
Generative Adversarial Networks for Creating Synthetic Nucleic Acid Sequences of Cat Genome



Abstract
:1. Introduction
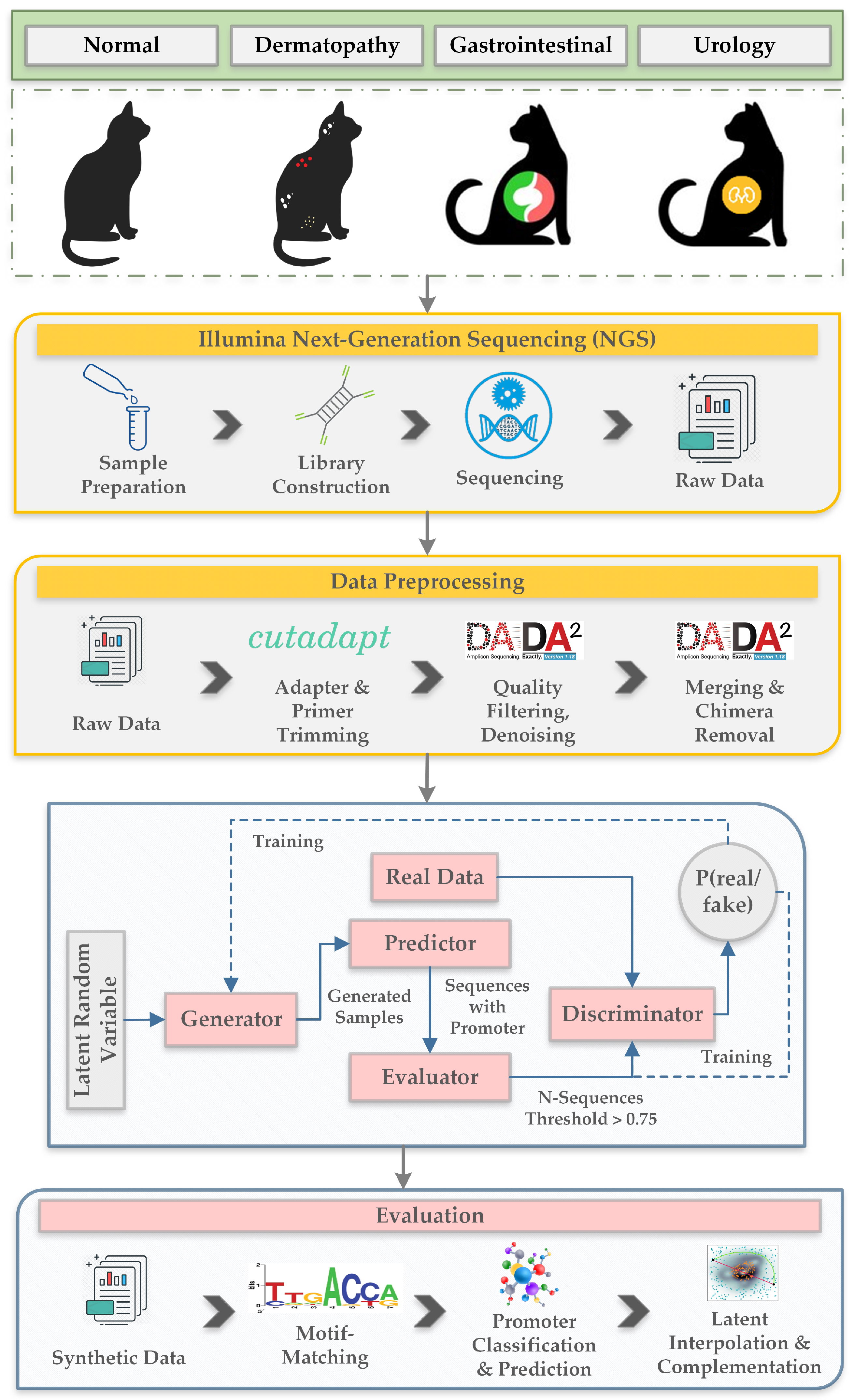
- We prepared a new dataset consisting of fecal samples collected from eight cats belonging to four categories according to their health condition. The categories are normal, dermatopathy, gastrointestinal and urology.
- The advantage of the proposed model is that it includes two separate networks in the GAN architecture itself. We propose a GAN model consisting of a predictor and an evaluator to generate synthetic nucleic acid sequences tuned to acquire desired properties of a DNA sequence. The inclusion of the predictor and the evaluator contributes to the generation of realistic samples more efficiently than existing GAN models.
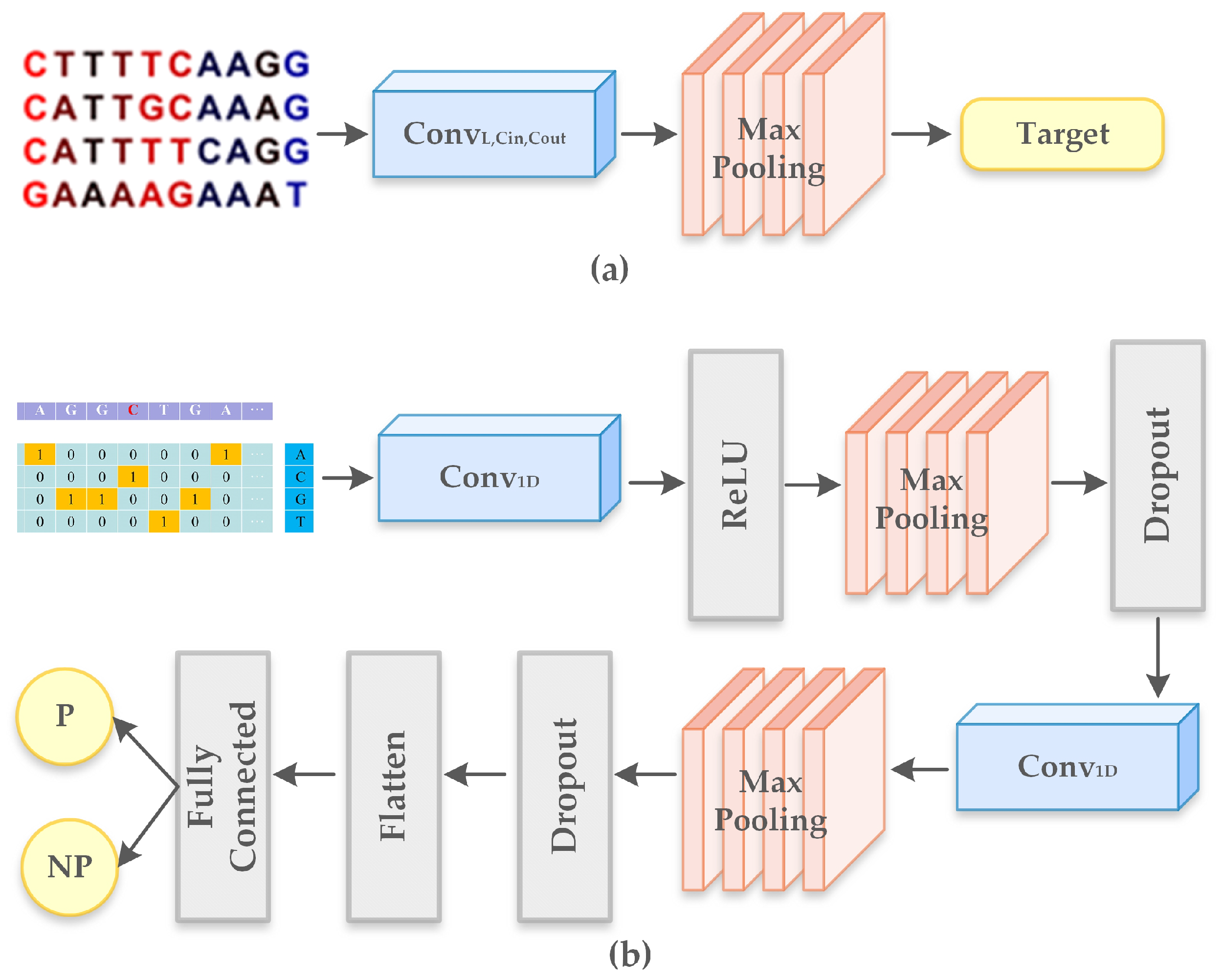
- We built a predictor that is trained to classify and recognize promoter sequences in the generated synthetic sequences. This helps to tune the sequences to have properties of the promoter.
- We also implemented an evaluator that matches motif sequences in the generated synthetic data. This model helps in tuning the synthetic sequences to exhibit properties of a motif.
- Our proposed model achieves a mean correlation coefficient of 93.7%, 2% more than our previously proposed TGAN-skip-WGAN-GP model and 6–10% more than CWGAN-GP, TGAN-WGAN-GP, and TGAN-skip models.
2. Proposed Methodology
2.1. Dataset
- Innovative sample preparation
- Rapid sequencing of whole genomes
- Targeted sequencing
- RNA sequencing that eliminates the expense and inefficiency of traditional technologies
- Studying tumor subclones and somatic variants through cancer sample sequencing
- Epigenetic factor analysis such as DNA-protein interactions
- Human microbiome study and novel pathogen identification.
- Sample ID: Sample name.
- Total read bases: Total number of bases sequenced.
- Total reads: Total number of reads. For Illumina paired-end sequencing, this value refers to the sum of read 1 and read 2 (Paired-end).
- GC(%): GC content.
- AT(%): AT content.
- Q20(%): Ratio of bases that have phred quality score of over 20.
- Q30(%): Ratio of bases that have phred quality score of over 30.
2.2. Proposed Generative Adversarial Networks Model for Creating Synthetic Nucleic Acid Sequences
2.2.1. Data Preprocessing
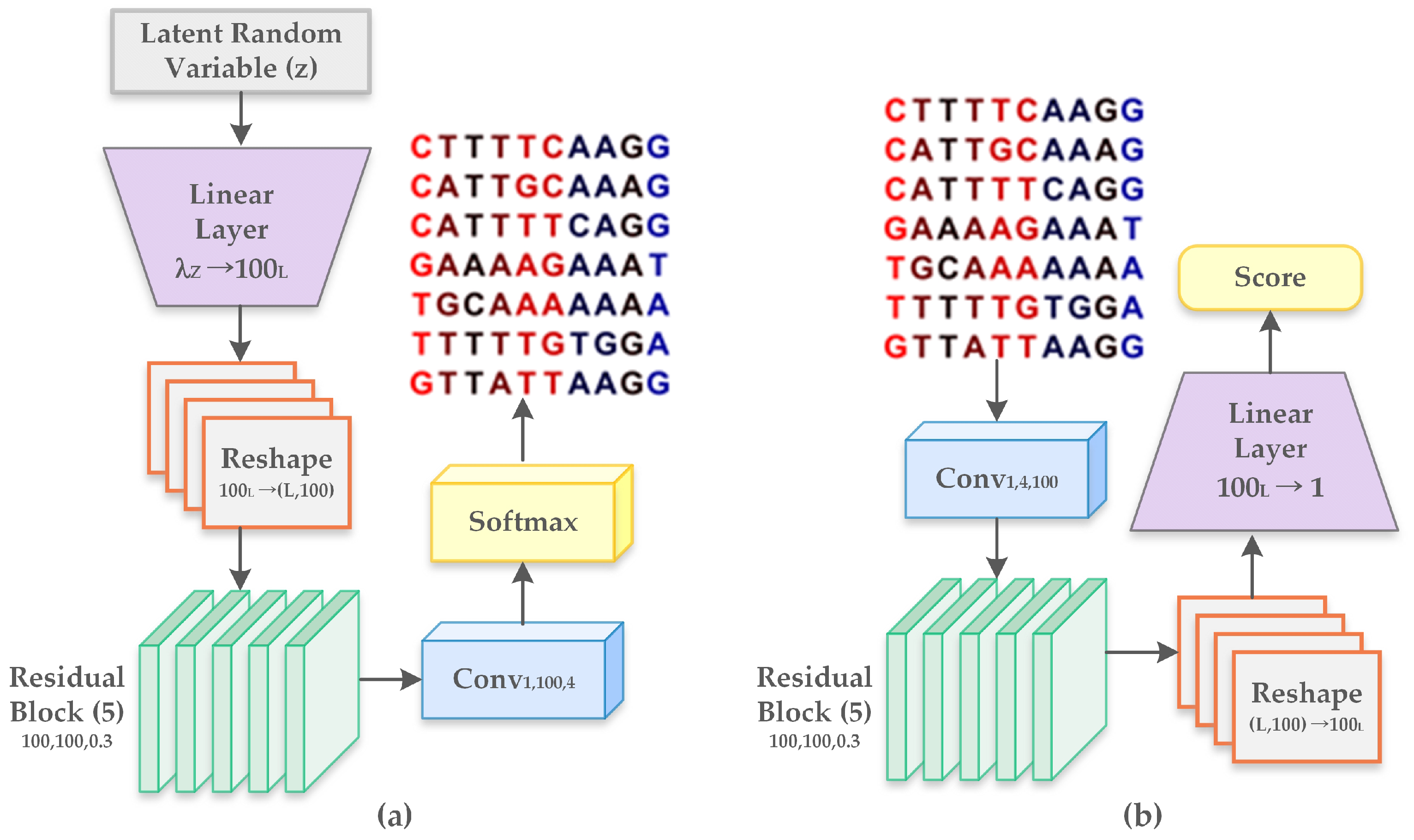
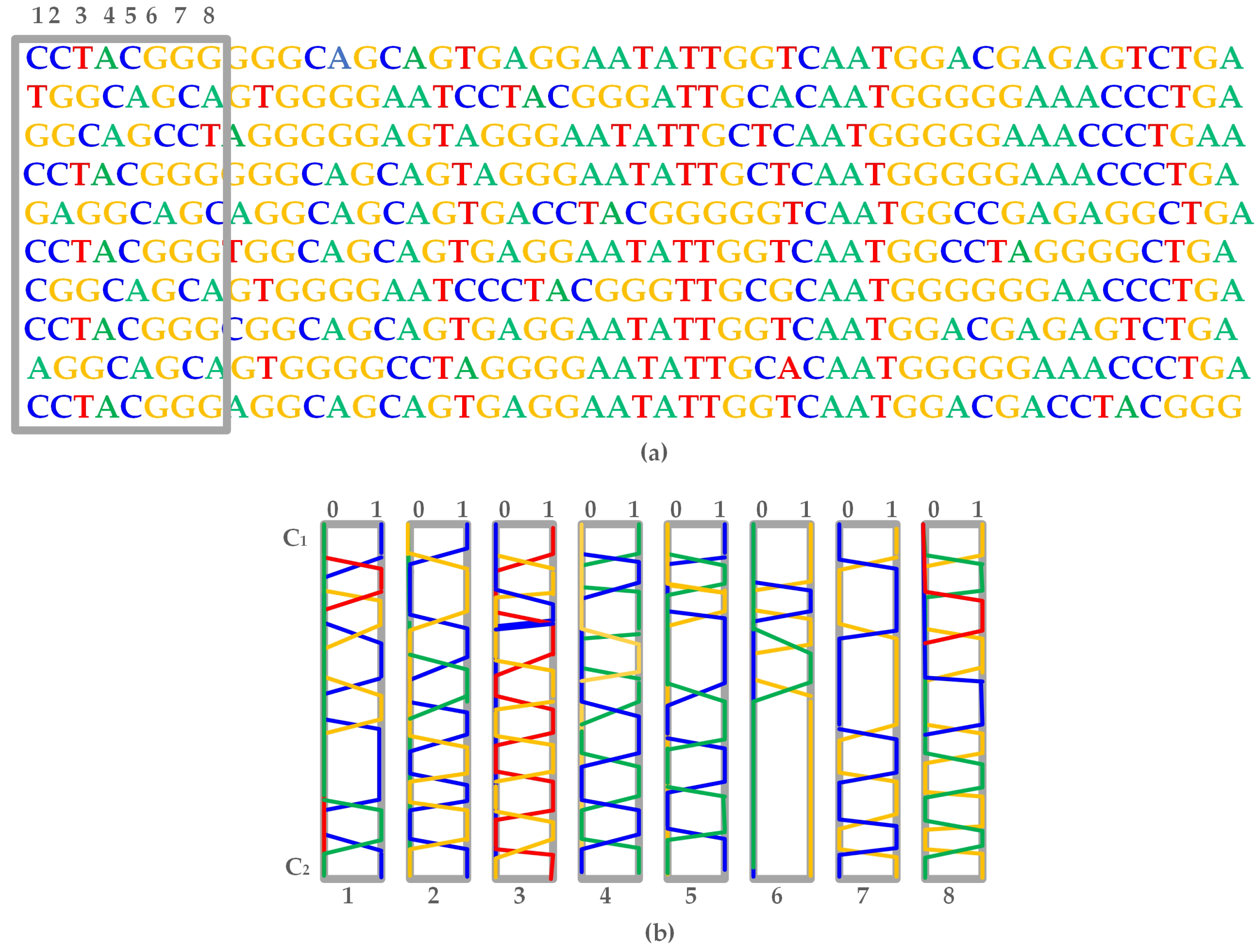
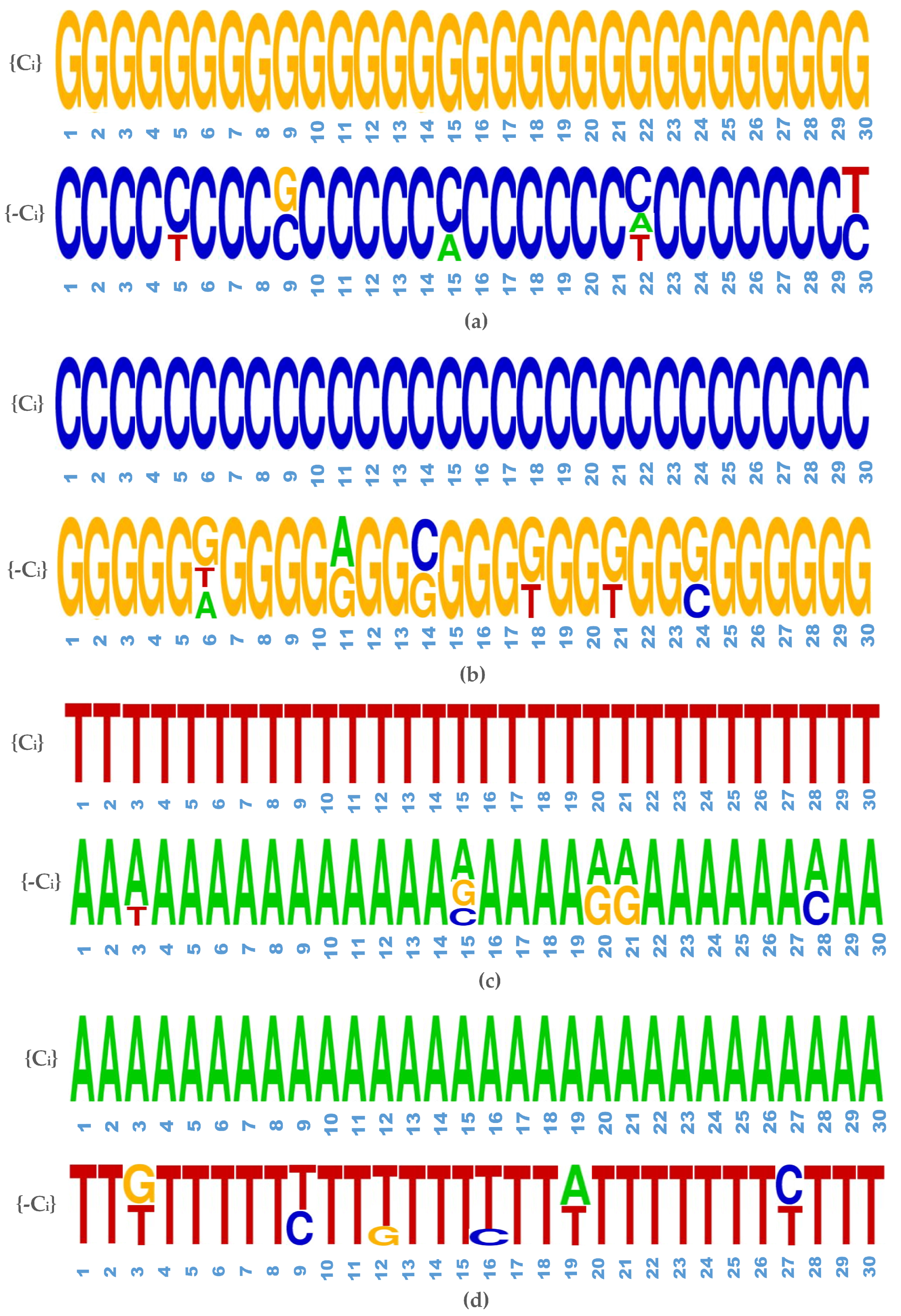
2.2.2. Generative Adversarial Networks
- Linear layer : It represents the multiplication by a weight matrix of shape and includes a bias parameter of dimension .
- Reshape : Altering shape of features from to .
- Residual block : It consists of 2 internal layers, with every layer comprising of a rectified linear unit (ReLU). It is followed by a transformation of one-dimensional convolution operation with filters of length L that is mapped from to channels. The output from the second layer is multiplied by and forwarded to the input of the residual block.
- Convolutional layer : Refers to one-dimensional convolution with filters of length L, mapped from to channels and followed by an inclusion of bias parameter of dimension .
- Softmax: Softmax is the final activation function representing four nucleotides with dimension four. It is applied to each position separately along the sequence length.
3. Results
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Griffin, H.G.; Griffin, A.M. DNA sequencing. Appl. Biochem. Biotechnol. 1993, 38, 147–159. [Google Scholar] [CrossRef]
- Church, G.M.; Gilbert, W. Genomic sequencing. Proc. Natl. Acad. Sci. USA 1984, 81, 1991–1995. [Google Scholar] [CrossRef] [Green Version]
- Nouws, S.; Bogaerts, B.; Verhaegen, B.; Denayer, S.; Piérard, D.; Marchal, K.; Roosens, N.H.; Vanneste, K.; De Keersmaecker, S.C. Impact of DNA extraction on whole genome sequencing analysis for characterization and relatedness of Shiga toxin-producing Escherichia coli isolates. Sci. Rep. 2020, 10, 14649. [Google Scholar] [CrossRef]
- Dias, R.; Torkamani, A. Artificial intelligence in clinical and genomic diagnostics. Genome Med. 2019, 11, 70. [Google Scholar] [CrossRef] [Green Version]
- Goodfellow, I.; Pouget-Abadie, J.; Mirza, M.; Xu, B.; Warde-Farley, D.; Ozair, S.; Courville, A.; Bengio, Y. Generative adversarial nets. Adv. Neural Inf. Process. Syst. 2014, 27. [Google Scholar] [CrossRef]
- Mirza, M.; Osindero, S. Conditional generative adversarial nets. arXiv 2014, arXiv:1411.1784. [Google Scholar]
- Denton, E.; Chintala, S.; Szlam, A.; Fergus, R. Deep generative image models using a laplacian pyramid of adversarial networks. arXiv 2015, arXiv:1506.05751. [Google Scholar]
- Isola, P.; Zhu, J.Y.; Zhou, T.; Efros, A.A. Image-to-image translation with conditional adversarial networks. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Honolulu, HI, USA, 21–26 July 2017; pp. 1125–1134. [Google Scholar]
- Zhu, J.Y.; Park, T.; Isola, P.; Efros, A.A. Unpaired image-to-image translation using cycle-consistent adversarial networks. In Proceedings of the IEEE International Conference on Computer Vision, Venice, Italy, 22–29 October 2017; pp. 2223–2232. [Google Scholar]
- Reed, S.; Akata, Z.; Yan, X.; Logeswaran, L.; Schiele, B.; Lee, H. Generative adversarial text to image synthesis. In Proceedings of the International Conference on Machine Learning, PMLR, New York, NY, USA, 20–22 June 2016; pp. 1060–1069. [Google Scholar]
- Zhang, H.; Xu, T.; Li, H.; Zhang, S.; Wang, X.; Huang, X.; Metaxas, D.N. Stackgan: Text to photo-realistic image synthesis with stacked generative adversarial networks. In Proceedings of the IEEE International Conference on Computer Vision, Venice, Italy, 22–29 October 2017; pp. 5907–5915. [Google Scholar]
- Choi, Y.; Choi, M.; Kim, M.; Ha, J.W.; Kim, S.; Choo, J. Stargan: Unified generative adversarial networks for multi-domain image-to-image translation. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Salt Lake City, UT, USA, 18–23 June 2018; pp. 8789–8797. [Google Scholar]
- Arjovsky, M.; Chintala, S.; Bottou, L. Wasserstein GAN. arXiv 2017, arXiv:stat.ML/1701.07875. [Google Scholar]
- Gulrajani, I.; Ahmed, F.; Arjovsky, M.; Dumoulin, V.; Courville, A. Improved training of wasserstein gans. arXiv 2017, arXiv:1704.00028. [Google Scholar]
- Larsen, A.B.L.; Sønderby, S.K.; Larochelle, H.; Winther, O. Autoencoding beyond pixels using a learned similarity metric. In Proceedings of the International Conference on Machine Learning, PMLR, New York, NY, USA, 20–22 June 2016; pp. 1558–1566. [Google Scholar]
- Munjal, P.; Paul, A.; Krishnan, N.C. Implicit discriminator in variational autoencoder. In Proceedings of the 2020 International Joint Conference on Neural Networks (IJCNN), Glasgow, UK, 19–24 July 2020; pp. 1–8. [Google Scholar]
- Kingma, D.P.; Welling, M. Auto-encoding variational bayes. arXiv 2013, arXiv:1312.6114. [Google Scholar]
- Chui, K.T.; Liu, R.W.; Zhao, M.; De Pablos, P.O. Predicting students’ performance with school and family tutoring using generative adversarial network-based deep support vector machine. IEEE Access 2020, 8, 86745–86752. [Google Scholar] [CrossRef]
- Li, Y.; Gan, Z.; Shen, Y.; Liu, J.; Cheng, Y.; Wu, Y.; Carin, L.; Carlson, D.; Gao, J. Storygan: A sequential conditional gan for story visualization. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, Long Beach, CA, USA, 16–20 June 2019; pp. 6329–6338. [Google Scholar]
- Kwon, Y.H.; Park, M.G. Predicting future frames using retrospective cycle gan. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, Long Beach, CA, USA, 16–20 June 2019; pp. 1811–1820. [Google Scholar]
- Rajeswar, S.; Subramanian, S.; Dutil, F.; Pal, C.; Courville, A. Adversarial generation of natural language. arXiv 2017, arXiv:1705.10929. [Google Scholar]
- Yu, L.; Zhang, W.; Wang, J.; Yu, Y. Seqgan: Sequence generative adversarial nets with policy gradient. In Proceedings of the AAAI Conference on Artificial Intelligence, San Francisco, CA, USA, 4–9 February 2017; Volume 31. [Google Scholar]
- Tuan, Y.L.; Lee, H.Y. Improving conditional sequence generative adversarial networks by stepwise evaluation. IEEE/ACM Trans. Audio Speech Lang. Process. 2019, 27, 788–798. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Zou, J. Feedback GAN for DNA optimizes protein functions. Nat. Mach. Intell. 2019, 1, 105–111. [Google Scholar] [CrossRef]
- Yelmen, B.; Decelle, A.; Ongaro, L.; Marnetto, D.; Tallec, C.; Montinaro, F.; Furtlehner, C.; Pagani, L.; Jay, F. Creating artificial human genomes using generative neural networks. PLoS Genet. 2021, 17, e1009303. [Google Scholar] [CrossRef]
- Yu, H.; Welch, J.D. MichiGAN: Sampling from disentangled representations of single-cell data using generative adversarial networks. Genome Biol. 2021, 22, 158. [Google Scholar] [CrossRef]
- Illumina, I. An Introduction to Next-Generation Sequencing Technology; Illumina, Inc.: San Diego, CA, USA, 2015. [Google Scholar]
- Alkhateeb, A.; Rueda, L. Zseq: An approach for preprocessing next-generation sequencing data. J. Comput. Biol. 2017, 24, 746–755. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High resolution sample inference from amplicon data. bioRxiv 2015, 024034. [Google Scholar] [CrossRef] [Green Version]
- Shujaat, M.; Wahab, A.; Tayara, H.; Chong, K.T. pcPromoter-CNN: A CNN-Based Prediction and Classification of Promoters. Genes 2020, 11, 1529. [Google Scholar] [CrossRef]
- Liu, B.; Yang, F.; Huang, D.S.; Chou, K.C. iPromoter-2L: A two-layer predictor for identifying promoters and their types by multi-window-based PseKNC. Bioinformatics 2018, 34, 33–40. [Google Scholar] [CrossRef]
- Zhang, M.; Li, F.; Marquez-Lago, T.T.; Leier, A.; Fan, C.; Kwoh, C.K.; Chou, K.C.; Song, J.; Jia, C. MULTiPly: A novel multi-layer predictor for discovering general and specific types of promoters. Bioinformatics 2019, 35, 2957–2965. [Google Scholar] [CrossRef]
- Xu, L.; Veeramachaneni, K. Synthesizing tabular data using generative adversarial networks. arXiv 2018, arXiv:1811.11264. [Google Scholar]
- Xu, L.; Skoularidou, M.; Cuesta-Infante, A.; Veeramachaneni, K. Modeling tabular data using conditional gan. arXiv 2019, arXiv:1907.00503. [Google Scholar]
- Brenninkmeijer, B.; de Vries, A.; Marchiori, E.; Hille, Y. On the Generation and Evaluation of Tabular Data Using GANs. Ph.D. Thesis, Radboud University, Nijmegen, The Netherlands, 2019. [Google Scholar]
- Wei, X.; Gong, B.; Liu, Z.; Lu, W.; Wang, L. Improving the improved training of wasserstein gans: A consistency term and its dual effect. arXiv 2018, arXiv:1803.01541. [Google Scholar]
- Zheng, M.; Li, T.; Zhu, R.; Tang, Y.; Tang, M.; Lin, L.; Ma, Z. Conditional Wasserstein generative adversarial network-gradient penalty-based approach to alleviating imbalanced data classification. Inf. Sci. 2020, 512, 1009–1023. [Google Scholar] [CrossRef]
- Hazra, D.; Byun, Y.C. Generating Synthetic Fermentation Data of Shindari, a Traditional Jeju Beverage, Using Multiple Imputation Ensemble and Generative Adversarial Networks. Appl. Sci. 2021, 11, 2787. [Google Scholar] [CrossRef]
- Mukaka, M.M. A guide to appropriate use of correlation coefficient in medical research. Malawi Med. J. 2012, 24, 69–71. [Google Scholar]
- Zhu, F.; Ye, F.; Fu, Y.; Liu, Q.; Shen, B. Electrocardiogram generation with a bidirectional LSTM-CNN generative adversarial network. Sci. Rep. 2019, 9, 6734. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Total Read Bases (bp) | Total Reads | GC (%) | AT (%) | Q20 (%) | Q30 (%) |
|---|---|---|---|---|---|---|
| drem_3 | 54,370,232 | 180,632 | 56.84 | 43.16 | 88.21 | 77.60 |
| drem_4 | 62,781,376 | 208,576 | 55.57 | 44.43 | 89.66 | 79.97 |
| gast_5 | 55,694,632 | 185,032 | 53.89 | 46.11 | 90.73 | 81.30 |
| gast_6 | 58,980,950 | 195,950 | 54.07 | 45.93 | 90.08 | 80.37 |
| norm_1 | 68,054,294 | 226,094 | 51.64 | 48.36 | 91.34 | 82.10 |
| norm_2 | 65,382,618 | 217,218 | 55.47 | 44.53 | 89.31 | 79.59 |
| urol_7 | 56,866,124 | 188,924 | 53.52 | 46.48 | 91.10 | 81.93 |
| urol_8 | 55,780,116 | 185,316 | 51.24 | 48.76 | 91.49 | 82.21 |
| Sample Name | Adapter & Primer Trimming | Quality Filter | DenoisedFor | DenoisedRev | MergedPair | Non-Chimeric |
|---|---|---|---|---|---|---|
| norm_1 | 112,078 | 91,286 | 90,092 | 90,575 | 86,784 | 66,605 |
| norm_2 | 107,715 | 84,245 | 82,726 | 83,449 | 78,310 | 59,086 |
| derm_3 | 89,403 | 68,194 | 67,967 | 68,075 | 67,408 | 63,361 |
| derm_4 | 103,322 | 81,146 | 79,791 | 80,394 | 74,674 | 56,874 |
| gast_5 | 91,749 | 74,340 | 72,938 | 73,598 | 70,158 | 62,708 |
| gast_6 | 97,252 | 76,664 | 75,368 | 75,863 | 71,881 | 52,800 |
| urol_7 | 93,669 | 76,908 | 76,833 | 76,855 | 76,702 | 75,019 |
| urol_8 | 91,949 | 75,438 | 75,150 | 75,197 | 74,187 | 66,993 |
| Correlation Values | Relation |
|---|---|
| 0 to 0.3 or 0 to −0.3 | Negligibly correlated |
| 0.3 to 0.5 or −0.3 to −0.5 | Low correlation |
| 0.5 to 0.7 or −0.5 to −0.7 | Moderately correlated |
| 0.7 to 0.9 or −0.7 to −0.9 | Highly correlated |
| 0.9 to 1 or −0.9 to 1 | Extensively correlated |
| Model | Mean Correlation Coefficient | RMSE | FD | MAE | Mirror Column Association | PRD |
|---|---|---|---|---|---|---|
| TGAN | 0.628 | 0.72 | 0.78 | 0.71 | 0.691 | 78.6 |
| CTGAN | 0.790 | 0.72 | 0.76 | 0.66 | 0.716 | 77.9 |
| TGAN-skip | 0.833 | 0.69 | 0.72 | 0.63 | 0.772 | 73.5 |
| TGAN-WGAN-GP | 0.863 | 0.66 | 0.68 | 0.58 | 0.783 | 71.0 |
| CWGAN-GP | 0.875 | 0.66 | 0.68 | 0.55 | 0.849 | 67.5 |
| TGAN-skip-WGAN-GP | 0.913 | 0.62 | 0.61 | 0.49 | 0.914 | 58.9 |
| Proposed Model | 0.937 | 0.57 | 0.58 | 0.44 | 0.939 | 53.1 |
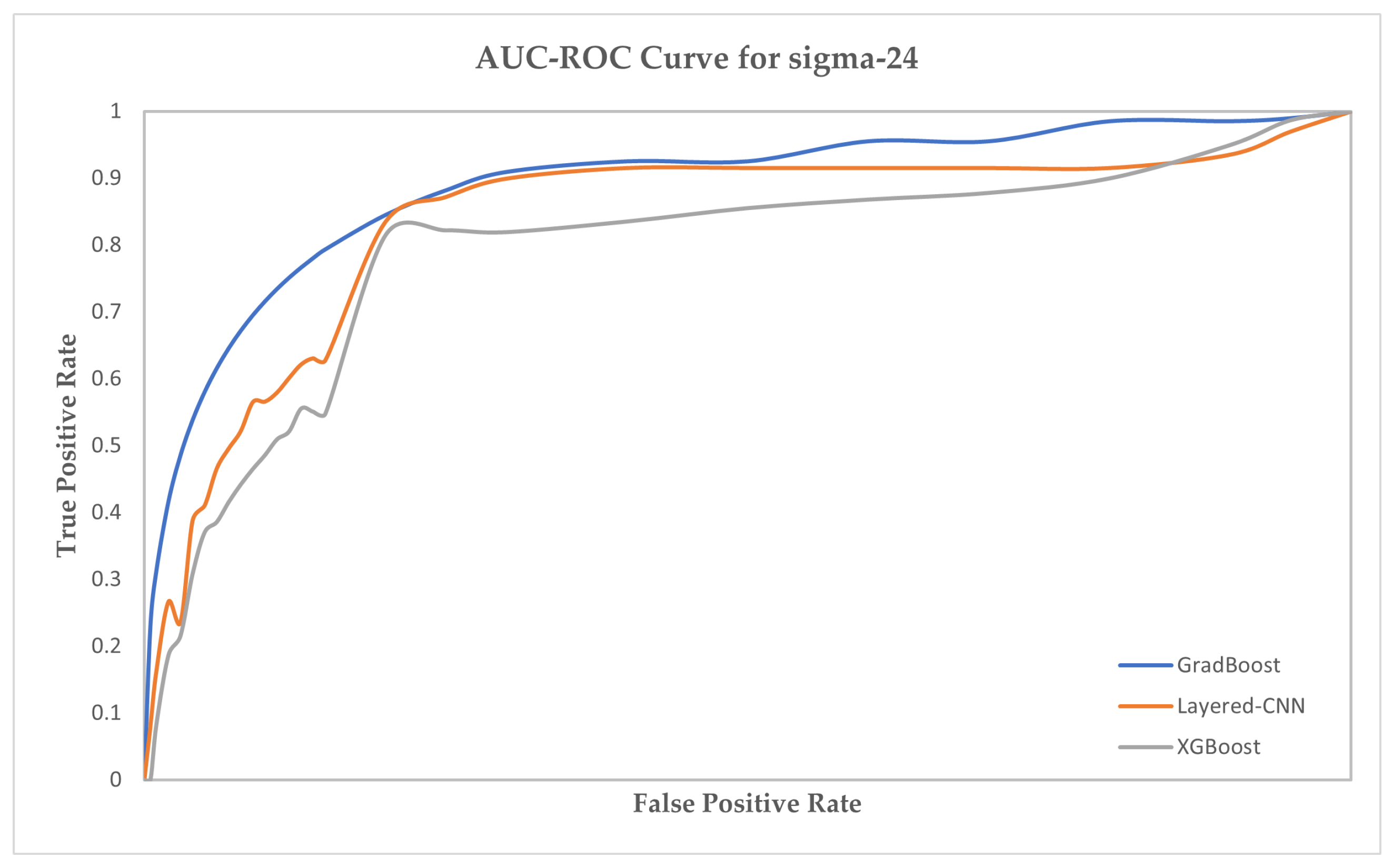
| sigma-24 | sigma-32 | sigma-54 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Model | Acc | MCC | Sen | Spc | Acc | MCC | Sen | Spc | Acc | MCC | Sen | Spc |
| BiLSTM | 65.23 | 72.13 | 76.52 | 74.26 | 66.11 | 70.41 | 78.47 | 74.89 | 65.41 | 73.62 | 72.47 | 71.84 |
| BiRNN | 66.78 | 74.56 | 76.71 | 74.55 | 65.72 | 72.55 | 78.61 | 73.74 | 65.79 | 74.55 | 77.82 | 73.58 |
| SVM | 69.91 | 80.23 | 79.16 | 75.83 | 67.83 | 78.91 | 80.16 | 78.11 | 67.46 | 78.02 | 77.91 | 74.60 |
| Ensemble | 73.12 | 83.81 | 81.88 | 78.92 | 72.90 | 80.83 | 82.88 | 78.50 | 69.92 | 85.39 | 81.47 | 77.92 |
| Naive Bayes | 76.51 | 85.66 | 83.45 | 81.65 | 77.36 | 84.57 | 84.66 | 83.22 | 71.47 | 88.40 | 86.92 | 82.49 |
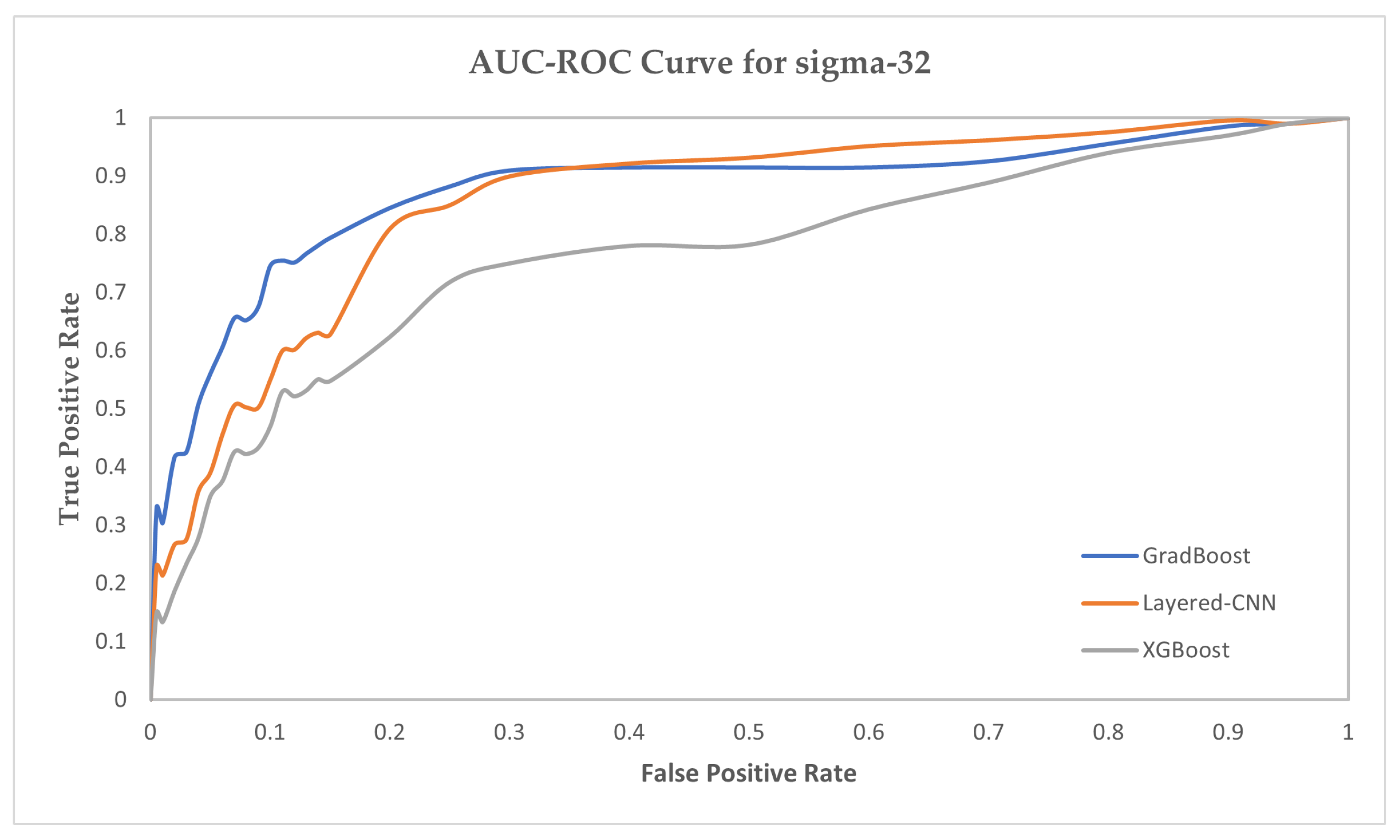
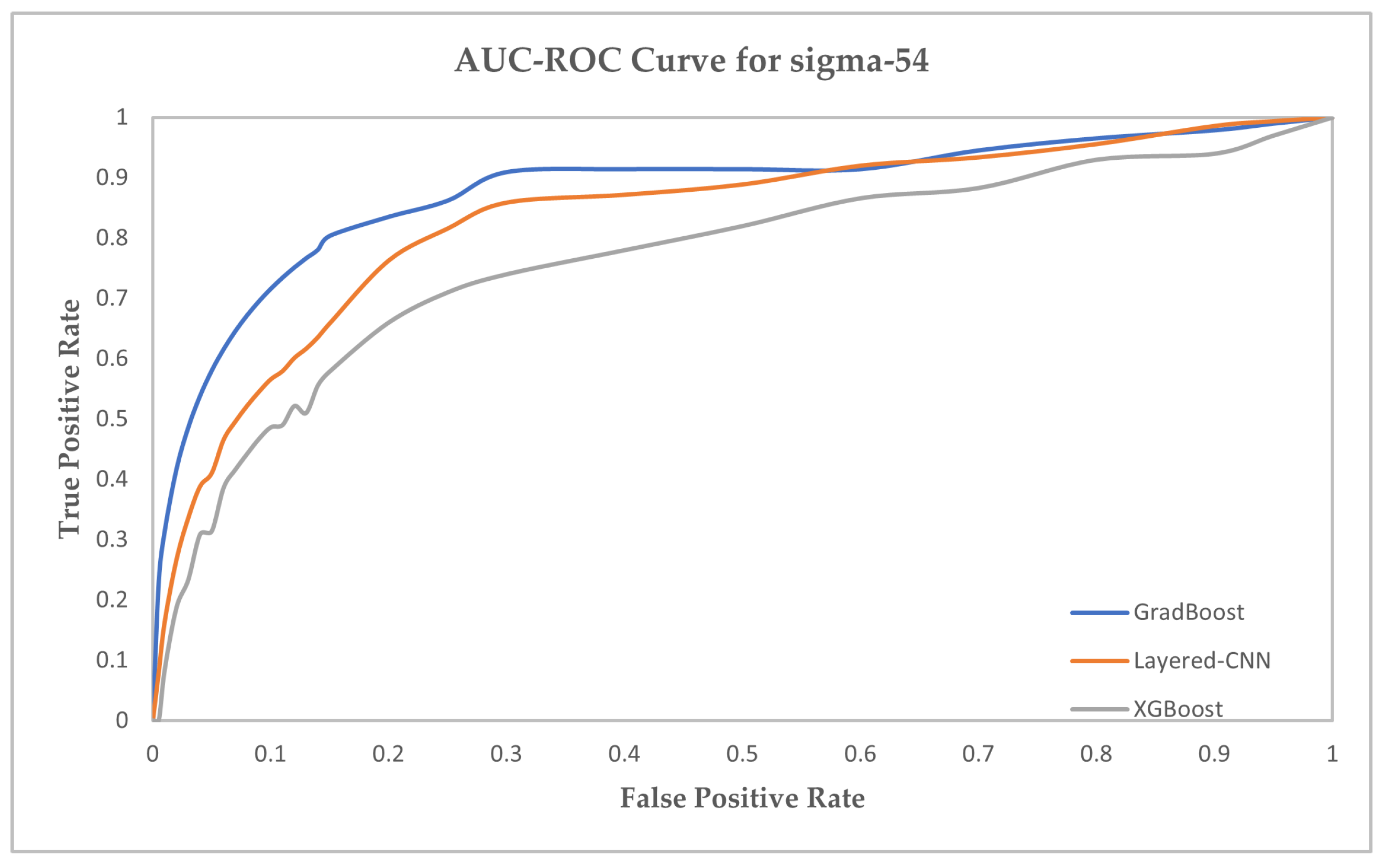
| XGBoost | 79.34 | 88.94 | 86.83 | 85.02 | 80.62 | 89.84 | 88.12 | 85.64 | 75.55 | 89.15 | 86.61 | 85.62 |
| GradBoost | 84.81 | 92.32 | 92.55 | 88.38 | 87.15 | 93.47 | 91.48 | 90.46 | 81.36 | 93.08 | 91.44 | 86.71 |
| Layered-CNN | 91.63 | 94.56 | 92.53 | 92.36 | 92.57 | 93.61 | 92.16 | 93.73 | 93.38 | 94.96 | 92.02 | 93.42 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hazra, D.; Kim, M.-R.; Byun, Y.-C. Generative Adversarial Networks for Creating Synthetic Nucleic Acid Sequences of Cat Genome. Int. J. Mol. Sci. 2022, 23, 3701. https://doi.org/10.3390/ijms23073701
Hazra D, Kim M-R, Byun Y-C. Generative Adversarial Networks for Creating Synthetic Nucleic Acid Sequences of Cat Genome. International Journal of Molecular Sciences. 2022; 23(7):3701. https://doi.org/10.3390/ijms23073701
Chicago/Turabian StyleHazra, Debapriya, Mi-Ryung Kim, and Yung-Cheol Byun. 2022. "Generative Adversarial Networks for Creating Synthetic Nucleic Acid Sequences of Cat Genome" International Journal of Molecular Sciences 23, no. 7: 3701. https://doi.org/10.3390/ijms23073701
APA StyleHazra, D., Kim, M.-R., & Byun, Y.-C. (2022). Generative Adversarial Networks for Creating Synthetic Nucleic Acid Sequences of Cat Genome. International Journal of Molecular Sciences, 23(7), 3701. https://doi.org/10.3390/ijms23073701