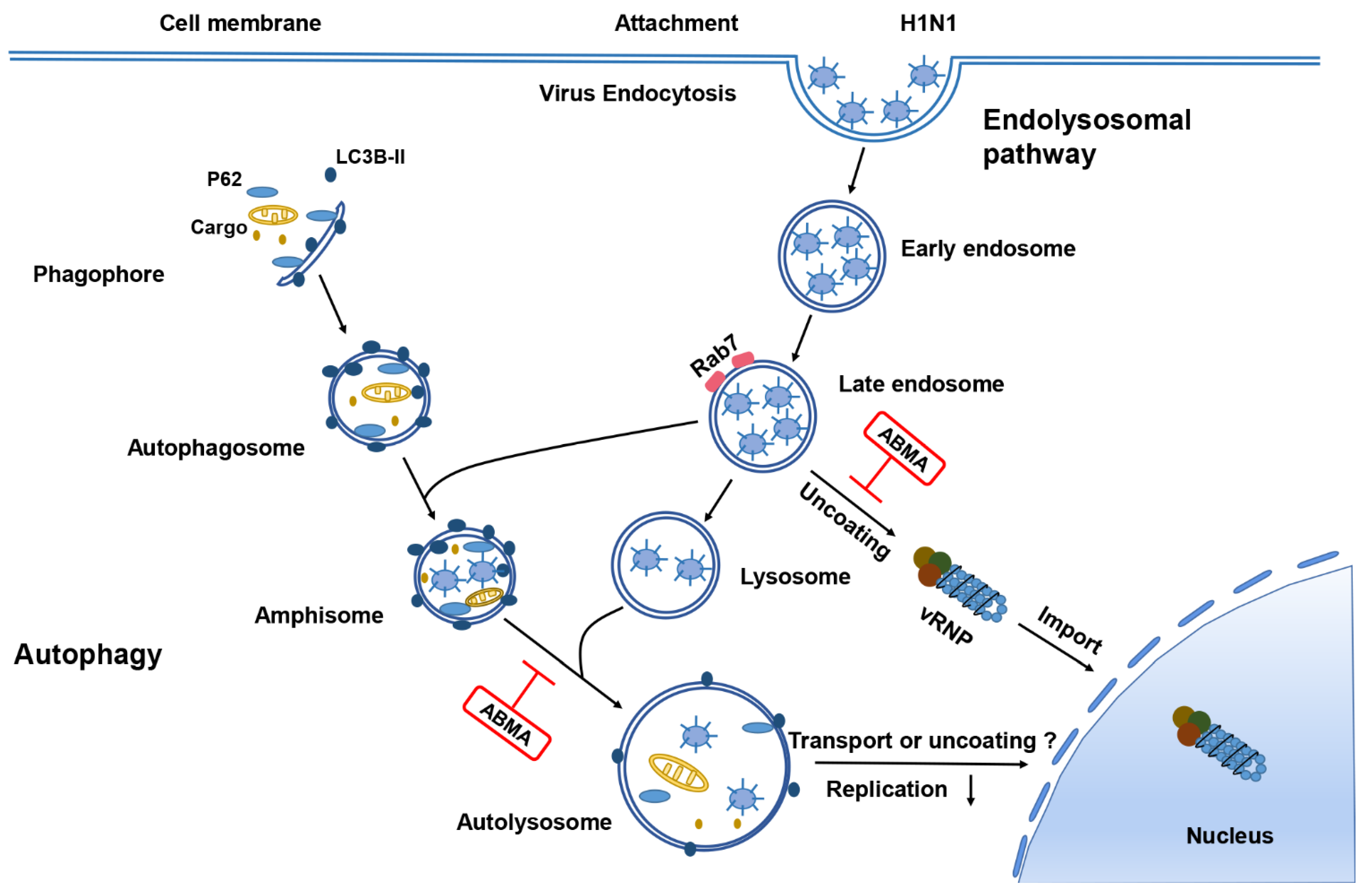
2.4. ABMA Induced Accumulation of H1N1 Virions in Late Endosomes at the Early Stage Post Infection
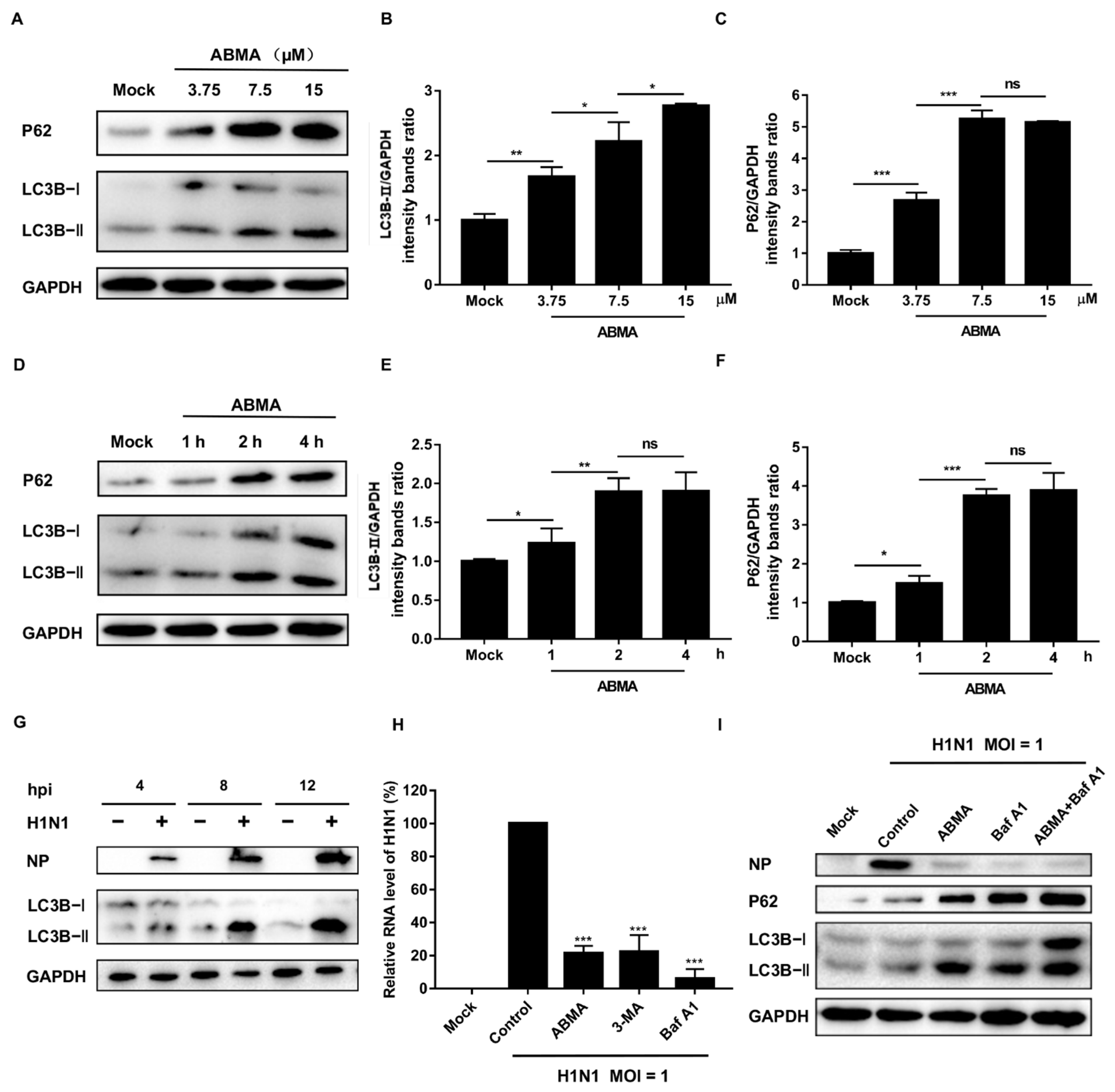
2.5. ABMA Inhibits H1N1 Replication by Interfering with Autophagy
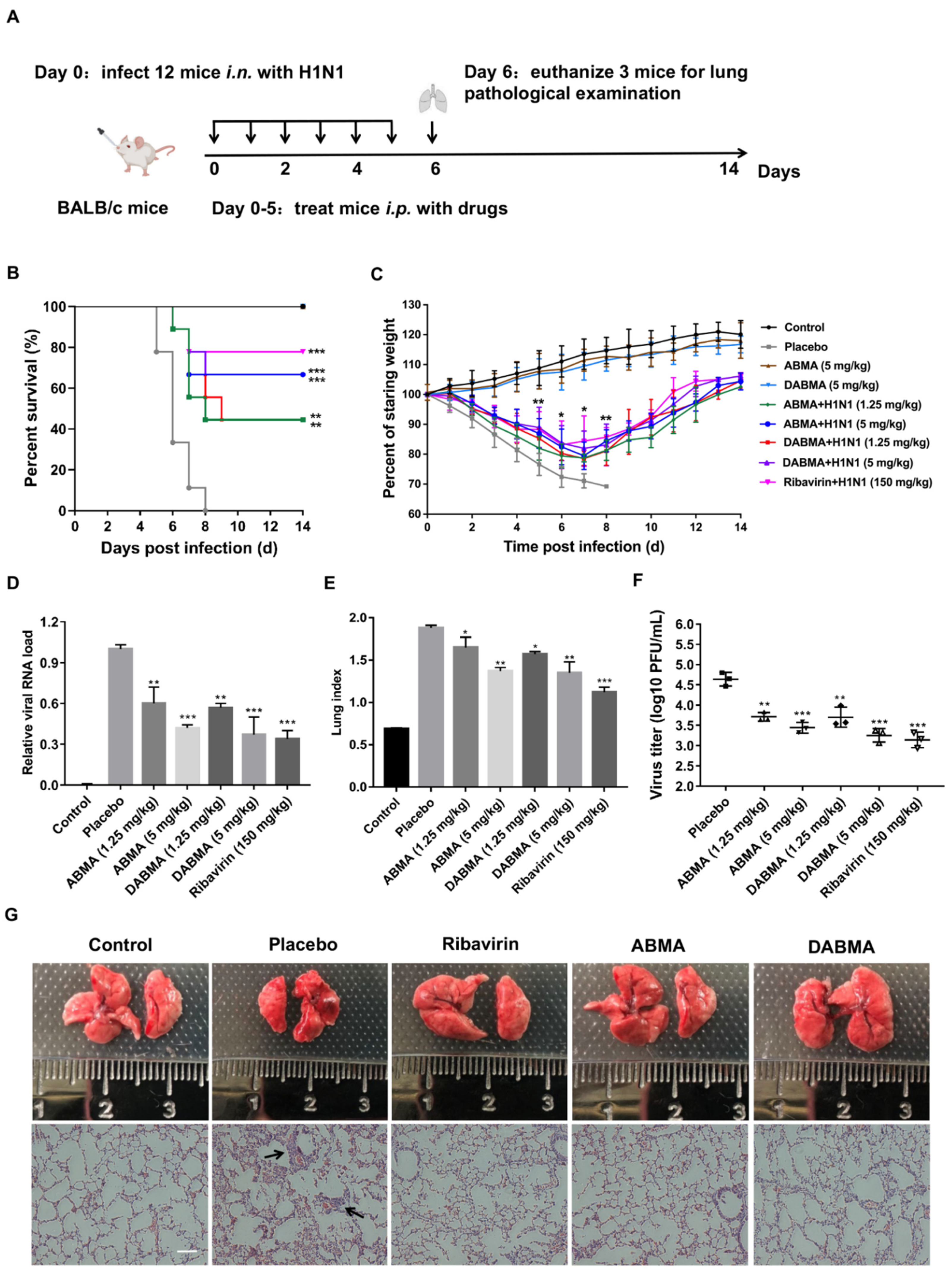
2.6. ABMA and DABMA Protect Mice from H1N1 Infection
3. Discussion
4. Materials and Methods
4.1. Reagents, Cells, Viruses, and Mice
4.2. Cytopathic Effect (CPE) Inhibition Assay on IAV Infection
4.3. Quantitative Reverse Transcription PCR (qRT-PCR)
4.4. Western Blotting Assay
4.5. Time of Addition Assay
4.6. Immunofluorescence Assay
4.7. Molecular Docking
4.8. Antiviral Activity of Compounds in H1N1-Inoculated Mice
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iuliano, A.D.; Roguski, K.M.; Chang, H.H.; Muscatello, D.J.; Palekar, R.; Tempia, S.; Cohen, C.; Gran, J.M.; Schanzer, D.; Cowling, B.J.; et al. Estimates of global seasonal influenza-associated respiratory mortality: A modelling study. Lancet 2018, 391, 1285–1300. [Google Scholar] [CrossRef]
- Paget, J.; Spreeuwenberg, P.; Charu, V.; Taylor, R.J.; Iuliano, A.D.; Bresee, J.; Simonsen, L.; Viboud, C. Global mortality associated with seasonal influenza epidemics: New burden estimates and predictors from the GLaMOR Project. J. Glob. Health 2019, 9, 020421. [Google Scholar] [CrossRef]
- Ison, M.G. Optimizing antiviral therapy for influenza: Understanding the evidence. Expert Rev. Anti-Infect. Ther. 2015, 13, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Palomba, E.; Castelli, V.; Renisi, G.; Bandera, A.; Lombardi, A.; Gori, A. Antiviral Treatments for Influenza. Semin. Respir. Crit. Care Med. 2021, 42, 859–872. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.K.; Leung, T.W.; Ho, E.C.; Leung, P.C.; Ng, A.Y.; Lai, M.Y.; Lim, W.W. Oseltamivir- and amantadine-resistant influenza viruses A (H1N1). Emerg. Infect. Dis. 2009, 15, 966–968. [Google Scholar] [CrossRef] [PubMed]
- Homma, M.; Jayewardene, A.L.; Gambertoglio, J.; Aweeka, F. High-performance liquid chromatographic determination of ribavirin in whole blood to assess disposition in erythrocytes. Antimicrob. Agents Chemother. 1999, 43, 2716–2719. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yoshino, R.; Yasuo, N.; Sekijima, M. Molecular Dynamics Simulation reveals the mechanism by which the Influenza Cap-dependent Endonuclease acquires resistance against Baloxavir marboxil. Sci. Rep. 2019, 9, 17464. [Google Scholar] [CrossRef]
- Wu, Y.; Pons, V.; Goudet, A.; Panigai, L.; Fischer, A.; Herweg, J.A.; Kali, S.; Davey, R.A.; Laporte, J.; Bouclier, C.; et al. ABMA, a small molecule that inhibits intracellular toxins and pathogens by interfering with late endosomal compartments. Sci. Rep. 2017, 7, 15567. [Google Scholar] [CrossRef]
- Dai, W.; Wu, Y.; Bi, J.; Wang, S.; Li, F.; Kong, W.; Barbier, J.; Cintrat, J.C.; Gao, F.; Gillet, D.; et al. Antiviral Effects of ABMA against Herpes Simplex Virus Type 2 In Vitro and In Vivo. Viruses 2018, 10, 119. [Google Scholar] [CrossRef] [PubMed]
- Kali, S.; Jallet, C.; Azebi, S.; Cokelaer, T.; Da Fonseca, J.P.; Wu, Y.; Barbier, J.; Cintrat, J.C.; Gillet, D.; Tordo, N. Broad spectrum compounds targeting early stages of rabies virus (RABV) infection. Antivir. Res. 2021, 188, 105016. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Pons, V.; Noël, R.; Kali, S.; Shtanko, O.; Davey, R.A.; Popoff, M.R.; Tordo, N.; Gillet, D.; Cintrat, J.C.; et al. DABMA: A Derivative of ABMA with Improved Broad-Spectrum Inhibitory Activity of Toxins and Viruses. ACS Med. Chem. Lett. 2019, 10, 1140–1147. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Boulogne, C.; Carle, S.; Podinovskaia, M.; Barth, H.; Spang, A.; Cintrat, J.C.; Gillet, D.; Barbier, J. Regulation of endo-lysosomal pathway and autophagic flux by broad-spectrum antipathogen inhibitor ABMA. FEBS J. 2020, 287, 3184–3199. [Google Scholar] [CrossRef]
- Yan, M.; Zheng, T. Role of the endolysosomal pathway and exosome release in tau propagation. Neurochem. Int. 2021, 145, 104988. [Google Scholar] [CrossRef] [PubMed]
- Stadler, K.; Ha, H.R.; Ciminale, V.; Spirli, C.; Saletti, G.; Schiavon, M.; Bruttomesso, D.; Bigler, L.; Follath, F.; Pettenazzo, A.; et al. Amiodarone alters late endosomes and inhibits SARS coronavirus infection at a post-endosomal level. Am. J. Respir. Cell Mol. Biol. 2008, 39, 142–149. [Google Scholar] [CrossRef]
- Hernáez, B.; Guerra, M.; Salas, M.L.; Andrés, G. African Swine Fever Virus Undergoes Outer Envelope Disruption, Capsid Disassembly and Inner Envelope Fusion before Core Release from Multivesicular Endosomes. PLoS Pathog. 2016, 12, e1005595. [Google Scholar] [CrossRef]
- Macovei, A.; Petrareanu, C.; Lazar, C.; Florian, P.; Branza-Nichita, N. Regulation of hepatitis B virus infection by Rab5, Rab7, and the endolysosomal compartment. J. Virol. 2013, 87, 6415–6427. [Google Scholar] [CrossRef] [PubMed]
- Drews, K.; Calgi, M.P.; Harrison, W.C.; Drews, C.M.; Costa-Pinheiro, P.; Shaw, J.J.P.; Jobe, K.A.; Nelson, E.A.; Han, J.D.; Fox, T.; et al. Glucosylceramidase Maintains Influenza Virus Infection by Regulating Endocytosis. J. Virol. 2019, 93, e00017-19. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Deng, X.; Xu, K. Increased expression of sterol regulatory element binding protein-2 alleviates autophagic dysfunction in NAFLD. Int. J. Mol. Med. 2018, 41, 1877–1886. [Google Scholar] [CrossRef]
- Zhang, M.L.; Zhao, G.L.; Hou, Y.; Zhong, S.M.; Xu, L.J.; Li, F.; Niu, W.R.; Yuan, F.; Yang, X.L.; Wang, Z.; et al. Rac1 conditional deletion attenuates retinal ganglion cell apoptosis by accelerating autophagic flux in a mouse model of chronic ocular hypertension. Cell Death Dis. 2020, 11, 734. [Google Scholar] [CrossRef]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Taisne, C.; Lussignol, M.; Hernandez, E.; Moris, A.; Mouna, L.; Esclatine, A. Human cytomegalovirus hijacks the autophagic machinery and LC3 homologs in order to optimize cytoplasmic envelopment of mature infectious particles. Sci. Rep. 2019, 9, 4560. [Google Scholar] [CrossRef] [PubMed]
- Corona, A.K.; Saulsbery, H.M.; Corona Velazquez, A.F.; Jackson, W.T. Enteroviruses Remodel Autophagic Trafficking through Regulation of Host SNARE Proteins to Promote Virus Replication and Cell Exit. Cell Rep. 2018, 22, 3304–3314. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhu, Y.; Zhao, J.; Ren, C.; Li, P.; Chen, H.; Jin, M.; Zhou, H. Autophagy Promotes Replication of Influenza A Virus In Vitro. J. Virol. 2019, 93, e01984-18. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, F.; Ma, C. Recent progress in designing inhibitors that target the drug-resistant M2 proton channels from the influenza A viruses. Biopolymers 2015, 104, 291–309. [Google Scholar] [CrossRef] [PubMed]
- Jalily, P.H.; Duncan, M.C.; Fedida, D.; Wang, J.; Tietjen, I. Put a cork in it: Plugging the M2 viral ion channel to sink influenza. Antivir. Res. 2020, 178, 104780. [Google Scholar] [CrossRef] [PubMed]
- Di Trani, L.; Savarino, A.; Campitelli, L.; Norelli, S.; Puzelli, S.; D’Ostilio, D.; Vignolo, E.; Donatelli, I.; Cassone, A. Different pH requirements are associated with divergent inhibitory effects of chloroquine on human and avian influenza A viruses. Virol. J. 2007, 4, 39. [Google Scholar] [CrossRef]
- Savarino, A.; Boelaert, J.R.; Cassone, A.; Majori, G.; Cauda, R. Effects of chloroquine on viral infections: An old drug against today’s diseases? Lancet Infect. Dis. 2003, 3, 722–727. [Google Scholar] [CrossRef]
- Simpson, C.; Yamauchi, Y. Microtubules in Influenza Virus Entry and Egress. Viruses 2020, 12, 117. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.E.; Johansen, T. Following autophagy step by step. BMC Biol. 2011, 9, 39. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Choi, A.M. Regulation of autophagy in oxygen-dependent cellular stress. Curr. Pharm. Des. 2013, 19, 2747–2756. [Google Scholar] [CrossRef]
- Zhao, Q.; Hu, Z.Y.; Zhang, J.P.; Jiang, J.D.; Ma, Y.Y.; Li, J.R.; Peng, Z.G.; Chen, J.H. Dual Roles of Two Isoforms of Autophagy-related Gene ATG10 in HCV-Subgenomic replicon Mediated Autophagy Flux and Innate Immunity. Sci. Rep. 2017, 7, 11250. [Google Scholar] [CrossRef]
- Wu, Y.T.; Tan, H.L.; Shui, G.; Bauvy, C.; Huang, Q.; Wenk, M.R.; Ong, C.N.; Codogno, P.; Shen, H.M. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem. 2010, 285, 10850–10861. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.; Tagawa, Y.; Yoshimori, T.; Moriyama, Y.; Masaki, R.; Tashiro, Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct. Funct. 1998, 23, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Uddback, I.E.; Pedersen, L.M.; Pedersen, S.R.; Steffensen, M.A.; Holst, P.J.; Thomsen, A.R.; Christensen, J.P. Combined local and systemic immunization is essential for durable T-cell mediated heterosubtypic immunity against influenza A virus. Sci. Rep. 2016, 6, 20137. [Google Scholar] [CrossRef] [PubMed]
- Vieira, S.I.; Rebelo, S.; Esselmann, H.; Wiltfang, J.; Lah, J.; Lane, R.; Small, S.A.; Gandy, S.; da Cruz, E.S.E.F.; da Cruz, E.S.O.A. Retrieval of the Alzheimer’s amyloid precursor protein from the endosome to the TGN is S655 phosphorylation state-dependent and retromer-mediated. Mol. Neurodegener. 2010, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, E.M.; Jarosinski, K.W.; Jackson, W.; Carpenter, J.E.; Grose, C. Exocytosis of Varicella-Zoster Virus Virions Involves a Convergence of Endosomal and Autophagy Pathways. J. Virol. 2016, 90, 8673–8685. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Zhang, J.; Si, X.; Gao, G.; Mao, I.; McManus, B.M.; Luo, H. Autophagosome supports coxsackievirus B3 replication in host cells. J. Virol. 2008, 82, 9143–9153. [Google Scholar] [CrossRef] [PubMed]
- Maassab, H.F. Biologic and immunologic characteristics of cold-adapted influenza virus. J. Immunol. 1969, 102, 728–732. [Google Scholar]
- Crouch, S.P.; Kozlowski, R.; Slater, K.J.; Fletcher, J. The use of ATP bioluminescence as a measure of cell proliferation and cytotoxicity. J. Immunol. Methods 1993, 160, 81–88. [Google Scholar] [CrossRef]
- Dai, W.; Bi, J.; Li, F.; Wang, S.; Huang, X.; Meng, X.; Sun, B.; Wang, D.; Kong, W.; Jiang, C.; et al. Antiviral Efficacy of Flavonoids against Enterovirus 71 Infection in Vitro and in Newborn Mice. Viruses 2019, 11, 625. [Google Scholar] [CrossRef] [PubMed]
- Thomaston, J.L.; Polizzi, N.F.; Konstantinidi, A.; Wang, J.; Kolocouris, A.; DeGrado, W.F. Inhibitors of the M2 Proton Channel Engage and Disrupt Transmembrane Networks of Hydrogen-Bonded Waters. J. Am. Chem. Soc. 2018, 140, 15219–15226. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | EC50 ± SD (μM) a | CC50 ± SD (μM) b | |||||
|---|---|---|---|---|---|---|---|
| A/NY/61/LV16A (H1N1) | A/17/HK/2014/8296 (H3N2) | A/Maonan/SWL1536/2019 (H1N1) | A/HK/2671/2019 (H3N2) | B/Washington/02/2019 | MDCK | A549 | |
| A/M2-S31 | A/M2-S31 | A/M2-S31 | A/M2-S31N | B/M2 | |||
| ABMA | 3.25 ± 0.17 | 5.58 ± 0.38 | 4.57 ± 0.33 | 2.83 ± 0.26 | 7.36 ± 0.65 | 72.30 ± 1.09 | 83.77 ± 1.92 |
| DABMA | 1.82 ± 0.11 | 6.73 ± 0.73 | 2.69 ± 0.26 | 2.95 ± 0.27 | 6.62 ± 0.46 | 42.71 ± 1.46 | 47.42 ± 1.68 |
| Chloroquine | 17.07 ± 2.27 | 11.97 ± 1.26 | ND | ND | ND | 76.71 ± 5.08 | ND |
| Oseltamivir | 7.97 ± 0.85 | 7.30 ± 0.79 | 2.95 ± 0.21 | 1.79 ± 0.64 | 8.53 ± 1.64 | 1893 ± 71.8 | ND |
| Amantadine | 9.97 ± 1.34 | 8.27 ± 0.66 | 20.19 ± 1.36 | >100 | >100 | 476.7 ± 39.49 | ND |
| Ribavirin | 1.61 ± 0.62 | 0.13 ± 0.04 | 3.20 ± 0.34 | 19.76 ± 3.26 | 3.02 ±0.39 | 5484 ± 255.10 | ND |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Jiang, C.; Wu, Y.; Wu, M.; Wu, J.; Zhao, G.; Sun, J.; Huang, X.; Li, J.; Sheng, R.; et al. Antiviral Effects of ABMA and DABMA against Influenza Virus In Vitro and In Vivo via Regulating the Endolysosomal Pathway and Autophagy. Int. J. Mol. Sci. 2022, 23, 3940. https://doi.org/10.3390/ijms23073940
Liu H, Jiang C, Wu Y, Wu M, Wu J, Zhao G, Sun J, Huang X, Li J, Sheng R, et al. Antiviral Effects of ABMA and DABMA against Influenza Virus In Vitro and In Vivo via Regulating the Endolysosomal Pathway and Autophagy. International Journal of Molecular Sciences. 2022; 23(7):3940. https://doi.org/10.3390/ijms23073940
Chicago/Turabian StyleLiu, Hongtao, Chunlai Jiang, Yu Wu, Min Wu, Jiaxin Wu, Guanshu Zhao, Jie Sun, Xinyu Huang, Jiemin Li, Rui Sheng, and et al. 2022. "Antiviral Effects of ABMA and DABMA against Influenza Virus In Vitro and In Vivo via Regulating the Endolysosomal Pathway and Autophagy" International Journal of Molecular Sciences 23, no. 7: 3940. https://doi.org/10.3390/ijms23073940
APA StyleLiu, H., Jiang, C., Wu, Y., Wu, M., Wu, J., Zhao, G., Sun, J., Huang, X., Li, J., Sheng, R., Barbier, J., Cintrat, J.-C., Gillet, D., & Su, W. (2022). Antiviral Effects of ABMA and DABMA against Influenza Virus In Vitro and In Vivo via Regulating the Endolysosomal Pathway and Autophagy. International Journal of Molecular Sciences, 23(7), 3940. https://doi.org/10.3390/ijms23073940