
Whole Genome Sequencing, Focused Assays and Functional Studies Increasing Understanding in Cryptic Inherited Retinal Dystrophies

 , ,
, ,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Molecular Diagnoses Obtained in Previously Unsolved IRD Families
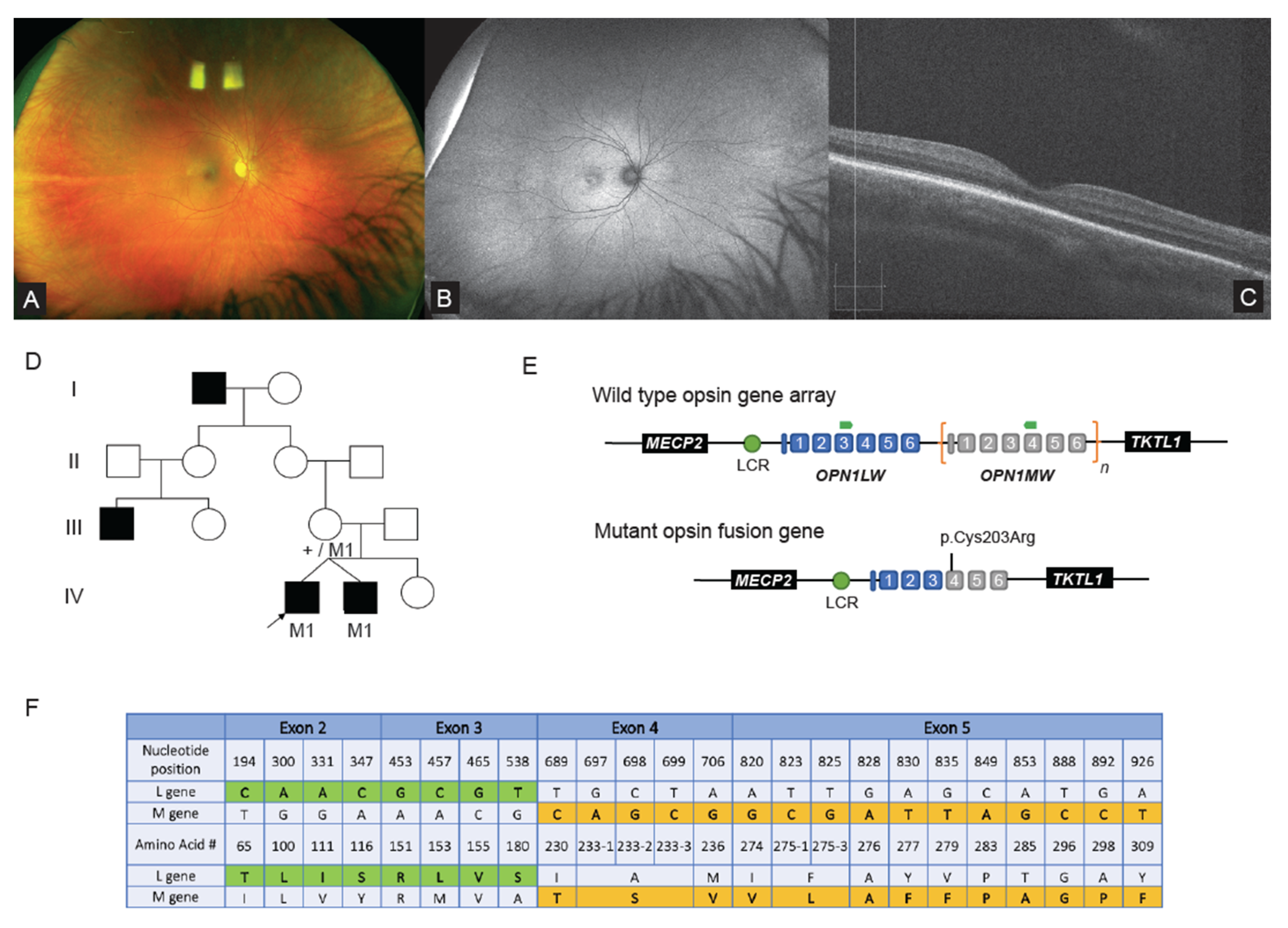
2.2. WGS Analysis Revealed Complex Hybrid Opsin SV and Missense Allele in Repetitive Region
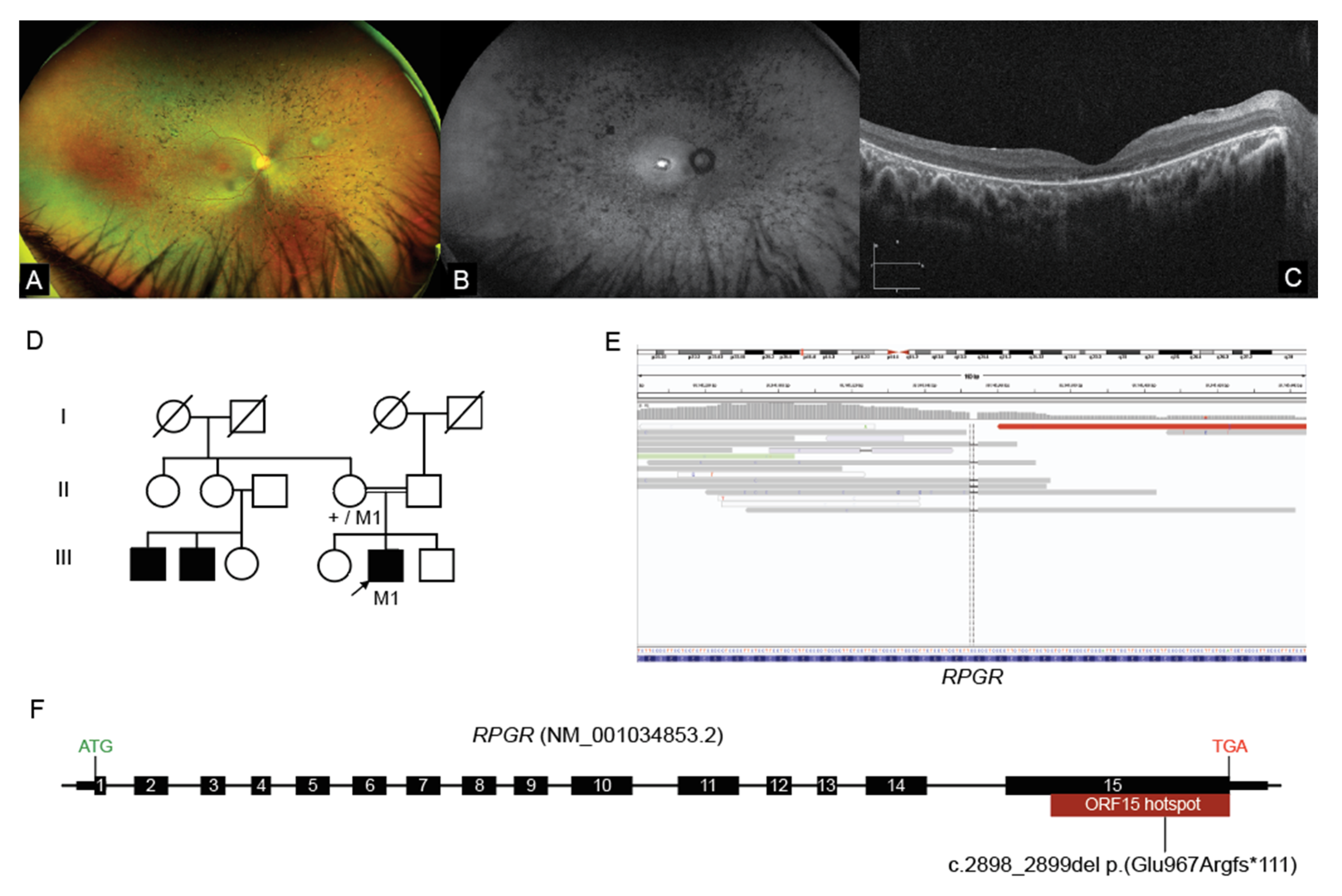
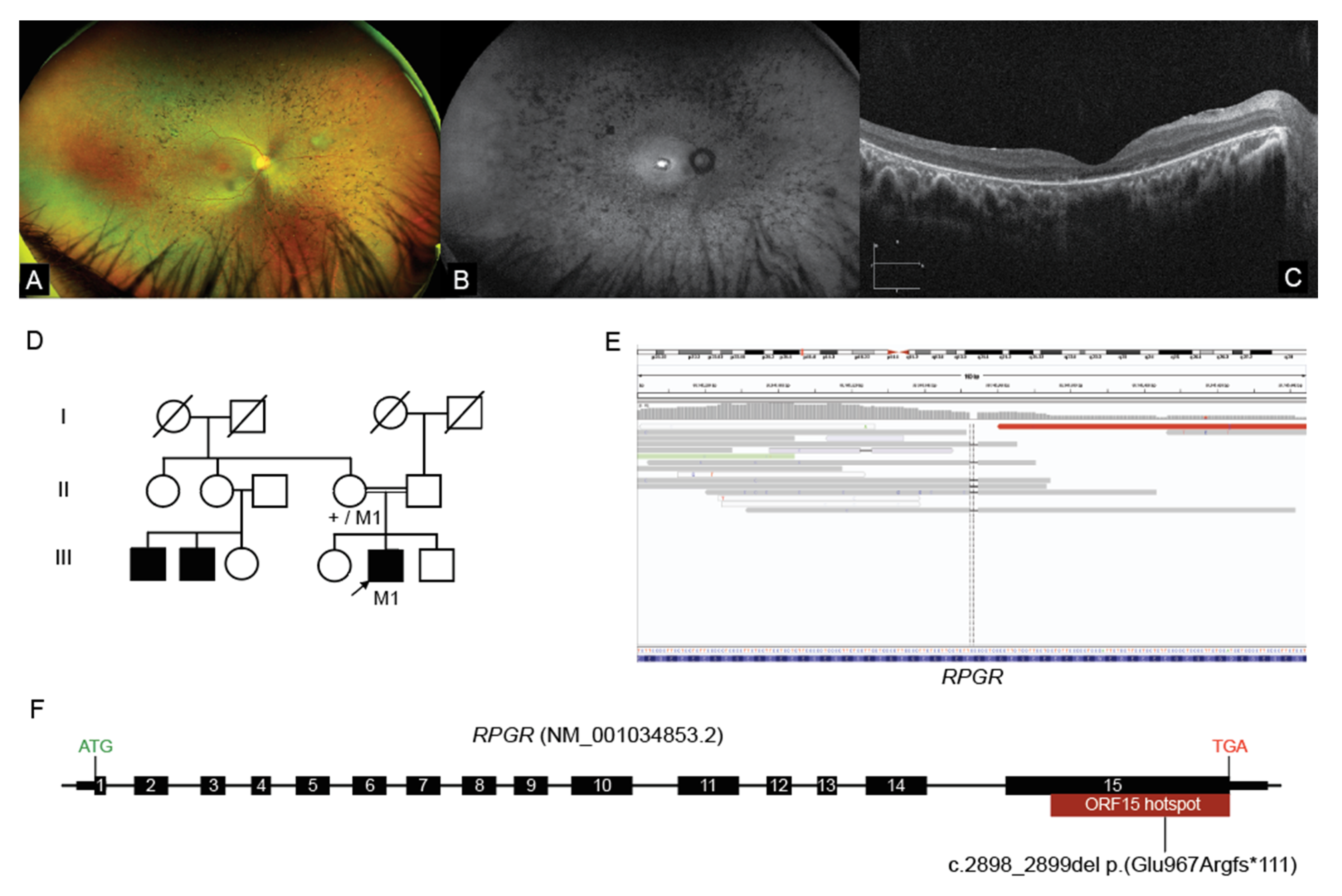
2.3. Interrogation of WGS Data Identifies an RPGR ORF15 Variant, in a Repetitive GC Rich Region
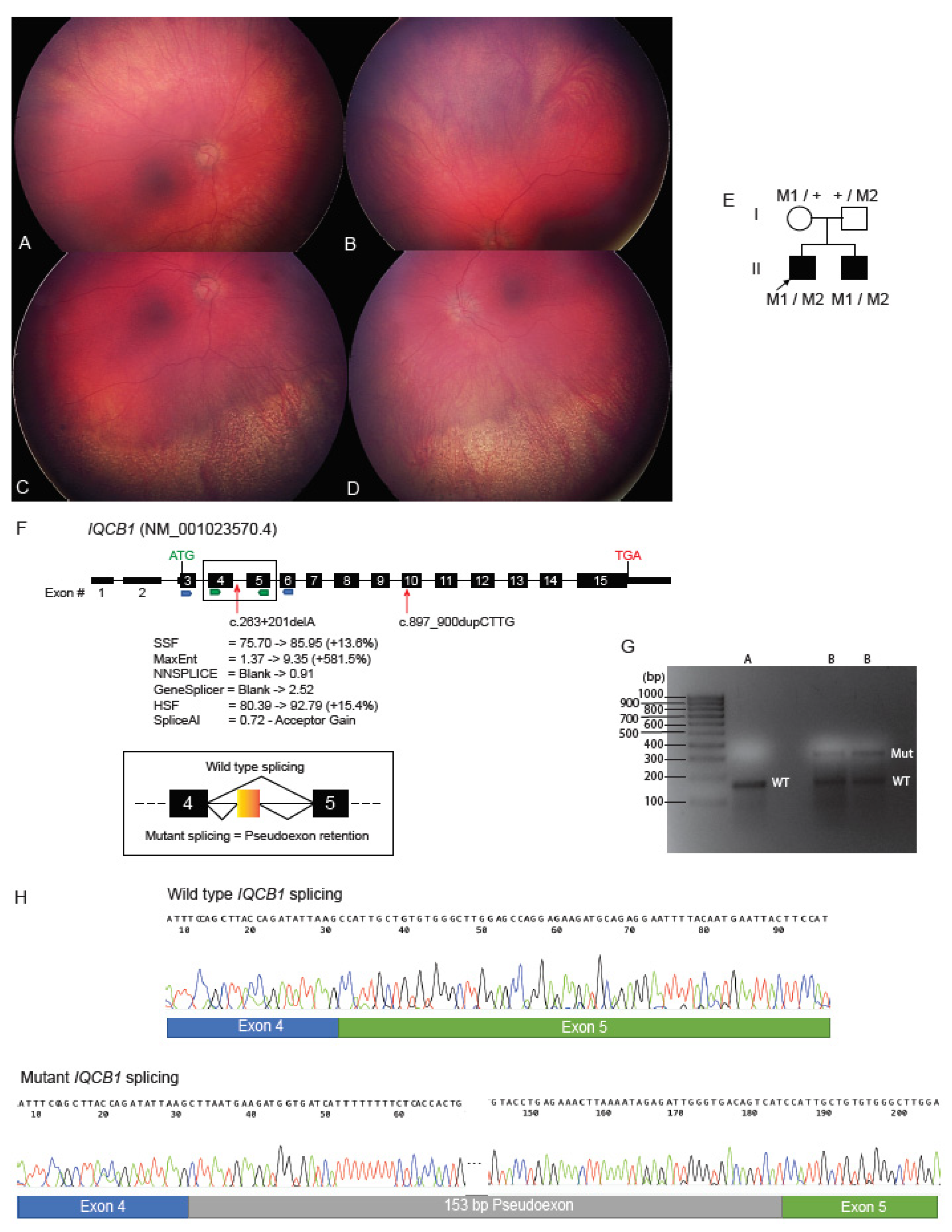
2.4. Deep Intronic Variant Identified on WGS Shown on RNA Studies to Create an IQCB1 Pseudoexon
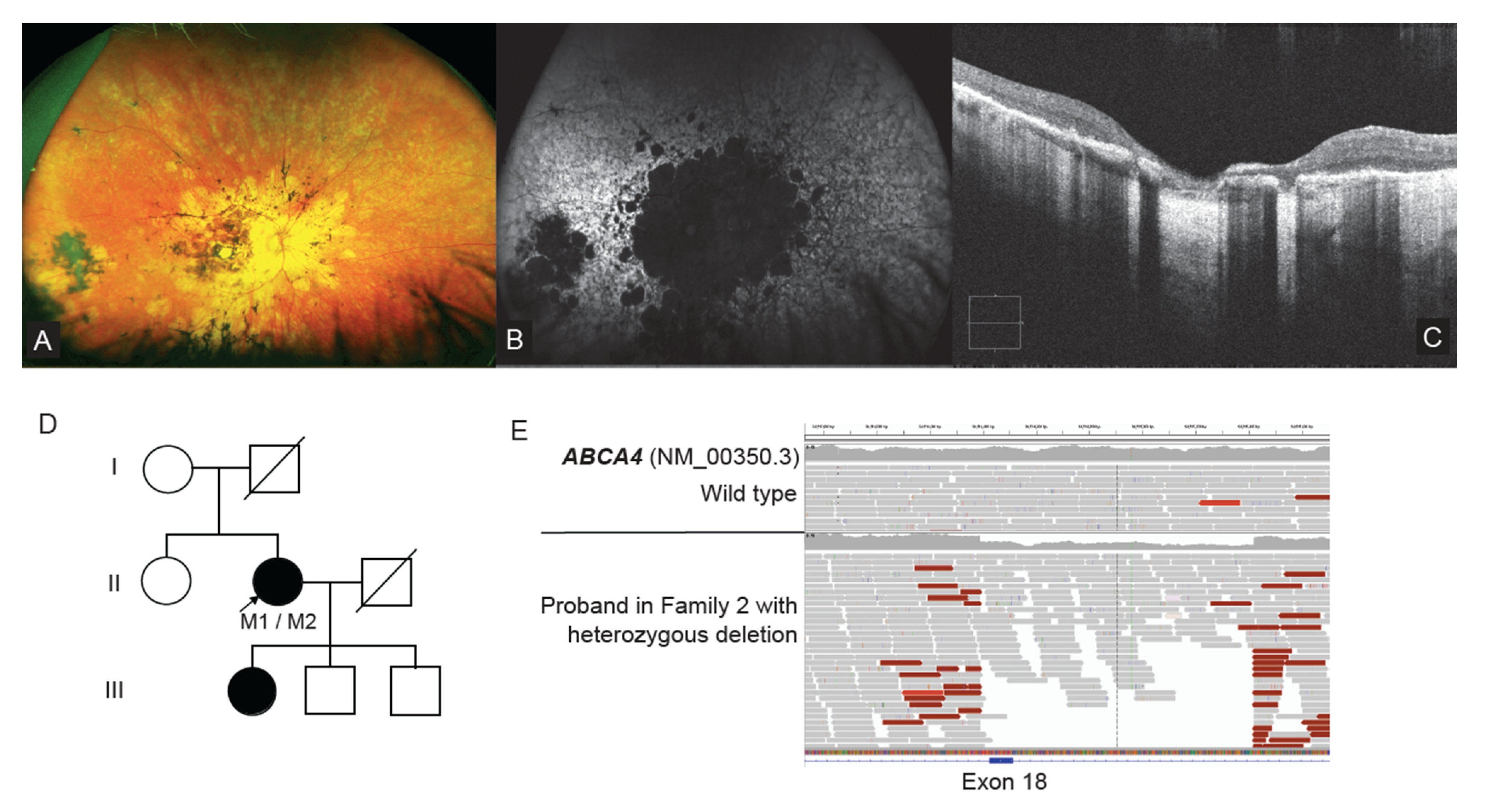
2.5. ABCA4 Deep Intronic Variant Identified on WGS
2.6. WGS Identifies Homozygous KCNV2 Deletion
2.7. WGS Identifies a Single Exon CNV in ABCA4
3. Discussion
4. Materials and Methods
4.1. Cohort and Patient Ascertainment
4.2. Ophthalmic Examination
4.3. Genomics and Bioinformatics
4.4. Genomic Examination of the OPN1LW and OPN1MW Genes
4.5. RNA Studies of Deep Intronic ICQB1 Variant
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanany, M.; Rivolta, C.; Sharon, D. Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc. Natl. Acad. Sci. USA 2020, 117, 2710–2716. [Google Scholar] [CrossRef] [PubMed]
- Liew, G.; Michaelides, M.; Bunce, C. A comparison of the causes of blindness certifications in England and Wales in working age adults (16–64 years), 1999–2000 with 2009–2010. BMJ Open 2014, 4, e004015. [Google Scholar] [CrossRef] [PubMed]
- Heath Jeffery, R.C.; Mukhtar, S.A.; McAllister, I.L.; Morgan, W.H.; Mackey, D.A.; Chen, F.K. Inherited retinal diseases are the most common cause of blindness in the working-age population in Australia. Ophthalmic Genet. 2021, 42, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Nash, B.M.; Wright, D.C.; Grigg, J.R.; Bennetts, B.; Jamieson, R.V. Retinal dystrophies, genomic applications in diagnosis and prospects for therapy. Transl. Pediatr. 2015, 4, 139–163. [Google Scholar] [CrossRef] [PubMed]
- Maquire, A.M.; Russell, S.; Wellman, J.A.; Chung, D.C.; Yu, Z.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; Marshall, K.A.; et al. Efficacy, Safety, and Durability of Voretigene Neparvovec-rzyl in RPE65 Mutations Associated Inherited Retinal Dystrophy. Ophthalmology 2019, 126, 1273–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeder, M.L.; Stefanidakis, M.; Wilson, C.J.; Baral, R.; Barrera, L.A.; Bounoutas, G.S.; Bumcrot, D.; Chao, H.; Ciulla, D.M.; DaSilva, J.A.; et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 2019, 25, 229–233. [Google Scholar] [CrossRef]
- Dulla, K.; Slijkerman, R.; van Diepen, H.C.; Albert, S.; Dona, M.; Beumer, W.; Turunen, J.J.; Chan, H.L.; Schulkens, I.A.; Vorthoren, L.; et al. Antisense oligonucleotide-based treatment of retinitis pigmentosa caused by USH2A exon 13 mutations. Mol. Ther. 2021, 29, 2441–2455. [Google Scholar] [CrossRef]
- Neveling, K.; Collin, R.W.; Gilissen, C.; van Huet, R.A.; Visser, L.; Kwint, M.P.; Gijsen, S.J.; Zonneveld, M.N.; Wieskamp, N.; de Ligt, J.; et al. Next-generation genetic testing for retinitis pigmentosa. Hum. Mutat. 2012, 33, 963–972. [Google Scholar] [CrossRef]
- Ellingford, J.M.; Barton, S.; Bhaskar, S.; O’Sullivan, J.; Williams, S.G.; Lamb, J.A.; Panda, B.; Sergouniotis, P.I.; Gillespie, R.L.; Daiger, S.P.; et al. Molecular findings from 537 individuals with inherited retinal disease. J. Med. Genet. 2016, 53, 761–767. [Google Scholar] [CrossRef] [Green Version]
- Stone, E.M.; Andorf, J.L.; Whitmore, S.S.; DeLuca, A.P.; Giacalone, J.C.; Streb, L.M.; Braun, T.A.; Mullins, R.F.; Scheetz, T.E.; Sheffield, V.C.; et al. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology 2017, 124, 1314–1331. [Google Scholar] [CrossRef]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perea-Romero, I.; Gordo, G.; Iancu, I.F.; Del Pozo-Valero, M.; Almoguera, B.; Blanco-Kelly, F.; Carreno, E.; Jimenez-Rolando, B.; Lopez-Rodriguez, R.; Lorda-Sanchez, I.; et al. Genetic landscape of 6089 inherited retinal dystrophies affected cases in Spain and their therapeutic and extended epidemiological implications. Sci. Rep. 2021, 11, 1526. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, S.; Iwasa, M.; Hayashi, T.; Hosono, K.; Yamashita, T.; Kuniyoshi, K.; Ueno, S.; Kondo, M.; Ueyama, H.; Ogita, H.; et al. Genotype determination of the OPN1LW/OPN1MW genes: Novel disease-causing mechanisms in Japanese patients with blue cone monochromacy. Sci. Rep. 2018, 8, 11507. [Google Scholar] [CrossRef] [PubMed]
- Nathans, J.; Maumenee, I.H.; Zrenner, E.; Sadowski, B.; Sharpe, L.T.; Lewis, R.A.; Hansen, E.; Rosenberg, T.; Schwartz, M.; Heckenlively, J.R.; et al. Genetic heterogeneity among Blue-Cone Monochromats. Am. J. Hum. Genet. 1993, 53, 987–1000. [Google Scholar]
- Michaelides, M.; Johnson, S.; Simunovic, M.P.; Bradshaw, K.; Holder, G.; Mollon, J.D.; Moore, A.T.; Hunt, D.M. Blue cone monochromatism: A phenotype and genotype assessment with evidence of progressive loss of cone function in older individuals. Eye (Lond.) 2005, 19, 2–10. [Google Scholar] [CrossRef] [Green Version]
- Stone, E.M.; Cideciyan, A.V.; Aleman, T.S.; Scheetz, T.E.; Sumaroka, A.; Ehlinger, M.A.; Schwartz, S.B.; Fishman, G.A.; Traboulsi, E.I.; Lam, B.L.; et al. Variations in NPHP5 in patients with nonsyndromic leber congenital amaurosis and Senior-Loken syndrome. Arch. Ophthalmol. 2011, 129, 81–87. [Google Scholar] [CrossRef] [Green Version]
- Halbritter, J.; Diaz, K.; Chaki, M.; Porath, J.D.; Tarrier, B.; Fu, C.; Innis, J.L.; Allen, S.J.; Lyons, R.H.; Stefanidis, C.J.; et al. High-throughput mutation analysis in patients with a nephronophthisis-associated ciliopathy applying multiplexed barcoded array-based PCR amplification and next-generation sequencing. J. Med. Genet. 2012, 49, 756–767. [Google Scholar] [CrossRef]
- Fujinami, K.; Strauss, R.W.; Chiang, J.P.; Audo, I.S.; Bernstein, P.S.; Birch, D.G.; Bomotti, S.M.; Cideciyan, A.V.; Ervin, A.M.; Marino, M.J.; et al. Detailed genetic characteristics of an international large cohort of patients with Stargardt disease: ProgStar study report 8. Br. J. Ophthalmol. 2019, 103, 390–397. [Google Scholar] [CrossRef]
- Braun, T.A.; Mullins, R.F.; Wagner, A.H.; Andorf, J.L.; Johnston, R.M.; Bakall, B.B.; Deluca, A.P.; Fishman, G.A.; Lam, B.L.; Weleber, R.G.; et al. Non-exomic and synonymous variants in ABCA4 are an important cause of Stargardt disease. Hum. Mol. Genet. 2013, 22, 5136–5145. [Google Scholar] [CrossRef]
- Wu, H.; Cowing, J.A.; Michaelides, M.; Wilkie, S.E.; Jeffery, G.; Jenkins, S.A.; Mester, V.; Bird, A.C.; Robson, A.G.; Holder, G.E.; et al. Mutations in the gene KCNV2 encoding a voltage-gated potassium channel subunit cause “cone dystrophy with supernormal rod electroretinogram” in humans. Am. J. Hum. Genet. 2006, 79, 574–579. [Google Scholar] [CrossRef] [Green Version]
- Wissinger, B.; Schaich, S.; Baumann, B.; Bonin, M.; Jagle, H.; Friedburg, C.; Varsanyi, B.; Hoyng, C.B.; Dollfus, H.; Heckenlively, J.R.; et al. Large deletions of the KCNV2 gene are common in patients with cone dystrophy with supernormal rod response. Hum. Mutat. 2011, 32, 1398–1406. [Google Scholar] [CrossRef] [PubMed]
- Grigg, J.R.; Holder, G.E.; Billson, F.A.; Korsakova, M.; Jamieson, R.V. The importance of electrophysiology in revealing a complete homozygous deletion of KCNV2. J. Am. Assoc. Pediatric Ophthalmol. Strabismus 2013, 17, 641–643. [Google Scholar] [CrossRef] [PubMed]
- Nash, B.M.; Symes, R.; Goel, H.; Dinger, M.E.; Bennetts, B.; Grigg, J.R.; Jamieson, R.V. NMNAT1 variants cause cone and cone-rod dystrophy. Eur. J. Hum. Genet. 2018, 26, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Nash, B.M.; Watson, C.J.G.; Hughes, E.; Hou, A.L.; Loi, T.H.; Bennetts, B.; Jelovic, D.; Polkinghorne, P.J.; Gorbatov, M.; Grigg, J.R.; et al. Heterozygous COL9A3 variants cause severe peripheral vitreoretinal degeneration and retinal detachment. Eur. J. Hum. Genet. 2021, 29, 881–886. [Google Scholar] [CrossRef]
- Chiang, J.P.W.; Lamey, T.M.; Wang, N.K.; Duan, J.; Zhou, W.; McLaren, T.L.; Thompson, J.A.; Ruddle, J.; De Roach, J.N. Development of High-Throughput Clinical Testing of RPGR ORF15 Using a Large Inherited Retinal Dystrophy Cohort. Investig. Ophthalmol. Vis. Sci. 2018, 59, 4434–4440. [Google Scholar] [CrossRef] [Green Version]
- Gardner, J.C.; Liew, G.; Quan, Y.H.; Ermetal, B.; Ueyama, H.; Davidson, A.E.; Schwarz, N.; Kanuga, N.; Chana, R.; Maher, E.R.; et al. Three different cone opsin gene array mutational mechanisms with genotype-phenotype correlation and functional investigation of cone opsin variants. Hum. Mutat. 2014, 35, 1354–1362. [Google Scholar] [CrossRef] [Green Version]
- Sangermano, R.; Garanto, A.; Khan, M.; Runhart, E.H.; Bauwens, M.; Bax, N.M.; van den Born, L.I.; Khan, M.I.; Cornelis, S.S.; Verheij, J.; et al. Deep-intronic ABCA4 variants explain missing heritability in Stargardt disease and allow correction of splice defects by antisense oligonucleotides. Genet. Med. 2019, 21, 1751–1760. [Google Scholar] [CrossRef] [Green Version]
- Small, K.W.; DeLuca, A.P.; Whitmore, S.S.; Rosenberg, T.; Silva-Garcia, R.; Udar, N.; Puech, B.; Garcia, C.A.; Rice, T.A.; Fishman, G.A.; et al. North Carolina Macular Dystrophy Is Caused by Dysregulation of the Retinal Transcription Factor PRDM13. Ophthalmology 2016, 123, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Kazmi, M.A.; Sakmar, T.P.; Ostrer, H. Mutation of a conserved cysteine in the X-linked cone opsins causes color vision deficiencies by distupting protein folding and stability. Investig. Ophthalmol. Vis. Sci. 1997, 38, 1074–1081. [Google Scholar]
- Ma, A.; Grigg, J.R.; Flaherty, M.; Smith, J.; Minoche, A.E.; Cowley, M.J.; Nash, B.M.; Ho, G.; Gayagay, T.; Lai, T.; et al. Genome sequencing in congenital cataracts improves diagnostic yield. Hum. Mutat. 2021, 42, 1173–1183. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef] [PubMed]
- Bedoni, N.; Quinodoz, M.; Pinelli, M.; Cappuccio, G.; Torella, A.; Nigro, V.; Testa, F.; Simonelli, F.; Tudp; Corton, M.; et al. An Alu-mediated duplication in NMNAT1, involved in NAD biosynthesis, causes a novel syndrome, SHILCA, affecting multiple tissues and organs. Hum. Mol. Genet. 2020, 29, 2250–2260. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nash, B.M.; Loi, T.H.; Fernando, M.; Sabri, A.; Robinson, J.; Cheng, A.; Eamegdool, S.S.; Farnsworth, E.; Bennetts, B.; Grigg, J.R.; et al. Evaluation for Retinal Therapy for RPE65 Variation Assessed in hiPSC Retinal Pigment Epithelial Cells. Stem Cells Int. 2021, 2021, 4536382. [Google Scholar] [CrossRef]
- Kalia, S.S.; Adelman, K.; Bale, S.J.; Chung, W.K.; Eng, C.; Evans, J.P.; Herman, G.E.; Hufnagel, S.B.; Klein, T.E.; Korf, B.R.; et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the American College of Medical Genetics and Genomics. Genet. Med. 2017, 19, 249–255. [Google Scholar] [CrossRef] [Green Version]
- de Wert, G.; Dondorp, W.; Clarke, A.; Dequeker, E.M.C.; Cordier, C.; Deans, Z.; van El, C.G.; Fellmann, F.; Hastings, R.; Hentze, S.; et al. Opportunistic genomic screening. Recommendations of the European Society of Human Genetics. Eur. J. Hum. Genet. 2021, 29, 365–377. [Google Scholar] [CrossRef]
- Royer-Bertrand, B.; Cisarova, K.; Niel-Butschi, F.; Mittaz-Crettol, L.; Fodstad, H.; Superti-Furga, A. CNV Detection from Exome Sequencing Data in Routine Diagnostics of Rare Genetic Disorders: Opportunities and Limitations. Genes 2021, 12, 1427. [Google Scholar] [CrossRef]
- Coutelier, M.; Holtgrewe, M.; Jager, M.; Flottman, R.; Mensah, M.A.; Spielmann, M.; Krawitz, P.; Horn, D.; Beule, D.; Mundlos, S. Combining callers improves the detection of copy number variants from whole-genome sequencing. Eur. J. Hum. Genet. 2022, 30, 178–186. [Google Scholar] [CrossRef]
- Ellingford, J.M.; Horn, B.; Campbell, C.; Arno, G.; Barton, S.; Tate, C.; Bhaskar, S.; Sergouniotis, P.I.; Taylor, R.L.; Carss, K.J.; et al. Assessment of the incorporation of CNV surveillance into gene panel next-generation sequencing testing for inherited retinal diseases. J. Med. Genet. 2018, 55, 114–121. [Google Scholar] [CrossRef] [Green Version]
- Minoche, A.E.; Lundie, B.; Peters, G.B.; Ohnesorg, T.; Pinese, M.; Thomas, D.M.; Zankl, A.; Roscioli, T.; Schonrock, N.; Kummerfeld, S.; et al. ClinSV: Clinical grade structural and copy number variant detection from whole genome sequencing data. Genome Med. 2021, 13, 32. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nash, B.M.; Ma, A.; Ho, G.; Farnsworth, E.; Minoche, A.E.; Cowley, M.J.; Barnett, C.; Smith, J.M.; Loi, T.H.; Wong, K.; et al. Whole Genome Sequencing, Focused Assays and Functional Studies Increasing Understanding in Cryptic Inherited Retinal Dystrophies. Int. J. Mol. Sci. 2022, 23, 3905. https://doi.org/10.3390/ijms23073905
Nash BM, Ma A, Ho G, Farnsworth E, Minoche AE, Cowley MJ, Barnett C, Smith JM, Loi TH, Wong K, et al. Whole Genome Sequencing, Focused Assays and Functional Studies Increasing Understanding in Cryptic Inherited Retinal Dystrophies. International Journal of Molecular Sciences. 2022; 23(7):3905. https://doi.org/10.3390/ijms23073905
Chicago/Turabian StyleNash, Benjamin M., Alan Ma, Gladys Ho, Elizabeth Farnsworth, Andre E. Minoche, Mark J. Cowley, Christopher Barnett, Janine M. Smith, To Ha Loi, Karen Wong, and et al. 2022. "Whole Genome Sequencing, Focused Assays and Functional Studies Increasing Understanding in Cryptic Inherited Retinal Dystrophies" International Journal of Molecular Sciences 23, no. 7: 3905. https://doi.org/10.3390/ijms23073905
APA StyleNash, B. M., Ma, A., Ho, G., Farnsworth, E., Minoche, A. E., Cowley, M. J., Barnett, C., Smith, J. M., Loi, T. H., Wong, K., St Heaps, L., Wright, D., Dinger, M. E., Bennetts, B., Grigg, J. R., & Jamieson, R. V. (2022). Whole Genome Sequencing, Focused Assays and Functional Studies Increasing Understanding in Cryptic Inherited Retinal Dystrophies. International Journal of Molecular Sciences, 23(7), 3905. https://doi.org/10.3390/ijms23073905