
Synthesis of Novel Derivatives of 5,6,7,8-Tetrahydroquinazolines Using α-Aminoamidines and In Silico Screening of Their Biological Activity


Abstract
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Molecular Docking
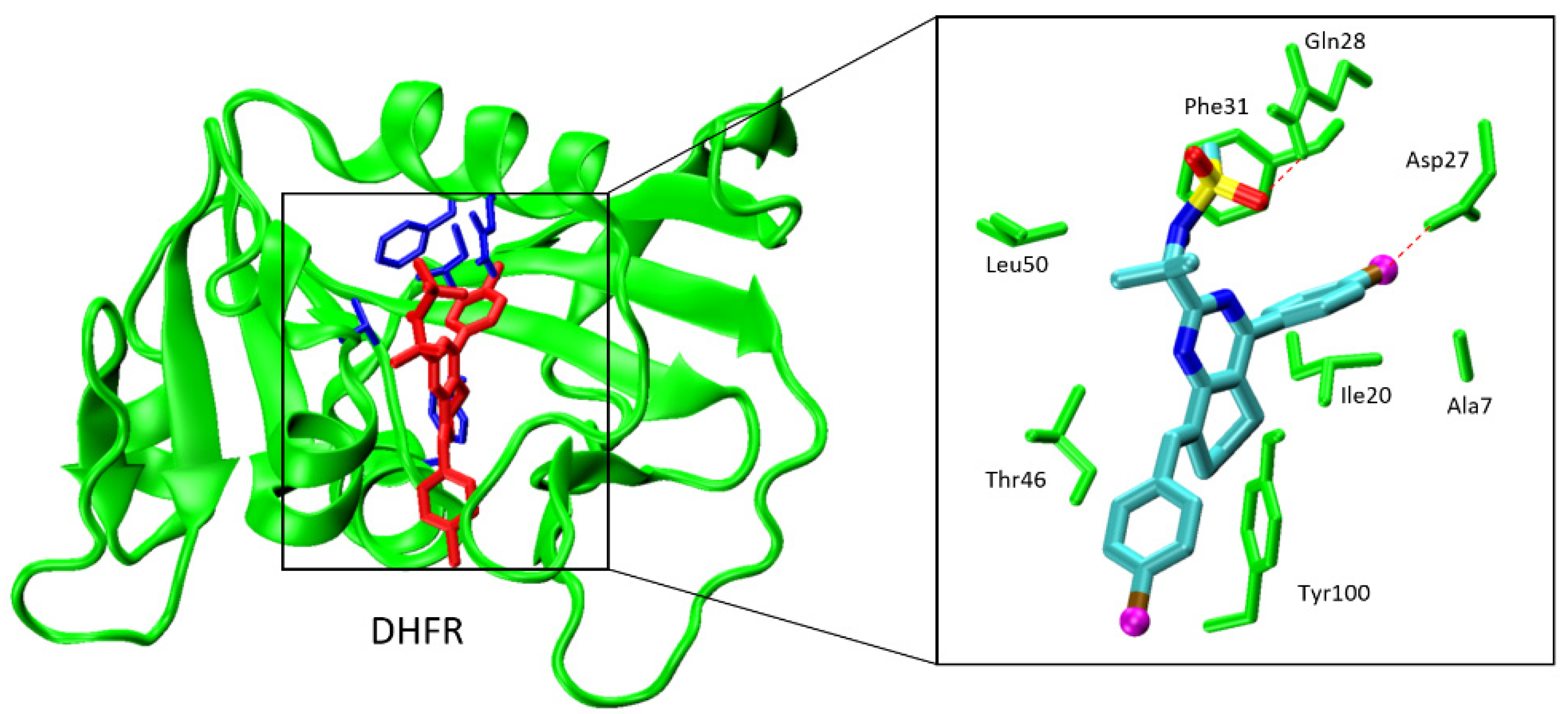
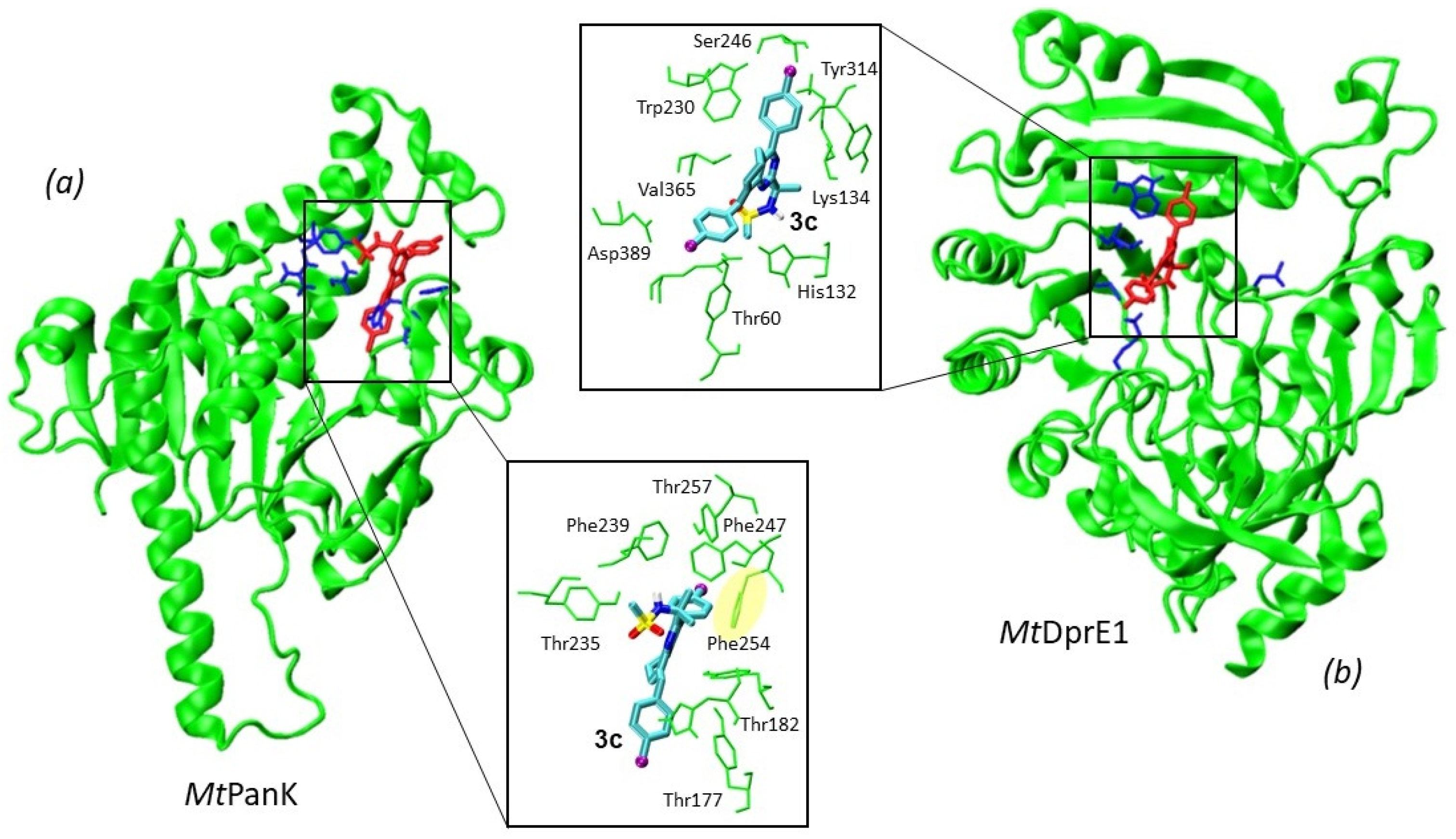
2.2.1. Molecular Docking against Mycobacterium tuberculosis Enzymes
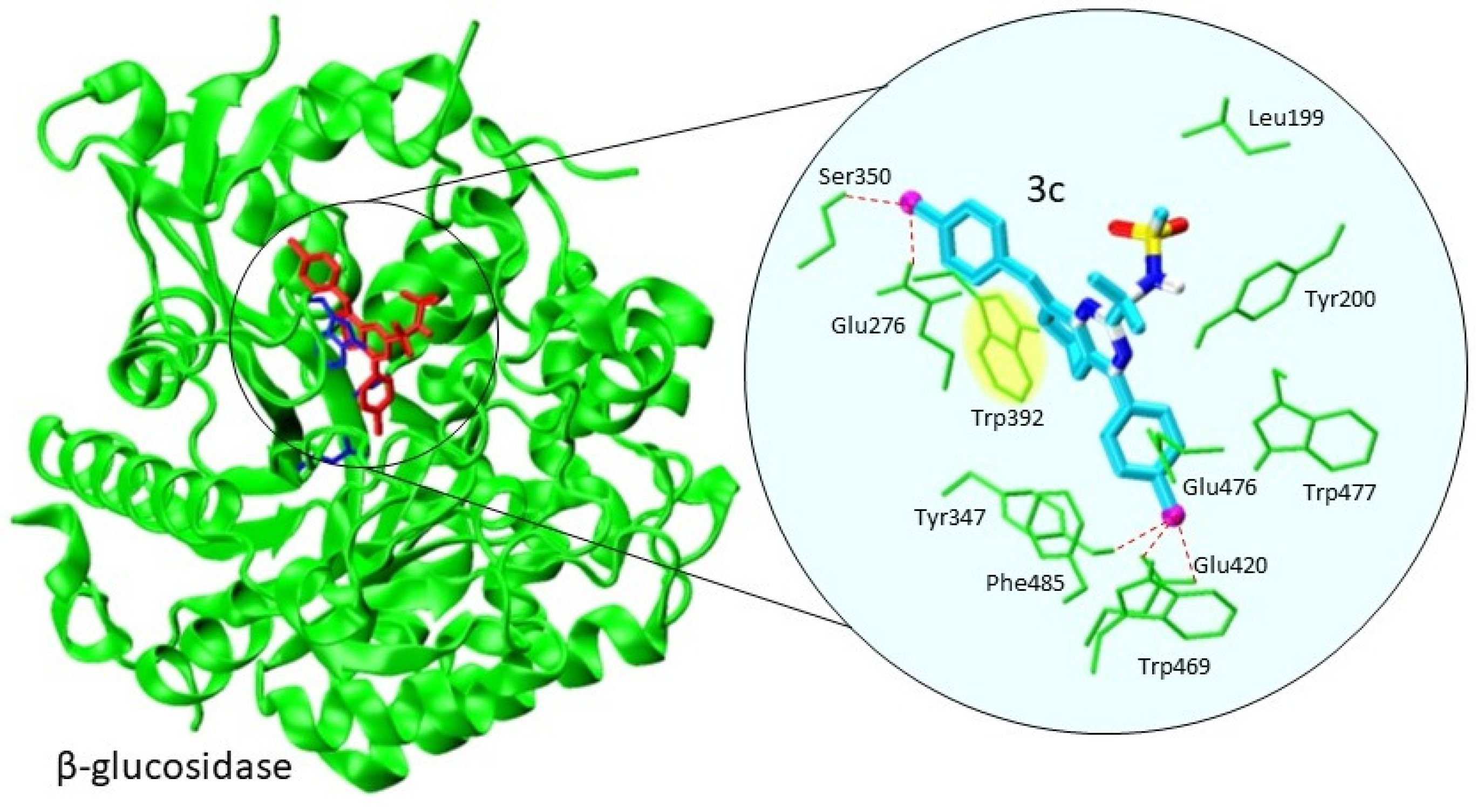
2.2.2. Molecular Docking against β-Glucosidase
3. Materials and Methods
3.1. General Procedure for the Preparation of Protected 2-(5,6,7,8-Tetrahydroquinazolin-2-yl)propan-2-amines 3a-g
3.1.1. N-(2-(8-Benzylidene-4-phenyl-5,6,7,8-tetrahydroquinazolin-2-yl)propan-2-yl)methanesulfonamide (3a)
3.1.2. N-(2-(8-(4-Methoxybenzylidene)-4-(4-methoxyphenyl)-5,6,7,8-tetrahydroquinazolin-2-yl)propan-2-yl)methanesulfonamide (3b)
3.1.3. N-(2-(8-(4-Chlorobenzylidene)-4-(4-chloromethoxyphenyl)-5,6,7,8-tetrahydroquinazolin-2-yl)propan-2-yl)methanesulfonamide (3c)
3.1.4. N-(2-(8-(4-Nitrobenzylidene)-4-(4-nitrophenyl)-5,6,7,8-tetrahydroquinazolin-2-yl)propan-2-yl)methanesulfonamide (3d)
3.1.5. tert-Butyl-(2-(8-benzylidene-4-phenyl-5,6,7,8-tetrahydroquinazolin-2-yl)butan2-yl)carbamate (3e)
3.1.6. tert-Butyl-(2-(8-(4-methoxybenzylidene)-4-(4-methoxyphenyl)-5,6,7,8tetrahydroquinazolin-2-yl)butan-2-yl)carbamate (3f)
3.1.7. tert-Butyl-(2-(8-(4-chlorobenzylidene)-4-(4-chlorophenyl)-5,6,7,8tetrahydroquinazolin-2-yl)butan-2-yl)carbamate (3g)
3.2. General Procedure for the Preparation of 2-(8-Aryliden-4-aryl-5,6,7,8-tetrahydroquinazolin-2-yl)butan-2-amine hydrochloride 4e-g
3.2.1. 2-(8-(4-Benzylidene)-4-(4-phenyl)-5,6,7,8-tetrahydroquinazolin-2-yl)butan-2-amine hydrochloride (4e)
3.2.2. 2-(8-(4-Methoxybenzylidene)-4-(4-methoxyphenyl)-5,6,7,8-tetrahydroquinazolin-2-yl)butan-2-amine hydrochloride (4f)
3.2.3. 2-(8-(4-Chlorobenzylidene)-4-(4-chlorophenyl)-5,6,7,8-tetrahydroquinazolin-2-yl)butan-2-amine hydrochloride (4g)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Foster, B.A.; Coffey, H.A.; Morin, M.J.; Rastinejad, F. Pharmacological rescue of mutant p53 conformation and function. Science 1999, 286, 2507. [Google Scholar] [CrossRef] [Green Version]
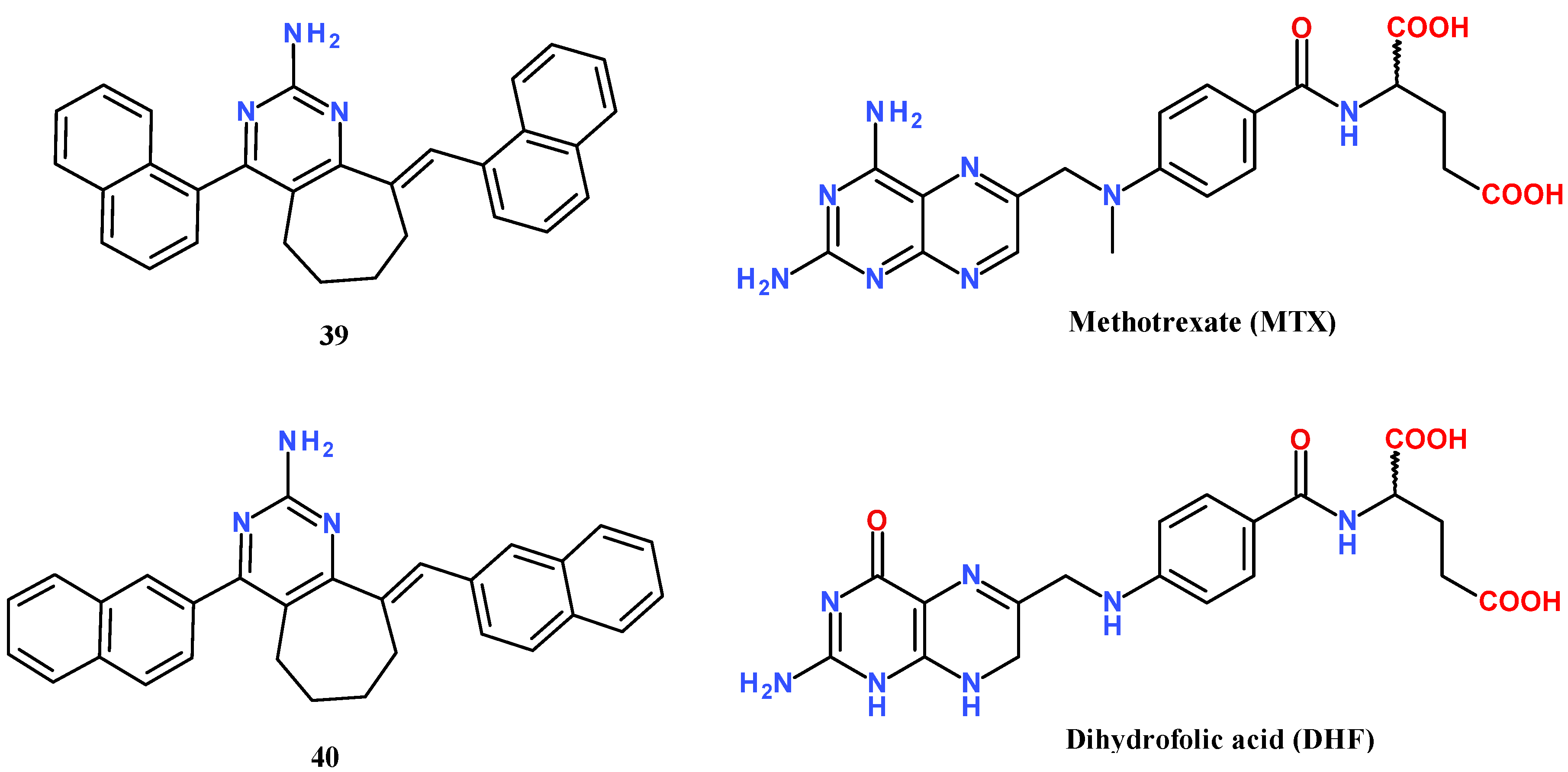
- Henderson, E.A.; Bavetsias, V.; Theti, D.S.; Wilson, S.C.; Clauss, R.; Jackman, A.L. Targeting the α-folate receptor with cyclopenta[g]quinazoline-based inhibitors of thymidylate synthase. Bioorg. Med. Chem. 2006, 14, 5020–5042. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Pasam, V.R.; Thakur, R.K.; Singh, M.; Singh, K.; Shukla, M.; Yadav, A.; Dogra, S.; Sona, C.; Umrao, D.; et al. Novel tetrahydroquinazolinamines as selective histamine 3 receptor antagonists for the treatment of obesity. J. Med. Chem. 2019, 62, 4638–4655. [Google Scholar] [CrossRef] [PubMed]
- Ortega, J.A.; Arencibia, J.M.; Minniti, E.; Byl, J.A.W.; Franco-Ulloa, S.; Borgogno, M.; Genna, V.; Summa, M.; Bertozzi, S.M.; Bertorelli, R.; et al. Novel, potent, and druglike tetrahydroquinazoline inhibitor that is highly selective for human topoisomerase ii α over β. J. Med. Chem. 2020, 63, 12873–12886. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Ma, S. Recent advances in the research of heterocyclic compounds as antitubercular agents. ChemMedChem 2012, 7, 2063–2075. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Pandey, S.K.; Anand, N.; Dwivedi, R.; Singh, S.; Sinha, S.K.; Chaturvedi, V.; Jaiswal, N.; Srivastava, A.K.; Shah, P.; et al. Synthesis, molecular modeling and bio-evaluation of cycloalkyl fused 2-aminopyrimidines as antitubercular and antidiabetic agents. Bioorg. Med. Chem. Lett. 2011, 21, 4404–4408. [Google Scholar] [CrossRef]
- El-Subbagh, H.I.; Hassan, G.S.; El-Messery, S.M.; Al-Rashood, S.T.; Al-Omary, F.A.M.; Abulfadl, Y.S.; Shabayek, M.I. Nonclassical antifolates, part 5. Benzodiazepine analogs as a new class of dhfr inhibitors: Synthesis, antitumor testing and molecular modeling study. Eur. J. Med. Chem. 2014, 74, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Rong, L.; Han, H.; Wang, H.; Jiang, H.; Tu, S.; Shi, D. An efficient and facile synthesis of pyrimidine and quinazoline derivatives via one-pot three-component reaction under solvent-free conditions. J. Heterocycl. Chem. 2009, 46, 152–157. [Google Scholar] [CrossRef]
- Zakeri, M.; Nasef, M.M.; Abouzari-Lotf, E. Eco-safe and expeditious approaches for synthesis of quinazoline and pyrimidine-2-amine derivatives using ionic liquids aided with ultrasound or microwave irradiation. J. Mol. Liq. 2014, 199, 267–274. [Google Scholar] [CrossRef]
- Al-Omary, F.A.M.; Hassan, G.S.; El-Messery, S.M.; El-Subbagh, H.I. Substituted thiazoles v. Synthesis and antitumor activity of novel thiazolo[2,3-b]quinazoline and pyrido[4,3-d]thiazolo[3,2-a]pyrimidine analogues. Eur. J. Med. Chem. 2012, 47, 65–72. [Google Scholar] [CrossRef]
- Gao, Q.; Wu, M.; Zhang, K.; Yang, N.; Liu, M.; Li, J.; Fang, L.; Bai, S.; Xu, Y. I2-catalyzed aerobic α,β-dehydrogenation and deamination of tertiary alkylamines: Highly selective synthesis of polysubstituted pyrimidines via hidden acyclic enamines. Org. Lett. 2020, 22, 5645–5649. [Google Scholar] [CrossRef] [PubMed]
- Rashid, H.u.; Martines, M.A.U.; Duarte, A.P.; Jorge, J.; Rasool, S.; Muhammad, R.; Ahmad, N.; Umar, M.N. Research developments in the syntheses, anti-inflammatory activities and structure–activity relationships of pyrimidines. RSC. Adv. 2021, 11, 6060–6098. [Google Scholar] [CrossRef]
- Guo, W.; Li, C.; Liao, J.; Ji, F.; Liu, D.; Wu, W.; Jiang, H. Transition metal free intermolecular direct oxidative C–N bond formation to polysubstituted pyrimidines using molecular oxygen as the sole oxidant. J. Org. Chem. 2016, 81, 5538–5546. [Google Scholar] [CrossRef]
- Zhan, J.-L.; Wu, M.-W.; Chen, F.; Han, B. Cu-catalyzed [3 + 3] annulation for the synthesis of pyrimidines via β-C(sp3)–H functionalization of saturated ketones. J. Org. Chem. 2016, 81, 11994–12000. [Google Scholar] [CrossRef]
- Chu, X.-Q.; Cao, W.-B.; Xu, X.-P.; Ji, S.-J. Iron catalysis for modular pyrimidine synthesis through β-ammoniation/cyclization of saturated carbonyl compounds with amidines. J. Org. Chem. 2017, 82, 1145–1154. [Google Scholar] [CrossRef]
- Kolomoitsev, O.O.; Gladkov, E.S.; Kotlyar, V.M.; Pedan, P.I.; Onipko, O.V.; Buravov, O.V.; Chebanov, V.A. Efficient synthesis of imidazole and pyrimidine derivatives. Chem. Heterocycl. Compd. 2020, 56, 1329–1334. [Google Scholar] [CrossRef]
- Lou, Z.; Zhang, X. Protein targets for structure-based anti-mycobacterium tuberculosis drug discovery. Protein Cell 2010, 1, 435–442. [Google Scholar] [CrossRef] [Green Version]
- Jackson, M.; McNeil, M.R.; Brennan, P.J. Progress in targeting cell envelope biogenesis in Mycobacterium Tuberculosis. Future Microbiol. 2013, 8, 855–875. [Google Scholar] [CrossRef] [Green Version]
- Mdluli, K.; Kaneko, T.; Upton, A. Tuberculosis drug discovery and emerging targets. Ann. N. Y. Acad. Sci. 2014, 1323, 56–75. [Google Scholar] [CrossRef]
- Batt, S.M.; Jabeen, T.; Bhowruth, V.; Quill, L.; Lund, P.A.; Eggeling, L.; Alderwick, L.J.; Fütterer, K.; Besra, G.S. Structural basis of inhibition of Mycobacterium Tuberculosis dpre1 by benzothiazinone inhibitors. Proc. Natl. Acad. Sci. USA 2012, 109, 11354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodsell, D.S.; Morris, G.M.; Olson, A.J. Automated docking of flexible ligands: Applications of autodock. J. Mol. Recognit. 1996, 9, 1–5. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. Autodock vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.T.; Blicharska, N.; Shilpi, J.A.; Seidel, V. Investigation of the anti-TB potential of selected propolis constituents using a molecular docking approach. Sci. Rep. 2018, 8, 12238. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, B.; Fu, L.; Guo, K.; Ma, C.; Wang, B.; Lin, Z.; Li, G.; Huang, H.; Lu, Y. Identification of novel benzothiopyranones with ester and amide motifs derived from active metabolite as promising leads against Mycobacterium Tuberculosis. Eur. J. Med. Chem. 2021, 222, 113603. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.; Wang, Y.; Chang, Z.; Yang, Y.; Pu, J.; Sun, T.; Kaur, S.; Sacchettini, J.C.; Jung, H.; Lin Wong, W.; et al. The identification of novel Mycobacterium Tuberculosis DHFR inhibitors and the investigation of their binding preferences by using molecular modelling. Sci. Rep. 2015, 5, 15328. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, M.V.; Randazzo, O.; La Franca, M.; Barone, G.; Vignoni, E.; Rossi, D.; Collina, S. Dhfr inhibitors: Reading the past for discovering novel anticancer agents. Molecules 2019, 24, 1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, K.; Neshat, N.; Sharma, S.; Giri, N.; Srivastava, A.; Almalki, F.; Saifullah, K.; Alam, M.M.; Shaquiquzzaman, M.; Akhter, M. Identification of novel selective Mtb-DHFR inhibitors as antitubercular agents through structure-based computational techniques. Arch. Pharm. 2020, 353, 1900287. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Sirawaraporn, R.; Chitnumsub, P.; Sirawaraporn, W.; Wooden, J.; Athappilly, F.; Turley, S.; Hol, W.G.J. Three-dimensional structure of M. Tuberculosis dihydrofolate reductase reveals opportunities for the design of novel tuberculosis drugs. J. Mol. Biol. 2000, 295, 307–323. [Google Scholar] [CrossRef] [Green Version]
- Wróbel, A.; Baradyn, M.; Ratkiewicz, A.; Drozdowska, D. Synthesis, biological activity, and molecular dynamics study of novel series of a trimethoprim analogs as multi-targeted compounds: Dihydrofolate reductase (DHFR) inhibitors and DNA-binding agents. Int. J. Mol. Sci. 2021, 22, 3685. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, I.; Loharch, S.; Gupta, P.; Madathil, R.; Parkesh, R. Structure, dynamics, and interaction of Mycobacterium Tuberculosis (Mtb) DprE1 and DprE2 examined by molecular modeling, simulation, and electrostatic studies. PLoS ONE 2015, 10, e0119771. [Google Scholar] [CrossRef]
- Makarov, V.; Lechartier, B.; Zhang, M.; Neres, J.; van der Sar, A.M.; Raadsen, S.A.; Hartkoorn, R.C.; Ryabova, O.B.; Vocat, A.; Decosterd, L.A.; et al. Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol. Med. 2014, 6, 372–383. [Google Scholar] [CrossRef]
- Borges de Melo, E.; da Silveira Gomes, A.; Carvalho, I. A- and β-glucosidase inhibitors: Chemical structure and biological activity. Tetrahedron 2006, 62, 10277–10302. [Google Scholar] [CrossRef]
- Li, T.; Guo, L.; Zhang, Y.; Wang, J.; Zhang, Z.; Li, J.; Zhang, W.; Lin, J.; Zhao, W.; Wang, P.G. Structure–activity relationships in a series of C2-substituted gluco-configured tetrahydroimidazopyridines as β-glucosidase inhibitors. Bioorg. Med. Chem. 2011, 19, 2136–2144. [Google Scholar] [CrossRef]
- Michelin, K.; Wajner, A.; Goulart, L.d.S.; Fachel, Â.A.; Pereira, M.L.S.; de Mello, A.S.; Souza, F.T.S.; Pires, R.F.; Giugliani, R.; Coelho, J.C. Biochemical study on β-glucosidase in individuals with gaucher’s disease and normal subjects. Clinica Chim. Acta 2004, 343, 145–153. [Google Scholar] [CrossRef]
- Chen, G.-Y.; Zhang, H.; Yang, F.-Q. A simple and portable method for β-glucosidase activity assay and its inhibitor screening based on a personal glucose meter. Analyt. Chim. Acta 2021, 1142, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Kazmi, M.; Zaib, S.; Ibrar, A.; Amjad, S.T.; Shafique, Z.; Mehsud, S.; Saeed, A.; Iqbal, J.; Khan, I. A new entry into the portfolio of α-glucosidase inhibitors as potent therapeutics for type 2 diabetes: Design, bioevaluation and one-pot multi-component synthesis of diamine-bridged coumarinyl oxadiazole conjugates. Bioorg. Chem. 2018, 77, 190–202. [Google Scholar] [CrossRef]
- Riaz, S.; Khan, I.U.; Yar, M.; Ashraf, M.; Rehman, T.U.; Shaukat, A.; Jamal, S.B.; Duarte, V.C.M.; Alves, M.J. Novel pyridine-2,4,6-tricarbohydrazide derivatives: Design, synthesis, characterization and in vitro biological evaluation as α- and β-glucosidase inhibitors. Bioorg. Chem. 2014, 57, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Parizadeh, H.; Garampalli, R.H. Evaluation of some lichen extracts for β-glucosidase inhibitory as a possible source of herbal anti-diabetic drugs. Am. J. Biochem. 2016, 6, 46–50. [Google Scholar] [CrossRef]
- Chu, C.; Deng, J.; Man, Y.; Qu, Y. Green tea extracts epigallocatechin-3-gallate for different treatments. BioMed Res. Intern. 2017, 2017, 5615647. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.G.; Liu, J.; Zeng, P.L.; Dong, Z.B. Synthesis of α, α′-bis(substituted benzylidene)ketones catalysed by a SOCl2/EtOH reagent. J. Chem. Res. 2004, 2004, 55–56. [Google Scholar] [CrossRef]
- Lüth, A.; Löwe, W. Syntheses of 4-(indole-3-yl)quinazolines—A new class of epidermal growth factor receptor tyrosine kinase inhibitors. Eur. J. Med. Chem. 2008, 43, 1478–1488. [Google Scholar] [CrossRef]
- Gundla, R.; Kazemi, R.; Sanam, R.; Muttineni, R.; Sarma, J.A.R.P.; Dayam, R.; Neamati, N. Discovery of novel small-molecule inhibitors of human epidermal growth factor receptor-2: Combined ligand and target-based approach. J. Med. Chem. 2008, 51, 3367–3377. [Google Scholar] [CrossRef] [PubMed]

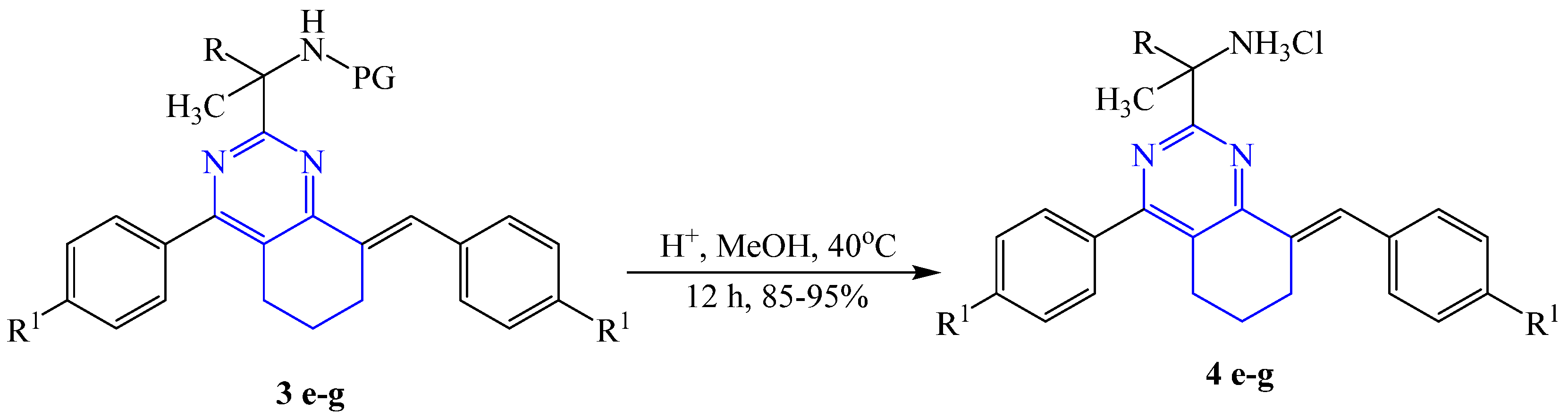


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Protecting Group (PG) | R | R1 | Yields, % |
|---|---|---|---|---|
| 1a | Ms | Me | - | - |
| 1e | Boc | Et | - | - |
| 2a | - | - | H | - |
| 2b | - | - | OCH3 | - |
| 2c | - | - | Cl | - |
| 2d | - | - | NO2 | - |
| 3a | Ms | Me | H | 70 |
| 3b | Ms | Me | OCH3 | 65 |
| 3c | Ms | Me | Cl | 50 |
| 3d | Ms | Me | NO2 | 80 |
| 3e | Boc | Et | H | 57 |
| 3f | Boc | Et | OCH3 | 47 |
| 3g | Boc | Et | Cl | 54 |
| 4e | H | Et | H | 88 |
| 4f | H | Et | OCH3 | 92 |
| 4g | H | Et | Cl | 95 |
| Compounds | Mw (g/mol) 1 | Docking Binding Energy (kcal/mol) | logP 2 | |||
|---|---|---|---|---|---|---|
| Mycobacterium Tuberculosis DHFR (PDB 1DF7) | Pantothenate Kinase (MtPanK) (PDB 4BFT) | FAD-Containing Oxidoreductase (MtDprE1) (PDB 4FF6) | β-Glucosidase (PDB 4A3Y) | |||
| studied ligands | ||||||
| 3a | 433.6 | −9.3 | −9.7 | −10.6 | −10.3 | 4.61 |
| 3b | 493.6 | −9.0 | −9.4 | −10.9 | −10.7 | 4.47 |
| 3c | 523.6 | −9.6 | −9.6 | −10.9 | −10.9 | 5.82 |
| 3d | 502.5 | −9.4 | −9.4 | −10.4 | −11.1 | 2.77 |
| 4e | 369.5 | −9.1 | −9.1 | −9.1 | −10.6 | 4.78 |
| 4f | 429.6 | −9.3 | −8.8 | −9.1 | −10.0 | 4.64 |
| 4g | 438.4 | −9.7 | −9.6 | −8.5 | −10.3 | 5.99 |
| reference inhibitors | ||||||
| 39 from [6] | 427.6 | −11.2 | −10.7 | -3 | −12.5 | 6.98 |
| 40 from [6] | 427.6 | −12.2 | −11.0 | −11.1 | −12.5 | 6.98 |
| methotrexate (MTX) | 454.5 | −9.0 | −8.5 | −9.4 | −8.7 | −1.23 |
| dihydrofolic acid (DHF) | 443.4 | −9.5 | −9.2 | −9.7 | −9.1 | −2.16 |
| macozinone (PBTZ169) | 456.5 | −10.3 | −8.8 | −9.8 | −9.2 | 4.42 |
| M-1 from [24] | 471.5 | −10.4 | −9.0 | −9.8 | −9.5 | 3.56 |
| TBA-7371 | 355.4 | −8.3 | −7.6 | −8.4 | −8.4 | 1.31 |
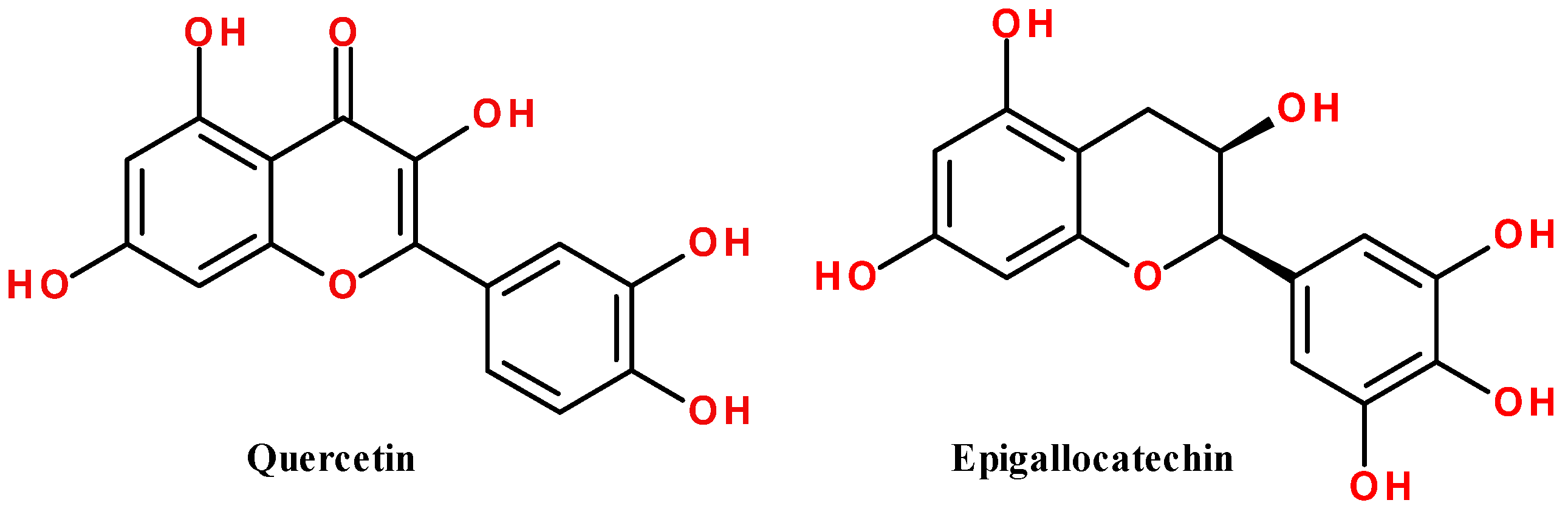
| quercetin | 302.2 | −8.4 | −8.3 | −9.1 | −9.2 | 0.35 |
| epigallocatechin (EGC) | 306.3 | −8.1 | −7.8 | −8.1 | −8.8 | 1.11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Snizhko, A.D.; Kyrychenko, A.V.; Gladkov, E.S. Synthesis of Novel Derivatives of 5,6,7,8-Tetrahydroquinazolines Using α-Aminoamidines and In Silico Screening of Their Biological Activity. Int. J. Mol. Sci. 2022, 23, 3781. https://doi.org/10.3390/ijms23073781
Snizhko AD, Kyrychenko AV, Gladkov ES. Synthesis of Novel Derivatives of 5,6,7,8-Tetrahydroquinazolines Using α-Aminoamidines and In Silico Screening of Their Biological Activity. International Journal of Molecular Sciences. 2022; 23(7):3781. https://doi.org/10.3390/ijms23073781
Chicago/Turabian StyleSnizhko, Arsenii D., Alexander V. Kyrychenko, and Eugene S. Gladkov. 2022. "Synthesis of Novel Derivatives of 5,6,7,8-Tetrahydroquinazolines Using α-Aminoamidines and In Silico Screening of Their Biological Activity" International Journal of Molecular Sciences 23, no. 7: 3781. https://doi.org/10.3390/ijms23073781
APA StyleSnizhko, A. D., Kyrychenko, A. V., & Gladkov, E. S. (2022). Synthesis of Novel Derivatives of 5,6,7,8-Tetrahydroquinazolines Using α-Aminoamidines and In Silico Screening of Their Biological Activity. International Journal of Molecular Sciences, 23(7), 3781. https://doi.org/10.3390/ijms23073781