
Nutraceuticals as Modulators of Autophagy: Relevance in Parkinson’s Disease


Abstract
:1. Introduction
1.1. Autophagy
1.2. Relevance of Autophagy Modulation in PD
2. Modulation of Autophagy by Nutraceuticals: Support for the Treatment of NDs
2.1. Carotenoids and Retinoids
2.2. Ascorbic Acid
2.3. Calciferol
2.4. Tocopherols and Tocotrienols
2.5. Coenzyme Q10
2.6. Curcumin
2.7. Ergothioneine
2.8. Lipoic Acid
2.9. N-Acetylcysteine
2.10. Polyunsaturated Fatty Acids (PUFAs)
3. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dorsey, E.R.; Bloem, B.R. The Parkinson Pandemic—A Call to Action. JAMA Neurol. 2018, 7, 9–10. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Watzlawik, J.O.; Fiesel, F.C.; Springer, W. Autophagy in Parkinson’s Disease. J. Mol. Biol. 2020, 432, 2651–2672. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson Disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, N.; Yan, J.; Yuasa, S.; Ueno, T.; Kominami, E.; Masuho, Y.; Koga, H.; Muramatsu, M. Interaction of the Unc-51-like Kinase and Microtubule-Associated Protein Light Chain 3 Related Proteins in the Brain: Possible Role of Vesicular Transport in Axonal Elongation. Mol. Brain Res. 2000, 85, 1–12. [Google Scholar] [CrossRef]
- Wei, Y.; Chiang, W.-C.; Sumpter, R.; Mishra, P.; Levine, B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 2017, 168, 224–238. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Jian, Y.; Sun, X.; Yang, C.; Gao, Z.; Zhang, Z.; Liu, X.; Li, Y.; Xu, J.; Jing, Y.; et al. Negative Regulation of Phosphatidylinositol 3-Phosphate Levels in Early-to-Late Endosome Conversion. J. Cell Biol. 2016, 212, 181–198. [Google Scholar] [CrossRef] [Green Version]
- Sagona, A.P.; Nezis, I.P.; Pedersen, N.M.; Liestøl, K.; Poulton, J.; Rusten, T.E.; Skotheim, R.I.; Raiborg, C.; Stenmark, H. PtdIns(3)P Controls Cytokinesis through KIF13A-Mediated Recruitment of FYVE-CENT to the Midbody. Nat. Cell Biol. 2010, 12, 362–371. [Google Scholar] [CrossRef] [Green Version]
- Thoresen, S.B.; Pedersen, N.M.; Liestøl, K.; Stenmark, H. A Phosphatidylinositol 3-Kinase Class III Sub-Complex Containing VPS15, VPS34, Beclin 1, UVRAG and BIF-1 Regulates Cytokinesis and Degradative Endocytic Traffic. Exp. Cell Res. 2010, 316, 3368–3378. [Google Scholar] [CrossRef] [Green Version]
- Maday, S.; Holzbaur, E.L.F. Autophagosome Biogenesis in Primary Neurons Follows an Ordered and Spatially Regulated Pathway. Dev. Cell 2014, 30, 71–85. [Google Scholar] [CrossRef] [Green Version]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Autophagosome Phagosome 2008, 445, 77–88. [Google Scholar] [CrossRef]
- Nakatogawa, H. Two Ubiquitin-like Conjugation Systems That Mediate Membrane Formation during Autophagy. Essays Biochem. 2013, 55, 39–50. [Google Scholar] [CrossRef]
- Martini-Stoica, H.; Xu, Y.; Ballabio, A.; Zheng, H. The Autophagy–Lysosomal Pathway in Neurodegeneration: A TFEB Perspective. Trends Neurosci. 2016, 39, 221–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.-C.; Chen, Y.; Chen, Y.; Park, J.; Zheng, M.; Surh, Y.-J.; Kim, U.-H.; Park, J.W.; Yu, R.; Chung, H.T.; et al. GSK-3β Inhibition by Curcumin Mitigates Amyloidogenesis via TFEB Activation and Anti-Oxidative Activity in Human Neuroblastoma Cells. Free Rad. Res. 2020, 54, 918–930. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, C.; Liu, J.; Chun-Kit Tong, B.; Zhu, Z.; Malampati, S.; Gopalkrishnashetty Sreenivasmurthy, S.; Cheung, K.-H.; Iyaswamy, A.; Su, C.; et al. A Curcumin Derivative Activates TFEB and Protects Against Parkinsonian Neurotoxicity in Vitro. Int. J. Mol. Sci. 2020, 21, 1515. [Google Scholar] [CrossRef] [Green Version]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A Gene Network Regulating Lysosomal Biogenesis and Function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; di Malta, C.; Polito, V.A.; Arencibia, M.G.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB Links Autophagy to Lysosomal Biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [Green Version]
- Medina, D.L.; Fraldi, A.; Bouche, V.; Annunziata, F.; Mansueto, G.; Spampanato, C.; Puri, C.; Pignata, A.; Martina, J.A.; Sardiello, M.; et al. Transcriptional Activation of Lysosomal Exocytosis Promotes Cellular Clearance. Dev. Cell 2011, 21, 421–430. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Dice, J.F. A Receptor for the Selective Uptake and Degradation of Proteins by Lysosomes. Science 1996, 273, 501–503. [Google Scholar] [CrossRef]
- Bandyopadhyay, U.; Kaushik, S.; Varticovski, L.; Cuervo, A.M. The Chaperone-Mediated Autophagy Receptor Organizes in Dynamic Protein Complexes at the Lysosomal Membrane. Mol. Cell. Biol. 2008, 28, 18. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M.; Dice, J.F. Regulation of Lamp2a Levels in the Lysosomal Membrane. Traffic 2000, 1, 570–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, M.C.; Bishop, J.F.; Leng, Y.; Chock, P.B.; Chase, T.N.; Mouradian, M.M. Degradation of α-Synuclein by Proteasome. J. Biol. Chem. 1999, 274, 33855–33858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired Degradation of Mutant α-Synuclein by Chaperone-Mediated Autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef]
- Ebrahimi-Fakhari, D.; Cantuti-Castelvetri, I.; Fan, Z.; Rockenstein, E.; Masliah, E.; Hyman, B.T.; McLean, P.J.; Unni, V.K. Distinct Roles In Vivo for the Ubiquitin-Proteasome System and the Autophagy-Lysosomal Pathway in the Degradation of-Synuclein. J. Neurosci. 2011, 31, 14508–14520. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.-H.; Guo, F.; Shelburne, J.; Watkins, S.; Chu, C.T. Localization of Phosphorylated ERK/MAP Kinases to Mitochondria and Autophagosomes in Lewy Body Diseases. Brain Pathol. 2006, 13, 473–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xilouri, M.; Brekk, O.R.; Stefanis, L. Autophagy and Alpha-Synuclein: Relevance to Parkinson’s Disease and Related Synucleopathies. Mov. Disord. 2016, 31, 178–192. [Google Scholar] [CrossRef]
- Crews, L.; Spencer, B.; Desplats, P.; Patrick, C.; Paulino, A.; Rockenstein, E.; Hansen, L.; Adame, A.; Galasko, D.; Masliah, E. Selective Molecular Alterations in the Autophagy Pathway in Patients with Lewy Body Disease and in Models of α-Synucleinopathy. PLoS ONE 2010, 5, e9313. [Google Scholar] [CrossRef] [Green Version]
- Song, J.-X.; Lu, J.-H.; Liu, L.-F.; Chen, L.-L.; Durairajan, S.S.K.; Yue, Z.; Zhang, H.-Q.; Li, M. HMGB1 Is Involved in Autophagy Inhibition Caused by SNCA/α-Synuclein Overexpression. Autophagy 2014, 10, 144–154. [Google Scholar] [CrossRef] [Green Version]
- Vogiatzi, T.; Xilouri, M.; Vekrellis, K.; Stefanis, L. Wild Type α-Synuclein Is Degraded by Chaperone-Mediated Autophagy and Macroautophagy in Neuronal Cells. J. Biol. Chem. 2008, 283, 23542–23556. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Vicente, M.; Talloczy, Z.; Kaushik, S.; Massey, A.C.; Mazzulli, J.; Mosharov, E.V.; Hodara, R.; Fredenburg, R.; Wu, D.-C.; Follenzi, A.; et al. Dopamine-Modified α-Synuclein Blocks Chaperone-Mediated Autophagy. J. Clin. Investig. 2008, 117, 777–788. [Google Scholar] [CrossRef] [Green Version]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Spiro, A.S.; Furuta, A.; Cooper, A.; Garner, B.; Kabuta, T.; Halliday, G.M. Lysosomal-Associated Membrane Protein 2 Isoforms Are Differentially Affected in Early Parkinson’s Disease. Mov. Disord. 2015, 30, 1639–1647. [Google Scholar] [CrossRef] [PubMed]
- Haddad, D.; Nakamura, K. Understanding the Susceptibility of Dopamine Neurons to Mitochondrial Stressors in Parkinson’s Disease. FEBS Lett. 2015, 589, 3702–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinta, S.J.; Mallajosyula, J.K.; Rane, A.; Andersen, J.K. Mitochondrial Alpha-Synuclein Accumulation Impairs Complex I Function in Dopaminergic Neurons and Results in Increased Mitophagy in Vivo. Neurosci. Lett. 2010, 486, 235–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 2016, 8, 342ra78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, Y.; Zhang, J.; Dong, M. Nrf2 as a Potential Target for Parkinson’s Disease Therapy. J. Mol. Med. 2021, 99, 917–931. [Google Scholar] [CrossRef]
- Jung, U.J.; Kim, S.R. Beneficial Effects of Flavonoids Against Parkinson’s Disease. J. Med. Food 2018, 21, 421–432. [Google Scholar] [CrossRef]
- Chen, J.-H.; Ou, H.-P.; Lin, C.-Y.; Lin, F.-J.; Wu, C.-R.; Chang, S.-W.; Tsai, C.-W. Carnosic Acid Prevents 6-Hydroxydopamine-Induced Cell Death in SH-SY5Y Cells via Mediation of Glutathione Synthesis. Chem. Res. Toxicol. 2012, 25, 1893–1901. [Google Scholar] [CrossRef]
- Bae, J.; Lee, D.; Kim, Y.K.; Gil, M.; Lee, J.-Y.; Lee, K.J. Berberine Protects 6-Hydroxydopamine-Induced Human Dopaminergic Neuronal Cell Death through the Induction of Heme Oxygenase-1. Mol. Cells 2013, 35, 151–157. [Google Scholar] [CrossRef] [Green Version]
- Gillard, G.O.; Collette, B.; Anderson, J.; Chao, J.; Scannevin, R.H.; Huss, D.J.; Fontenot, J.D. DMF, but Not Other Fumarates, Inhibits NF-κB Activity in Vitro in an Nrf2-Independent Manner. J. Neuroimmunol. 2015, 283, 74–85. [Google Scholar] [CrossRef] [Green Version]
- Lastres-Becker, I.; García-Yagüe, A.J.; Scannevin, R.H.; Casarejos, M.J.; Kügler, S.; Rábano, A.; Cuadrado, A. Repurposing the NRF2 Activator Dimethyl Fumarate as Therapy Against Synucleinopathy in Parkinson’s Disease. Antioxid. Redox Signal. 2016, 25, 61–77. [Google Scholar] [CrossRef] [Green Version]
- Dolatshahi, M.; Ranjbar Hameghavandi, M.H.; Sabahi, M.; Rostamkhani, S. Nuclear Factor-kappa B (NF-κB) in Pathophysiology of Parkinson Disease: Diverse Patterns and Mechanisms Contributing to Neurodegeneration. Eur. J. Neur. 2021, 54, 4101–4123. [Google Scholar] [CrossRef] [PubMed]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting Molecular Cross-Talk between Nrf2 and NF-κB Response Pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Innamorato, N.G.; Jazwa, A.; Rojo, A.I.; García, C.; Fernández-Ruiz, J.; Grochot–Przeczek, A.; Stachurska, A.; Jozkowicz, A.; Dulak, J.; Cuadrado, A. Different Susceptibility to the Parkinson’s Toxin MPTP in Mice Lacking the Redox Master Regulator Nrf2 or Its Target Gene Heme Oxygenase-1. PLoS ONE 2010, 5, e11838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, K.; Kim, J.; Jang, Y.J.; Yoon, K.; Cha, Y.; Lee, H.J.; Kim, J. Transcription Factor Nrf2 Suppresses LPS-Induced Hyperactivation of BV-2 Microglial Cells. J. Neuroimmunol. 2011, 233, 160–167. [Google Scholar] [CrossRef]
- Ciulla, M.; Marinelli, L.; Cacciatore, I.; Di Stefano, A. Role of Dietary Supplements in the Management of Parkinson’s Disease. Biomolecules 2019, 9, 271. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Wolk, A.; Håkansson, N.; Pedersen, N.L.; Wirdefeldt, K. Dietary Antioxidants and Risk of Parkinson’s Disease in Two Population-Based Cohorts. Mov. Disord. 2017, 32, 1631–1636. [Google Scholar] [CrossRef]
- Etminan, M.; Gill, S.S.; Samii, A. Intake of Vitamin E, Vitamin C, and Carotenoids and the Risk of Parkinson’s Disease: A Meta-Analysis. Lancet Neurol. 2005, 4, 362–365. [Google Scholar] [CrossRef]
- Hantikainen, E.; Lagerros, Y.T.; Ye, W.; Serafini, M.; Adami, H.-O.; Bellocco, R.; Bonn, S. Dietary Antioxidants and the Risk of Parkinson Disease. Neurology 2021, 96, 895–903. [Google Scholar] [CrossRef]
- Brigger, D.; Schläfli, A.M.; Garattini, E.; Tschan, M.P. Activation of RARα Induces Autophagy in SKBR3 Breast Cancer Cells and Depletion of Key Autophagy Genes Enhances ATRA Toxicity. Cell Death Dis. 2015, 6, e1861. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Xu, J.; Huang, X.; Sun, G.; Jiang, R.; Wu, H.; Shan, X.; Bao, K.; Wu, Q.; Wu, H.; et al. Crocin Induces Anti-Ischemia in Middle Cerebral Artery Occlusion Rats and Inhibits Autophagy by Regulating the Mammalian Target of Rapamycin. Eur. J. Pharmacol. 2019, 857, 172424. [Google Scholar] [CrossRef]
- Yang, Y.; Li, R.; Hui, J.; Li, L.; Zheng, X. β-Carotene Attenuates LPS-Induced Rat Intestinal Inflammation via Modulating Autophagy and Regulating the JAK2/STAT3 and JNK/P38 MAPK Signaling Pathways. J. Food Biochem. 2021, 45, e13544. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Xing, S.; Li, Z. Antagonistic Effects of Lycopene on Cadmium-Induced Hippocampal Dysfunctions in Autophagy, Calcium Homeostatis and Redox. Oncotarget 2017, 8, 44720–44731. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.-C.; Peng, L.-S.; Zou, L.; Huang, S.-F.; Xie, Y.; Mu, G.-P.; Zeng, X.-H.; Zhou, X.-L.; Zeng, Y.-C. Protective Effect and Mechanism of Lycopene on Endothelial Progenitor Cells (EPCs) from Type 2 Diabetes Mellitus Rats. Biomed. Pharmacother. 2017, 92, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Bayomy, N.A.; Elbakary, R.H.; Ibrahim, M.A.A.; Abdelaziz, E.Z. Effect of Lycopene and Rosmarinic Acid on Gentamicin Induced Renal Cortical Oxidative Stress, Apoptosis, and Autophagy in Adult Male Albino Rat. Anat. Rec. 2017, 300, 1137–1149. [Google Scholar] [CrossRef] [Green Version]
- Catanese, A.; Olde Heuvel, F.; Mulaw, M.; Demestre, M.; Higelin, J.; Barbi, G.; Freischmidt, A.; Weishaupt, J.H.; Ludolph, A.C.; Roselli, F.; et al. Retinoic Acid Worsens ATG10-Dependent Autophagy Impairment in TBK1-Mutant HiPSC-Derived Motoneurons through SQSTM1/P62 Accumulation. Autophagy 2019, 15, 1719–1737. [Google Scholar] [CrossRef]
- Lee, H.; Lim, J.W.; Kim, H. Effect of Astaxanthin on Activation of Autophagy and Inhibition of Apoptosis in Helicobacter Pylori-Infected Gastric Epithelial Cell Line AGS. Nutrients 2020, 12, 1750. [Google Scholar] [CrossRef]
- Fung, F.K.C.; Law, B.Y.K.; Lo, A.C.Y. Lutein Attenuates Both Apoptosis and Autophagy upon Cobalt (II) Chloride-Induced Hypoxia in Rat Műller Cells. PLoS ONE 2016, 11, e0167828. [Google Scholar] [CrossRef]
- Chang, C.-J.; Lin, J.-F.; Hsiao, C.-Y.; Chang, H.-H.; Li, H.-J.; Chang, H.-H.; Lee, G.-A.; Hung, C.-F. Lutein Induces Autophagy via Beclin-1 Upregulation in IEC-6 Rat Intestinal Epithelial Cells. Am. J. Chin. Med. 2017, 45, 1273–1291. [Google Scholar] [CrossRef]
- Sheu, S.-J.; Chen, J.-L.; Bee, Y.-S.; Chen, Y.-A.; Lin, S.-H.; Shu, C.-W. Differential Autophagic Effects of Vital Dyes in Retinal Pigment Epithelial ARPE-19 and Photoreceptor 661W Cells. PLoS ONE 2017, 12, e0174736. [Google Scholar] [CrossRef]
- Long, Y.; Cao, X.; Zhao, R.; Gong, S.; Jin, L.; Feng, C. Fucoxanthin Treatment Inhibits Nasopharyngeal Carcinoma Cell Proliferation through Induction of Autophagy Mechanism. Environ. Toxicol. 2020, 35, 1082–1090. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, H.; Fan, Y.; Gao, Y.; Li, X.; Hu, Z.; Ding, K.; Wang, Y.; Wang, X. Fucoxanthin Provides Neuroprotection in Models of Traumatic Brain Injury via the Nrf2-ARE and Nrf2-Autophagy Pathways. Sci. Rep. 2017, 7, 46763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Zhang, H.; Li, Y.-X.; Cai, L.; Huang, J.; Zhao, C.; Jia, L.; Buchanan, R.; Yang, T.; Jiang, L.-J. Crocin and Geniposide Profiles and Radical Scavenging Activity of Gardenia Fruits (Gardenia Jasminoides Ellis) from Different Cultivars and at the Various Stages of Maturation. Fitoterapia 2010, 81, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Unlu, N.Z.; Bohn, T.; Francis, D.M.; Nagaraja, H.N.; Clinton, S.K.; Schwartz, S.J. Lycopene from Heat-Induced Cis-Isomer-Rich Tomato Sauce Is More Bioavailable than from All-Rich Tomato Sauce in Human Subjects. Br. J. Nutr. 2007, 98, 140–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.; Kim, H. The Remedial Potential of Lycopene in Pancreatitis through Regulation of Autophagy. Int. J. Mol. Sci. 2020, 21, 5775. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, H. Astaxanthin Modulation of Signaling Pathways That Regulate Autophagy. Mar. Drugs 2019, 17, 546. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.-Q.; Sun, X.-B.; Xu, Y.-X.; Zhao, H.; Zhu, Q.-Y.; Zhu, C.-Q. Astaxanthin Upregulates Heme Oxygenase-1 Expression through ERK1/2 Pathway and Its Protective Effect against Beta-Amyloid-Induced Cytotoxicity in SH-SY5Y Cells. Brain Res. 2010, 1360, 159–167. [Google Scholar] [CrossRef]
- Ikeda, Y.; Tsuji, S.; Satoh, A.; Ishikura, M.; Shirasawa, T.; Shimizu, T. Protective Effects of Astaxanthin on 6-Hydroxydopamine-Induced Apoptosis in Human Neuroblastoma SH-SY5Y Cells. J. Neurochem. 2008, 107, 1730–1740. [Google Scholar] [CrossRef]
- Cho, K.S.; Shin, M.; Kim, S.; Lee, S.B. Recent Advances in Studies on the Therapeutic Potential of Dietary Carotenoids in Neurodegenerative Diseases. Oxidative Med. Cell. Longev. 2018, 2018, 4120458. [Google Scholar] [CrossRef]
- Barmaki, H.; Morovati, A.; Eydivandi, Z.; Jafari Naleshkenani, F.; Saedi, S.; Musavi, H.; Abbasi, M.; Hemmati-Dinarvand, M. The Association between Serum Oxidative Stress Indexes and Pathogenesis of Parkinson’s Disease in the Northwest of Iran. Iran. J. Public Health 2021, 50, 606. [Google Scholar] [CrossRef]
- Chang, M.C.; Kwak, S.G.; Kwak, S. Effect of Dietary Vitamins C and E on the Risk of Parkinson’s Disease: A Meta-Analysis. Clin. Nutr. 2021, 40, 3922–3930. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, Y.; Zhang, H.; Wang, L.; Wang, T.; Han, Z.; Wu, L.; Liu, G. Effect of Plasma Vitamin C Levels on Parkinson’s Disease and Age at Onset: A Mendelian Randomization Study. J. Transl. Med. 2021, 19, 221. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.-S.; Luan, J.; Sofianopoulou, E.; Imamura, F.; Stewart, I.D.; Day, F.R.; Pietzner, M.; Wheeler, E.; Lotta, L.A.; Gundersen, T.E.; et al. Plasma Vitamin C and Type 2 Diabetes: Genome-Wide Association Study and Mendelian Randomization Analysis in European Populations. Diabetes Care 2021, 44, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Wang, S.; Zhang, T.; Zhao, X.; Liu, X.; Cao, L.; Chi, Z. Ascorbic Acid Ameliorates Seizures and Brain Damage in Rats through Inhibiting Autophagy. Brain Res. 2013, 1535, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Sangani, R.; Periyasamy-Thandavan, S.; Pathania, R.; Ahmad, S.; Kutiyanawalla, A.; Kolhe, R.; Bhattacharyya, M.H.; Chutkan, N.; Hunter, M.; Hill, W.D.; et al. The Crucial Role of Vitamin C and Its Transporter (SVCT2) in Bone Marrow Stromal Cell Autophagy and Apoptosis. Stem Cell Res. 2015, 15, 312–321. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.-N.; Yang, L.-Y.; Wang, J.-Y.; Lai, C.-C.; Chiu, C.-T.; Wang, J.-Y. L-Ascorbate Protects Against Methamphetamine-Induced Neurotoxicity of Cortical Cells via Inhibiting Oxidative Stress, Autophagy, and Apoptosis. Mol. Neurobiol. 2017, 54, 125–136. [Google Scholar] [CrossRef]
- Martin, A.; Joseph, J.A.; Cuervo, A.M. Stimulatory Effect of Vitamin C on Autophagy in Glial Cells. J. Neurochem. 2002, 82, 538–549. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-Y.; Wu, J.-N.; Cherng, T.-L.; Hoffer, B.J.; Chen, H.-H.; Borlongan, C.V.; Wang, Y. Vitamin D3 Attenuates 6-Hydroxydopamine-Induced Neurotoxicity in Rats. Brain Res. 2001, 904, 67–75. [Google Scholar] [CrossRef]
- Jang, W.; Kim, H.J.; Li, H.; Jo, K.D.; Lee, M.K.; Song, S.H.; Yang, H.O. 1,25-Dyhydroxyvitamin D3 Attenuates Rotenone-Induced Neurotoxicity in SH-SY5Y Cells through Induction of Autophagy. Biochem. Biophys. Res. Commun. 2014, 451, 142–147. [Google Scholar] [CrossRef]
- Tao, S.; Zhang, H.; Xue, L.; Jiang, X.; Wang, H.; Li, B.; Tian, H.; Zhang, Z. Vitamin D Protects against Particles-caused Lung Injury through Induction of Autophagy in an Nrf2-dependent Manner. Environ. Toxicol. 2019, 34, 594–609. [Google Scholar] [CrossRef]
- Niu, L.; Chen, S.; Yang, X.; Ma, C.; Pan, C.; Wang, H.; Li, Q.; Geng, F.; Tang, X. Vitamin D Decreases Porphyromonas Gingivalis Internalized into Macrophages by Promoting Autophagy. Oral Dis. 2021, 27, 1775–1788. [Google Scholar] [CrossRef]
- Wang, Y.; He, D.; Ni, C.; Zhou, H.; Wu, S.; Xue, Z.; Zhou, Z. Vitamin D Induces Autophagy of Pancreatic β-Cells and Enhances Insulin Secretion. Mol. Med. Rep. 2016, 14, 2644–2650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavera-Mendoza, L.E.; Westerling, T.; Libby, E.; Marusyk, A.; Cato, L.; Cassani, R.; Cameron, L.A.; Ficarro, S.B.; Marto, J.A.; Klawitter, J.; et al. Vitamin D Receptor Regulates Autophagy in the Normal Mammary Gland and in Luminal Breast Cancer Cells. Proc. Natl. Acad. Sci. USA 2017, 114, E2186–E2194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, L.M.; Binko, A.M.; Traylor, Z.P.; Peng, H.; Lu, K.Q. Vitamin D Improves Sunburns by Increasing Autophagy in M2 Macrophages. Autophagy 2019, 15, 813–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, J.; Liang, Y.; Li, H.; Zhou, W.; Wang, B.; Gong, A.; Zhang, R. Vitamin D Enhances Resistance to Aspergillus Fumigatus in Mice via Inhibition of Excessive Autophagy. Am. J. Transl. Res. 2018, 10, 381–391. [Google Scholar] [PubMed]
- Abdel-Mohsen, M.A.; El-Braky, A.A.-A.; Ghazal, A.A.E.-R.; Shamseya, M.M. Autophagy, Apoptosis, Vitamin D, and Vitamin D Receptor in Hepatocellular Carcinoma Associated with Hepatitis C Virus. Medicine 2018, 97, e0172. [Google Scholar] [CrossRef] [PubMed]
- Evatt, M.L.; DeLong, M.R.; Khazai, N.; Rosen, A.; Triche, S.; Tangpricha, V. Prevalence of Vitamin D Insufficiency in Patients With Parkinson Disease and Alzheimer Disease. Arch. Neurol. 2008, 65, 1348–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knekt, P.; Kilkkinen, A.; Rissanen, H.; Marniemi, J.; Sääksjärvi, K.; Heliövaara, M. Serum Vitamin D and the Risk of Parkinson Disease. Arch. Neurol. 2010, 67, 808–811. [Google Scholar] [CrossRef] [Green Version]
- Periyasamy, K.M.; Ranganathan, U.D.; Tripathy, S.P.; Bethunaickan, R. Vitamin D—A Host Directed Autophagy Mediated Therapy for Tuberculosis. Mol. Immunol. 2020, 127, 238–244. [Google Scholar] [CrossRef]
- Høyer-Hansen, M.; Nordbrandt, S.P.S.; Jäättelä, M. Autophagy as a Basis for the Health-Promoting Effects of Vitamin D. Trends Mol. Med. 2010, 16, 295–302. [Google Scholar] [CrossRef]
- Ogura, H.; Hatip-Al-Khatib, I.; Suenaga, M.; Hatip, F.B.; Mishima, T.; Fujioka, S.; Ouma, S.; Matsunaga, Y.; Tsuboi, Y. Circulatory 25(OH)D and 1,25(OH)2D as Differential Biomarkers between Multiple System Atrophy and Parkinson’s Disease Patients. eNeurologicalSci 2021, 25, 100369. [Google Scholar] [CrossRef]
- Zhang, S.M.; Hernan, M.A.; Chen, H.; Spiegelman, D.; Willett, W.C.; Ascherio, A. Intakes of Vitamins E and C, Carotenoids, Vitamin Supplements, and PD Risk. Neurology 2002, 59, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Scheider, W.L.; Hershey, L.A.; Vena, J.E.; Holmlund, T.; Marshall, J.R.; Freudenheim, J.L. Dietary Antioxidants and Other Dietary Factors in the Etiology of Parkinson’s Disease. Mov. Disord. 1997, 12, 190–196. [Google Scholar] [CrossRef] [PubMed]
- DATATOP: A Multicenter Controlled Clinical Trial in Early Parkinson’s Disease. Archiv. Neurol. 1989, 46, 1052–1060. [CrossRef] [PubMed]
- Fukui, K.; Kawakami, H.; Honjo, T.; Ogasawara, R.; Takatsu, H.; Shinkai, T.; Koike, T.; Urano, S. Vitamin E Deficiency Induces Axonal Degeneration in Mouse Hippocampal Neurons. J. Nutr. Sci. Vitaminol. 2012, 58, 377–383. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.; Xu, J.; Lin, Y.; Zhao, X.; Liu, X.; Chi, Z. Autophagy Is Upregulated in Rats with Status Epilepticus and Partly Inhibited by Vitamin E. Biochem. Biophys. Res. Commun. 2009, 379, 949–953. [Google Scholar] [CrossRef]
- Cao, L.; Chen, R.; Xu, J.; Lin, Y.; Wang, R.; Chi, Z. Vitamin E Inhibits Activated Chaperone-Mediated Autophagy in Rats with Status Epilepticus. Neuroscience 2009, 161, 73–77. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, W.; Jia, Q.; Feng, Z.; Guo, J.; Han, X.; Liu, Y.; Shang, H.; Wang, Y.; Liu, W.J. High Dose Vitamin E Attenuates Diabetic Nephropathy via Alleviation of Autophagic Stress. Front. Physiol. 2019, 9, 1939. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Wu, H.; Jiang, R.; Sun, G.; Shen, J.; Ma, M.; Ma, C.; Zhang, S.; Huang, Z.; Wu, Q.; et al. The Antidepressant Effects of α-Tocopherol Are Related to Activation of Autophagy via the AMPK/MTOR Pathway. Eur. J. Pharmacol. 2018, 833, 1–7. [Google Scholar] [CrossRef]
- Karim, M.R.; Fujimura, S.; Kadowaki, M. Vitamin E as a Novel Enhancer of Macroautophagy in Rat Hepatocytes and H4-II-E Cells. Biochem. Biophys. Res. Commun. 2010, 394, 981–987. [Google Scholar] [CrossRef]
- Lekli, I.; Ray, D.; Mukherjee, S.; Gurusamy, N.; Ahsan, M.K.; Juhasz, B.; Bak, I.; Tosaki, A.; Gherghiceanu, M.; Popescu, L.M.; et al. Co-Ordinated Autophagy with Resveratrol and γ-Tocotrienol Confers Synergetic Cardioprotection. J. Cell. Mol. Med. 2010, 14, 2506–2518. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Q.; Rao, X.; Kim, C.Y.; Freiser, H.; Zhang, Q.; Jiang, Z.; Li, G. Gamma-Tocotrienol Induces Apoptosis and Autophagy in Prostate Cancer Cells by Increasing Intracellular Dihydrosphingosine and Dihydroceramide. Int. J. Cancer 2012, 130, 685–693. [Google Scholar] [CrossRef] [Green Version]
- Jang, Y.; Rao, X.; Jiang, Q. Gamma-Tocotrienol Profoundly Alters Sphingolipids in Cancer Cells by Inhibition of Dihydroceramide Desaturase and Possibly Activation of Sphingolipid Hydrolysis during Prolonged Treatment. J. Nutr. Biochem. 2017, 46, 49–56. [Google Scholar] [CrossRef]
- Tiwari, R.V.; Parajuli, P.; Sylvester, P.W. γ-Tocotrienol-Induced Autophagy in Malignant Mammary Cancer Cells. Exp. Biol. Med. 2014, 239, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Rickmann, M.; Vaquero, E.C.; Malagelada, J.R.; Molero, X. Tocotrienols Induce Apoptosis and Autophagy in Rat Pancreatic Stellate Cells Through the Mitochondrial Death Pathway. Gastroenterology 2007, 132, 2518–2532. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hahn, T.; Garrison, K.; Cui, Z.-H.; Thorburn, A.; Thorburn, J.; Hu, H.-M.; Akporiaye, E.T. The Vitamin E Analogue α-TEA Stimulates Tumor Autophagy and Enhances Antigen Cross-Presentation. Cancer Res. 2012, 72, 3535–3545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukui, K.; Masuda, A.; Hosono, A.; Suwabe, R.; Yamashita, K.; Shinkai, T.; Urano, S. Changes in Microtubule-Related Proteins and Autophagy in Long-Term Vitamin E-Deficient Mice. Free. Radic. Res. 2014, 48, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Wen, X.; Jiang, H.; Wang, J.; Song, N.; Xie, J. Interactions between Iron and α-Synuclein Pathology in Parkinson’s Disease. Free Rad. Biol. Med. 2019, 141, 253–260. [Google Scholar] [CrossRef]
- Wan, W.; Jin, L.; Wang, Z.; Wang, L.; Fei, G.; Ye, F.; Pan, X.; Wang, C.; Zhong, C. Iron Deposition Leads to Neuronal α-Synuclein Pathology by Inducing Autophagy Dysfunction. Front. Neurol. 2017, 8, 1. [Google Scholar] [CrossRef] [Green Version]
- Shults, C.W. Effects of Coenzyme Q10 in Early Parkinson Disease. Arch. Neurol. 2002, 59, 1541–1550. [Google Scholar] [CrossRef]
- Müller, T.; Büttner, T.; Gholipour, A.-F.; Kuhn, W. Coenzyme Q10 Supplementation Provides Mild Symptomatic Benefit in Patients with Parkinson’s Disease. Neurosci. Lett. 2003, 341, 201–204. [Google Scholar] [CrossRef]
- Storch, A. Randomized, Double-Blind, Placebo-Controlled Trial on Symptomatic Effects of Coenzyme Q10 in Parkinson Disease. Arch. Neurol. 2007, 64, 938–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.-G.; Sun, M.-X.; Zhang, W.-L.; Wang, W.-W.; Jin, Y.-M.; Xie, C.-L. The Efficacy and Safety of Coenzyme Q10 in Parkinson’s Disease: A Meta-Analysis of Randomized Controlled Trials. Neurol. Sci. 2017, 38, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, D.I.; Khairy, E.; Tawfek, S.S.; Habib, E.K.; Fetouh, M.A. Coenzyme Q10 Attenuates Lung and Liver Fibrosis via Modulation of Autophagy in Methotrexate Treated Rat. Biomed. Pharmacother. 2019, 109, 892–901. [Google Scholar] [CrossRef]
- Liu, Y.; Yao, Y.; Tao, W.; Liu, F.; Yang, S.; Zhao, A.; Song, D.; Li, X. Coenzyme Q10 Ameliorates BPA-Induced Apoptosis by Regulating Autophagy-Related Lysosomal Pathways. Ecotoxicol. Environ. Saf. 2021, 221, 112450. [Google Scholar] [CrossRef] [PubMed]
- Villanueva-Paz, M.; Povea-Cabello, S.; Villalón-García, I.; Álvarez-Córdoba, M.; Suárez-Rivero, J.M.; Talaverón-Rey, M.; Jackson, S.; Falcón-Moya, R.; Rodríguez-Moreno, A.; Sánchez-Alcázar, J.A. Parkin-Mediated Mitophagy and Autophagy Flux Disruption in Cellular Models of MERRF Syndrome. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165726. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Chen, S.; Tang, H.; Fang, W.; Chen, K.; Chen, X. CoQ10 Protects against Acetaminophen-Induced Liver Injury by Enhancing Mitophagy. Toxicol. Appl. Pharmacol. 2021, 410, 115355. [Google Scholar] [CrossRef]
- Xu, J.; Huang, B.; Tang, S.; Sun, J.; Bao, E. Co-Enzyme Q10 Protects Primary Chicken Myocardial Cells from Heat Stress by Upregulating Autophagy and Suppressing the PI3K/AKT/MTOR Pathway. Cell Stress Chaperones 2019, 24, 1067–1078. [Google Scholar] [CrossRef]
- Liang, S.; Ping, Z.; Ge, J. Coenzyme Q10 Regulates Antioxidative Stress and Autophagy in Acute Myocardial Ischemia-Reperfusion Injury. Oxidative Med. Cell. Longev. 2017, 2017, 9863181. [Google Scholar] [CrossRef]
- Cotán, D.; Cordero, M.D.; Garrido-Maraver, J.; Oropesa-Avila, M.; Rodríguez-Hernández, Á.; Izquierdo, L.G.; de la Mata, M.; de Miguel, M.; Lorite, J.B.; Infante, E.R.; et al. Secondary Coenzyme Q 10 Deficiency Triggers Mitochondria Degradation by Mitophagy in MELAS Fibroblasts. FASEB J. 2011, 25, 2669–2687. [Google Scholar] [CrossRef]
- Xue, S.Y.; Hebert, V.Y.; Hayes, D.M.; Robinson, C.N.; Glover, M.; Dugas, T.R. Nucleoside Reverse Transcriptase Inhibitors Induce a Mitophagy-Associated Endothelial Cytotoxicity That Is Reversed by Coenzyme Q10 Cotreatment. Toxicol. Sci. 2013, 134, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Xue, R.; Wang, J.; Yang, L.; Liu, X.; Gao, Y.; Pang, Y.; Wang, Y.; Hao, J. Coenzyme Q10 Ameliorates Pancreatic Fibrosis via the ROS-Triggered MTOR Signaling Pathway. Oxidative Med. Cell. Longev. 2019, 2019, 8039694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, R.; Yang, J.; Wu, J.; Meng, Q.; Hao, J. Coenzyme Q10 Inhibits the Activation of Pancreatic Stellate Cells through PI3K/AKT/MTOR Signaling Pathway. Oncotarget 2017, 8, 92300–92311. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Ren, T.; Zeng, J. Mitochondrial Coenzyme Q Protects Sepsis-Induced Acute Lung Injury by Activating PI3K/Akt/GSK-3 β/MTOR Pathway in Rats. BioMed Res. Int. 2019, 2019, 5240898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Hernández, Á.; Cordero, M.D.; Salviati, L.; Artuch, R.; Pineda, M.; Briones, P.; Gómez Izquierdo, L.; Cotán, D.; Navas, P.; Sánchez-Alcázar, J.A. Coenzyme Q Deficiency Triggers Mitochondria Degradation by Mitophagy. Autophagy 2009, 5, 19–32. [Google Scholar] [CrossRef] [Green Version]
- He, P.K.; Gao, Y.Y.; Lyu, F.-J.; Chen, J.N.; Zhang, Y.H.; Nie, K.; Zhang, Q.X.; Huang, R.; Duan, Q.R.; Guo, M.L.; et al. Idebenone-Activating Autophagic Degradation of α-Synuclein via Inhibition of AKT-MTOR Pathway in a SH-SY5Y-A53T Model of Parkinson’s Disease: A Network Pharmacological Approach. Evid. Based Complement. Altern. Med. 2021, 2021, 8548380. [Google Scholar] [CrossRef]
- Yu, C.-C.; Chiang, P.-C.; Lu, P.-H.; Kuo, M.-T.; Wen, W.-C.; Chen, P.; Guh, J.-H. Antroquinonol, a Natural Ubiquinone Derivative, Induces a Cross Talk between Apoptosis, Autophagy and Senescence in Human Pancreatic Carcinoma Cells. J. Nutr. Biochem. 2012, 23, 900–907. [Google Scholar] [CrossRef]
- Wang, X.-S.; Zhang, Z.-R.; Zhang, M.-M.; Sun, M.-X.; Wang, W.-W.; Xie, C.-L. Neuroprotective Properties of Curcumin in Toxin-Base Animal Models of Parkinson’s Disease: A Systematic Experiment Literatures Review. BMC Complementary Altern. Med. 2017, 17, 1186. [Google Scholar] [CrossRef]
- Di Tu, Q.; Jin, J.; Hu, X.; Ren, Y.; Zhao, L.; He, Q. Curcumin Improves the Renal Autophagy in Rat Experimental Membranous Nephropathy via Regulating the PI3K/AKT/MTOR and Nrf2/HO-1 Signaling Pathways. BioMed Res. Int. 2020, 2020, 7069052. [Google Scholar] [CrossRef]
- Tu, Q.; Li, Y.; Jin, J.; Jiang, X.; Ren, Y.; He, Q. Curcumin Alleviates Diabetic Nephropathy via Inhibiting Podocyte Mesenchymal Transdifferentiation and Inducing Autophagy in Rats and MPC5 Cells. Pharmaceut. Biol. 2019, 57, 778–786. [Google Scholar] [CrossRef]
- Kong, D.; Zhang, F.; Shao, J.; Wu, L.; Zhang, X.; Chen, L.; Lu, Y.; Zheng, S. Curcumin Inhibits Cobalt Chloride-Induced Epithelial-to-Mesenchymal Transition Associated with Interference with TGF-β/Smad Signaling in Hepatocytes. Lab. Investig. 2015, 95, 1234–1245. [Google Scholar] [CrossRef] [Green Version]
- Kong, D.; Zhang, Z.; Chen, L.; Huang, W.; Zhang, F.; Wang, L.; Wang, Y.; Cao, P.; Zheng, S. Curcumin blunts epithelial-mesenchymal transition of hepatocytes to alleviate hepatic fibrosis through regulating oxidative stress and autophagy. Redox Biol. 2020, 36, 101600. [Google Scholar] [CrossRef] [PubMed]
- Heebkaew, N.; Rujanapun, N.; Kunhorm, P.; Jaroonwitchawan, T.; Chaicharoenaudomrung, N.; Promjantuek, W.; Noisa, P. Curcumin Induces Neural Differentiation of Human Pluripotent Embryonal Carcinoma Cells through the Activation of Autophagy. BioMed Res. Int. 2019, 2019, 4378710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-E.; Yoon, S.S.; Moon, E.-Y. Curcumin-Induced Autophagy Augments Its Antitumor Effect against A172 Human Glioblastoma Cells. Biomol. Ther. 2019, 27, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Ma, X.; Wang, Z.; Zeng, X.; Hu, Z.; Ye, Z.; Shen, G. Curcumin Induces Apoptosis and Protective Autophagy in Castration-Resistant Prostate Cancer Cells through Iron Chelation. Drug Des. Dev. Ther. 2017, 11, 431–439. [Google Scholar] [CrossRef] [Green Version]
- Rahaman, M.S.; Banik, S.; Akter, M.; Rahman, M.M.; Sikder, M.T.; Hosokawa, T.; Saito, T.; Kurasaki, M. Curcumin Alleviates Arsenic-Induced Toxicity in PC12 Cells via Modulating Autophagy/Apoptosis. Ecotoxicol. Environ. Saf. 2020, 200, 110756. [Google Scholar] [CrossRef]
- Wang, J.; Wang, J.; Cai, Z.; Xu, C. The Effect of Curcumin on the Differentiation, Apoptosis and Cell Cycle of Neural Stem Cells Is Mediated through Inhibiting Autophagy by the Modulation of Atg7 and P62. Int. J. Mol. Med. 2018, 42, 2481–2488. [Google Scholar] [CrossRef]
- Jaroonwitchawan, T.; Chaicharoenaudomrung, N.; Namkaew, J.; Noisa, P. Curcumin Attenuates Paraquat-Induced Cell Death in Human Neuroblastoma Cells through Modulating Oxidative Stress and Autophagy. Neur. Lett. 2017, 636, 40–47. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, X.; Li, Q. Curcumin Accelerates the Repair of Sciatic Nerve Injury in Rats through Reducing Schwann Cells Apoptosis and Promoting Myelinization. Biomed. Pharmacoth. 2017, 92, 1103–1110. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, X.; Teng, Z.; Zhang, T.; Li, Y. Downregulation of PI3K/Akt/MTOR Signaling Pathway in Curcumin-Induced Autophagy in APP/PS1 Double Transgenic Mice. Eur. J. Pharmacol. 2014, 740, 312–320. [Google Scholar] [CrossRef]
- Hou, Y.; Wang, J.; Feng, J. The Neuroprotective Effects of Curcumin Are Associated with the Regulation of the Reciprocal Function between Autophagy and HIF-1α in Cerebral Ischemia-Reperfusion Injury. Drug Design Develop. Ther. 2019, 13, 1135–1144. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.-F.; Zhang, Y.-J.; Zhou, H.-Y.; Wang, H.-M.; Tian, L.-P.; Liu, J.; Ding, J.-Q.; Chen, S.-D. Curcumin Ameliorates the Neurodegenerative Pathology in A53T α-Synuclein Cell Model of Parkinson’s Disease Through the Downregulation of MTOR/P70S6K Signaling and the Recovery of Macroautophagy. J. Neuroimm. Pharmacol. 2013, 8, 356–369. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Pang, Y.; Zhao, X.; Li, R.; Jin, C.; Xue, J.; Dong, R.; Liu, P. Curcumin Induces Apoptotic Cell Death and Protective Autophagy by Inhibiting AKT/MTOR/P70S6K Pathway in Human Ovarian Cancer Cells. Arch. Gynecol. Obstet. 2019, 299, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Maiti, P.; Scott, J.; Sengupta, D.; Al-Gharaibeh, A.; Dunbar, G. Curcumin and Solid Lipid Curcumin Particles Induce Autophagy, but Inhibit Mitophagy and the PI3K-Akt/MTOR Pathway in Cultured Glioblastoma Cells. Int. J. Mol. Sci. 2019, 20, 399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Li, J.; Wu, L.; Zhou, C.; Wang, Q.; Li, X.; Zhou, M.; Wang, H. Tetrahydrocurcumin Provides Neuroprotection in Rats after Traumatic Brain Injury: Autophagy and the PI3K/AKT Pathways as a Potential Mechanism. J. Surgic. Res. 2016, 206, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhuang, Z.; Gao, S.; Li, X.; Zhang, Z.; Ye, Z.; Li, L.; Tang, C.; Zhou, M.; Han, X.; et al. Tetrahydrocurcumin Reduces Oxidative Stress-Induced Apoptosis via the Mitochondrial Apoptotic Pathway by Modulating Autophagy in Rats after Traumatic Brain Injury. Am. J. Transl. Res. 2017, 9, 887–899. [Google Scholar]
- Shi, Q.; Zhang, Q.; Peng, Y.; Zhang, X.; Wang, Y.; Shi, L. A Natural Diarylheptanoid Protects Cortical Neurons against Oxygen–Glucose Deprivation-Induced Autophagy and Apoptosis. J. Pharm. Pharmacol. 2019, 71, 1110–1118. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, F. Iron Homeostasis and Tumorigenesis: Molecular Mechanisms and Therapeutic Opportunities. Prot. Cell 2015, 6, 88–100. [Google Scholar] [CrossRef] [Green Version]
- Franco, R.; Li, S.; Rodriguez-Rocha, H.; Burns, M.; Panayiotidis, M.I. Molecular Mechanisms of Pesticide-Induced Neurotoxicity: Relevance to Parkinson’s Disease. Chemico-Biologic. Interact. 2010, 188, 289–300. [Google Scholar] [CrossRef] [Green Version]
- Fei, Q.; McCormack, A.L.; di Monte, D.A.; Ethell, D.W. Paraquat Neurotoxicity Is Mediated by a Bak-Dependent Mechanism. J. Biol. Chem. 2008, 283, 3357–3364. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.-F.; Block, M.L.; Zhang, W.; Qin, L.; Wilson, B.; Zhang, W.-Q.; Veronesi, B.; Hong, J.-S. The Role of Microglia in Paraquat-Induced Dopaminergic Neurotoxicity. Antioxid. Redox Signal. 2005, 7, 654–661. [Google Scholar] [CrossRef]
- Perrone, L.; Squillaro, T.; Napolitano, F.; Terracciano, C.; Sampaolo, S.; Melone, M.A.B. The Autophagy Signaling Pathway: A Potential Multifunctional Therapeutic Target of Curcumin in Neurological and Neuromuscular Diseases. Nutrients 2019, 11, 1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forouzanfar, F.; Read, M.I.; Barreto, G.E.; Sahebkar, A. Neuroprotective Effects of Curcumin through Autophagy Modulation. IUBMB Life 2020, 72, 652–664. [Google Scholar] [CrossRef] [PubMed]
- Borodina, I.; Kenny, L.C.; McCarthy, C.M.; Paramasivan, K.; Pretorius, E.; Roberts, T.J.; van der Hoek, S.A.; Kell, D.B. The Biology of Ergothioneine, an Antioxidant Nutraceutical. Nutr. Res. Rev. 2020, 33, 190–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halliwell, B.; Cheah, I.K.; Tang, R.M.Y. Ergothioneine—A Diet-Derived Antioxidant with Therapeutic Potential. FEBS Lett. 2018, 592, 3357–3366. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.-H.; Aruoma, O.I.; Jen, L.-S.; Chung, H.Y.; Surh, Y.-J. Ergothioneine Rescues PC12 Cells from β-Amyloid-Induced Apoptotic Death. Free. Radic. Biol. Med. 2004, 36, 288–299. [Google Scholar] [CrossRef]
- Song, T.-Y.; Chen, C.-L.; Liao, J.-W.; Ou, H.-C.; Tsai, M.-S. Ergothioneine Protects against Neuronal Injury Induced by Cisplatin Both in Vitro and in Vivo. Food Chem. Toxicol. 2010, 48, 3492–3499. [Google Scholar] [CrossRef]
- Moncaster, J.A.; Walsh, D.T.; Gentleman, S.M.; Jen, L.-S.; Aruoma, O.I. Ergothioneine Treatment Protects Neurons against N -Methyl- d -Aspartate Excitotoxicity in an in Vivo Rat Retinal Model. Neurosci. Lett. 2002, 328, 55–59. [Google Scholar] [CrossRef]
- Sugiura, T.; Kato, S.; Shimizu, T.; Wakayama, T.; Nakamichi, N.; Kubo, Y.; Iwata, D.; Suzuki, K.; Soga, T.; Asano, M.; et al. Functional Expression of Carnitine/Organic Cation Transporter OCTN1/SLC22A4 in Mouse Small Intestine and Liver. Drug Metab. Dispos. 2010, 38, 1665–1672. [Google Scholar] [CrossRef] [Green Version]
- Yee, S.W.; Buitrago, D.; Stecula, A.; Ngo, H.X.; Chien, H.; Zou, L.; Koleske, M.L.; Giacomini, K.M. Deorphaning a Solute Carrier 22 Family Member, SLC22A15, through Functional Genomic Studies. FASEB J. 2020, 34, 15734–15752. [Google Scholar] [CrossRef]
- Tang, R.M.Y.; Cheah, I.K.-M.; Yew, T.S.K.; Halliwell, B. Distribution and Accumulation of Dietary Ergothioneine and Its Metabolites in Mouse Tissues. Sci. Rep. 2018, 8, 1601. [Google Scholar] [CrossRef]
- Hang, L.; Basil, A.H.; Lim, K.-L. Nutraceuticals in Parkinson’s Disease. NeuroMolecular Med. 2016, 18, 306–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, Y.; Le, W. Recent Advances and Perspectives of Metabolomics-Based Investigations in Parkinson’s Disease. Mol. Neurodegener. 2019, 14, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.P.; Duda, J.E. Dietary Modifications in Parkinson’s Disease: A Neuroprotective Intervention? Med. Hypotheses 2015, 85, 1002–1005. [Google Scholar] [CrossRef]
- Mori, K.; Inatomi, S.; Ouchi, K.; Azumi, Y.; Tuchida, T. Improving Effects of the Mushroom Yamabushitake (Hericium Erinaceus) on Mild Cognitive Impairment: A Double-Blind Placebo-Controlled Clinical Trial. Phytother. Res. 2009, 23, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Roda, E.; Priori, E.C.; Ratto, D.; de Luca, F.; di Iorio, C.; Angelone, P.; Locatelli, C.A.; Desiderio, A.; Goppa, L.; Savino, E.; et al. Neuroprotective Metabolites of Hericium Erinaceus Promote Neuro-Healthy Aging. Int. J. Mol. Sci. 2021, 22, 6379. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Cheah, I.K.-M.; Ng, M.M.-X.; Li, J.; Chan, S.M.; Lim, S.L.; Mahendran, R.; Kua, E.-H.; Halliwell, B. The Association between Mushroom Consumption and Mild Cognitive Impairment: A Community-Based Cross-Sectional Study in Singapore. J. Alzheimer’s Dis. 2019, 68, 197–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, X.; Cioccoloni, G.; Sier, J.H.; Naseem, K.M.; Thorne, J.L.; Moore, J.B. Ergothioneine Supplementation in People with Metabolic Syndrome (ErgMS): Protocol for a Randomised, Double-Blind, Placebo-Controlled Pilot Study. Pilot Feasibility Stud. 2021, 7, 193. [Google Scholar] [CrossRef]
- Ishimoto, T.; Kato, Y. Ergothioneine in the Brain. FEBS Lett. 2022. [Google Scholar] [CrossRef]
- Ishimoto, T.; Masuo, Y.; Kato, Y.; Nakamichi, N. Ergothioneine-Induced Neuronal Differentiation Is Mediated through Activation of S6K1 and Neurotrophin 4/5-TrkB Signaling in Murine Neural Stem Cells. Cell. Signal. 2019, 53, 269–280. [Google Scholar] [CrossRef]
- Lamhonwah, A.-M.; Tein, I. Novel Localization of OCTN1, an Organic Cation/Carnitine Transporter, to Mammalian Mitochondria. Biochem. Biophys. Res. Commun. 2006, 345, 1315–1325. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H. The Unusual Amino Acid L-Ergothioneine Is a Physiologic Cytoprotectant. Cell Death Differ. 2010, 17, 1134–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamichi, N.; Nakayama, K.; Ishimoto, T.; Masuo, Y.; Wakayama, T.; Sekiguchi, H.; Sutoh, K.; Usumi, K.; Iseki, S.; Kato, Y. Food-derived Hydrophilic Antioxidant Ergothioneine Is Distributed to the Brain and Exerts Antidepressant Effect in Mice. Brain Behav. 2016, 6, e00477. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Ozawa, N.; Shinozaki, T.; Wakabayashi, K.; Suzuki, K.; Kawano, Y.; Ohtsu, I.; Tatebayashi, Y. Ergothioneine, a Metabolite of the Gut Bacterium Lactobacillus Reuteri, Protects against Stress-Induced Sleep Disturbances. Transl. Psychiatry 2020, 10, 170. [Google Scholar] [CrossRef] [PubMed]
- Kitsanayanyong, L.; Ohshima, T. Ergothioneine: A Potential Antioxidative and Antimelanosis Agent for Food Quality Preservation. FEBS Lett. 2022. [Google Scholar] [CrossRef] [PubMed]
- Beelman, R.B.; Kalaras, M.D.; Richie, J.P. Micronutrients and Bioactive Compounds in Mushrooms. Nutr. Today 2019, 54, 16–22. [Google Scholar] [CrossRef] [Green Version]
- Beelman, R.B.; Kalaras, M.D.; Phillips, A.T.; Richie, J.P. Is Ergothioneine a ‘Longevity Vitamin’ Limited in the American Diet? J. Nutr. Sci. 2020, 9, e52. [Google Scholar] [CrossRef]
- Zhang, S.; Xie, C.; Lin, J.; Wang, M.; Wang, X.; Liu, Z. Lipoic Acid Alleviates L-DOPA-induced Dyskinesia in 6-OHDA Parkinsonian Rats via Anti-oxidative Stress. Mol. Med. Rep. 2017, 17, 1118–1124. [Google Scholar] [CrossRef]
- Li, Y.-H.; He, Q.; Yu, J.; Liu, C.; Feng, L.; Chai, Z.; Wang, Q.; Zhang, H.; Zhang, G.-X.; Xiao, B.; et al. Lipoic Acid Protects Dopaminergic Neurons in LPS-Induced Parkinson’s Disease Model. Metab. Brain Dis. 2015, 30, 1217–1226. [Google Scholar] [CrossRef]
- Park, J.S.; Choi, H.I.; Kim, D.-H.; Kim, C.S.; Bae, E.H.; Ma, S.K.; Kim, S.W. Alpha-Lipoic Acid Attenuates p-Cresyl Sulfate-Induced Renal Tubular Injury through Suppression of Apoptosis and Autophagy in Human Proximal Tubular Epithelial Cells. Biomed. Pharmacother. 2019, 112, 108679. [Google Scholar] [CrossRef]
- El-Maadawy, W.; Hammam, O.; Seif el-Din, S.; El-Lakkany, N. α-Lipoic Acid Modulates Liver Fibrosis: A Cross Talk between TGF-Β1, Autophagy, and Apoptosis. Hum. Exp. Toxicol. 2020, 39, 440–450. [Google Scholar] [CrossRef]
- Göder, A.; Nagel, G.; Kraus, A.; Dörsam, B.; Seiwert, N.; Kaina, B.; Fahrer, J. Lipoic Acid Inhibits the DNA Repair Protein O6-Methylguanine-DNA Methyltransferase (MGMT) and Triggers Its Depletion in Colorectal Cancer Cells with Concomitant Autophagy Induction. Carcinogenesis 2015, 36, 817–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.; Chen, A.; Yang, P.; Song, X.; Liu, Y.; Li, Z.; Wang, X.; Wang, L.; Li, Y. Alpha-Lipoic Acid Protects Cardiomyocytes against Hypoxia/Reoxygenation Injury by Inhibiting Autophagy. Biochem. Biophys. Res. Commun. 2013, 441, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Liu, K.; Xiao, L.; Jin, S.; Dong, J.; Teng, X.; Guo, Q.; Chen, Y.; Wu, Y. Alpha-Lipoic Acid Regulates the Autophagy of Vascular Smooth Muscle Cells in Diabetes by Elevating Hydrogen Sulfide Level. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2018, 1864, 3723–3738. [Google Scholar] [CrossRef]
- Jia, J.; Gong, X.; Zhao, Y.; Yang, Z.; Ji, K.; Luan, T.; Zang, B.; Li, G. Autophagy Enhancing Contributes to the Organ Protective Effect of Alpha-Lipoic Acid in Septic Rats. Front. Immunol. 2019, 10, 1491. [Google Scholar] [CrossRef]
- Hahm, J.R.; Noh, H.S.; Ha, J.H.; Roh, G.S.; Kim, D.R. Alpha-Lipoic Acid Attenuates Adipocyte Differentiation and Lipid Accumulation in 3T3-L1 Cells via AMPK-Dependent Autophagy. Life Sci. 2014, 100, 125–132. [Google Scholar] [CrossRef]
- Banaclocha, M.M. Therapeutic Potential of N-Acetylcysteine in Age-Related Mitochondrial Neurodegenerative Diseases. Med. Hypotheses 2001, 56, 472–477. [Google Scholar] [CrossRef]
- Holmay, M.J.; Terpstra, M.; Coles, L.D.; Mishra, U.; Ahlskog, M.; Öz, G.; Cloyd, J.C.; Tuite, P.J. N-Acetylcysteine Boosts Brain and Blood Glutathione in Gaucher and Parkinson Diseases. Clin. Neuropharmacol. 2013, 36, 103. [Google Scholar] [CrossRef] [Green Version]
- Monti, D.A.; Zabrecky, G.; Kremens, D.; Liang, T.-W.; Wintering, N.A.; Cai, J.; Wei, X.; Bazzan, A.J.; Zhong, L.; Bowen, B.; et al. N-Acetyl Cysteine May Support Dopamine Neurons in Parkinson’s Disease: Preliminary Clinical and Cell Line Data. PLoS ONE 2016, 11, e0157602. [Google Scholar] [CrossRef]
- Xiong, G.; Zhao, L.; Yan, M.; Wang, X.; Zhou, Z.; Chang, X. N-acetylcysteine Alleviated Paraquat-induced Mitochondrial Fragmentation and Autophagy in Primary Murine Neural Progenitor Cells. J. Appl. Toxicol. 2019, 39, 1557–1567. [Google Scholar] [CrossRef]
- Boz, Z.; Hu, M.; Yu, Y.; Huang, X.-F. N-Acetylcysteine Prevents Olanzapine-Induced Oxidative Stress in MHypoA-59 Hypothalamic Neurons. Sci. Rep. 2020, 10, 19185. [Google Scholar] [CrossRef]
- Li, B.; Sun, Y.; Wang, J.-P.; Chi, R.-F.; Wang, K.; Yang, Z.-J.; Qin, F.-Z.; Fan, B. Antioxidant N-Acetylcysteine Inhibits Maladaptive Myocyte Autophagy in Pressure Overload Induced Cardiac Remodeling in Rats. Eur. J. Pharmacol. 2018, 839, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, C.; Peng, M.; Wang, L.; Zhao, D.; Wu, T.; Yi, D.; Hou, Y.; Wu, G. N-Acetylcysteine Improves Intestinal Function and Attenuates Intestinal Autophagy in Piglets Challenged with β-Conglycinin. Sci. Rep. 2021, 11, 1261. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kang, S.U.; Lee, Y.S.; Jang, J.Y.; Kang, H.; Kim, C.-H. Protective Effects of N-Acetylcysteine against Radiation-Induced Oral Mucositis In Vitro and In Vivo. Cancer Res. Treat. 2020, 52, 1019. [Google Scholar] [CrossRef]
- Tripathi, A.; Thangaraj, A.; Chivero, E.T.; Periyasamy, P.; Burkovetskaya, M.E.; Niu, F.; Guo, M.-L.; Buch, S. N-Acetylcysteine Reverses Antiretroviral-Mediated Microglial Activation by Attenuating Autophagy-Lysosomal Dysfunction. Front. Neurol. 2020, 11, 840. [Google Scholar] [CrossRef]
- Wang, S.; Wang, C.; Yan, F.; Wang, T.; He, Y.; Li, H.; Xia, Z.; Zhang, Z. N-Acetylcysteine Attenuates Diabetic Myocardial Ischemia Reperfusion Injury through Inhibiting Excessive Autophagy. Mediat. Inflamm. 2017, 2017, 9257291. [Google Scholar] [CrossRef]
- Di Miceli, M.; Bosch-Bouju, C.; Layé, S. PUFA and Their Derivatives in Neurotransmission and Synapses: A New Hallmark of Synaptopathies. Proc. Nutr. Soc. 2020, 79, 388–403. [Google Scholar] [CrossRef]
- Taghizadeh, M.; Tamtaji, O.R.; Dadgostar, E.; Daneshvar Kakhaki, R.; Bahmani, F.; Abolhassani, J.; Aarabi, M.H.; Kouchaki, E.; Memarzadeh, M.R.; Asemi, Z. The Effects of Omega-3 Fatty Acids and Vitamin E Co-Supplementation on Clinical and Metabolic Status in Patients with Parkinson’s Disease: A Randomized, Double-Blind, Placebo-Controlled Trial. Neurochem. Int. 2017, 108, 183–189. [Google Scholar] [CrossRef]
- Chen, X.; Pan, Z.; Fang, Z.; Lin, W.; Wu, S.; Yang, F.; Li, Y.; Fu, H.; Gao, H.; Li, S. Omega-3 Polyunsaturated Fatty Acid Attenuates Traumatic Brain Injury-Induced Neuronal Apoptosis by Inducing Autophagy through the Upregulation of SIRT1-Mediated Deacetylation of Beclin-1. J. Neuroinflamm. 2018, 15, 310. [Google Scholar] [CrossRef]
- Yang, B.; Zhou, Y.; Wu, M.; Li, X.; Mai, K.; Ai, Q. ω-6 Polyunsaturated Fatty Acids (Linoleic Acid) Activate Both Autophagy and Antioxidation in a Synergistic Feedback Loop via TOR-Dependent and TOR-Independent Signaling Pathways. Cell Death Dis. 2020, 11, 607. [Google Scholar] [CrossRef]
- Chen, Y.; Xu, C.; Yan, T.; Yu, C.; Li, Y. ω-3 Fatty Acids Reverse Lipotoxity through Induction of Autophagy in Nonalcoholic Fatty Liver Disease. Nutrition 2015, 31, 1423–1429. [Google Scholar] [CrossRef]
- Hwang, W.-M.; Bak, D.-H.; Kim, D.H.; Hong, J.Y.; Han, S.-Y.; Park, K.-Y.; Lim, K.; Lim, D.-M.; Kang, J.G. Omega-3 Polyunsaturated Fatty Acids May Attenuate Streptozotocin-Induced Pancreatic β-Cell Death via Autophagy Activation in Fat1 Transgenic Mice. Endocrinol. Metab. 2015, 30, 569. [Google Scholar] [CrossRef] [Green Version]
- Fang, X.; Ge, K.; Song, C.; Ge, Y.; Zhang, J. Effects of N-3PUFAs on Autophagy and Inflammation of Hypothalamus and Body Weight in Mice. Biochem. Biophys. Res. Commun. 2018, 501, 927–932. [Google Scholar] [CrossRef]
- Bak, D.H.; Zhang, E.; Yi, M.-H.; Kim, D.-K.; Lim, K.; Kim, J.-J.; Kim, D.W. High Ω3-Polyunsaturated Fatty Acids in Fat-1 Mice Prevent Streptozotocin-Induced Purkinje Cell Degeneration through BDNF-Mediated Autophagy. Sci. Rep. 2015, 5, 15465. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Tang, Y.; Wang, S.; Zhou, J.; Zhou, J.; Lu, X.; Bai, X.; Wang, X.-Y.; Chen, Z.; Zuo, D. Endogenous N-3 Polyunsaturated Fatty Acids Attenuate T Cell-Mediated Hepatitis via Autophagy Activation. Front. Immunol. 2016, 7, 350. [Google Scholar] [CrossRef] [Green Version]
- Gwon, D.; Hwang, T.; Ro, J.-Y.; Kang, Y.-J.; Jeong, J.; Kim, D.-K.; Lim, K.; Kim, D.; Choi, D.; Kim, J.-J. High Endogenous Accumulation of ω-3 Polyunsaturated Fatty Acids Protect against Ischemia-Reperfusion Renal Injury through AMPK-Mediated Autophagy in Fat-1 Mice. Int. J. Mol. Sci. 2017, 18, 2081. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.W.; Lee, J.; Lee, J.H.; Park, B.J.; Lee, E.J.; Shin, S.; Cha, G.-H.; Lee, Y.-H.; Lim, K.; Yuk, J.-M. Omega-3 Polyunsaturated Fatty Acids Prevent Toxoplasma Gondii Infection by Inducing Autophagy via AMPK Activation. Nutrients 2019, 11, 2137. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Jing, K.; Shin, S.; Jeong, S.; Han, S.-H.; Oh, H.; Yoo, Y.-S.; Han, J.; Jeon, Y.-J.; Heo, J.-Y.; et al. Ω3-Polyunsaturated Fatty Acids Induce Cell Death through Apoptosis and Autophagy in Glioblastoma Cells: In Vitro and in Vivo. Oncol. Rep. 2017, 39, 239–246. [Google Scholar] [CrossRef] [Green Version]
- Sharon, R.; Goldberg, M.S.; Bar-Josef, I.; Betensky, R.A.; Shen, J.; Selkoe, D.J. Synuclein Occurs in Lipid-Rich High Molecular Weight Complexes, Binds Fatty Acids, and Shows Homology to the Fatty Acid-Binding Proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 9110–9115. [Google Scholar] [CrossRef] [Green Version]
- Davidson, W.S.; Jonas, A.; Clayton, D.F.; George, J.M. Stabilization of α-Synuclein Secondary Structure upon Binding to Synthetic Membranes. J. Biol. Chem. 1998, 273, 9443–9449. [Google Scholar] [CrossRef] [Green Version]
- Shamoto-Nagai, M.; Hisaka, S.; Naoi, M.; Maruyama, W. Modification of α-Synuclein by Lipid Peroxidation Products Derived from Polyunsaturated Fatty Acids Promotes Toxic Oligomerization: Its Relevance to Parkinson Disease. J. Clin. Biochem. Nutr. 2018, 62, 207–212. [Google Scholar] [CrossRef] [Green Version]
- Almandoz-Gil, L.; Welander, H.; Ihse, E.; Khoonsari, P.E.; Musunuri, S.; Lendel, C.; Sigvardson, J.; Karlsson, M.; Ingelsson, M.; Kultima, K.; et al. Low Molar Excess of 4-Oxo-2-Nonenal and 4-Hydroxy-2-Nonenal Promote Oligomerization of Alpha-Synuclein through Different Pathways. Free. Radic. Biol. Med. 2017, 110, 421–431. [Google Scholar] [CrossRef]
- Näsström, T.; Fagerqvist, T.; Barbu, M.; Karlsson, M.; Nikolajeff, F.; Kasrayan, A.; Ekberg, M.; Lannfelt, L.; Ingelsson, M.; Bergström, J. The Lipid Peroxidation Products 4-Oxo-2-Nonenal and 4-Hydroxy-2-Nonenal Promote the Formation of α-Synuclein Oligomers with Distinct Biochemical, Morphological, and Functional Properties. Free. Radic. Biol. Med. 2011, 50, 428–437. [Google Scholar] [CrossRef]
- Woźniak, Ł.; Skąpska, S.; Marszałek, K. Ursolic Acid—A Pentacyclic Triterpenoid with a Wide Spectrum of Pharmacological Activities. Molecules 2015, 20, 20614–20641. [Google Scholar] [CrossRef] [Green Version]
- Tsai, S.-J.; Yin, M.-C. Antioxidative and Anti-Inflammatory Protection of Oleanolic Acid and Ursolic Acid in PC12 Cells. J. Food Sci. 2008, 73, 174–178. [Google Scholar] [CrossRef]
- Ramos-Hryb, A.B.; Pazini, F.L.; Kaster, M.P.; Rodrigues, A.L.S. Therapeutic Potential of Ursolic Acid to Manage Neurodegenerative and Psychiatric Diseases. CNS Drugs 2017, 31, 1029–1041. [Google Scholar] [CrossRef]
- Heitman, E.; Ingram, D.K. Cognitive and Neuroprotective Effects of Chlorogenic Acid. Nutr. Neur. 2017, 20, 32–39. [Google Scholar] [CrossRef]
- Ito, H.; Sun, X.-L.; Watanabe, M.; Okamoto, M.; Hatano, T. Chlorogenic Acid and Its Metabolite m-Coumaric Acid Evoke Neurite Outgrowth in Hippocampal Neuronal Cells. Biosci. Biotechnol. Biochem. 2008, 72, 885–888. [Google Scholar] [CrossRef] [Green Version]
- Teraoka, M.; Nakaso, K.; Kusumoto, C.; Katano, S.; Tajima, N.; Yamashita, A.; Zushi, T.; Ito, S.; Matsura, T. Cytoprotective Effect of Chlorogenic Acid against Alpha-Synuclein-Related Toxicity in Catecholaminergic PC12 Cells. J. Clinic. Biochem. Nutr. 2012, 51, 122–127. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.S.; Rai, S.N.; Birla, H.; Zahra, W.; Kumar, G.; Gedda, M.R.; Tiwari, N.; Patnaik, R.; Singh, R.K.; Singh, S.P. Effect of Chlorogenic Acid Supplementation in MPTP-Intoxicated Mouse. Front. Pharmacol. 2018, 9, 757. [Google Scholar] [CrossRef] [Green Version]
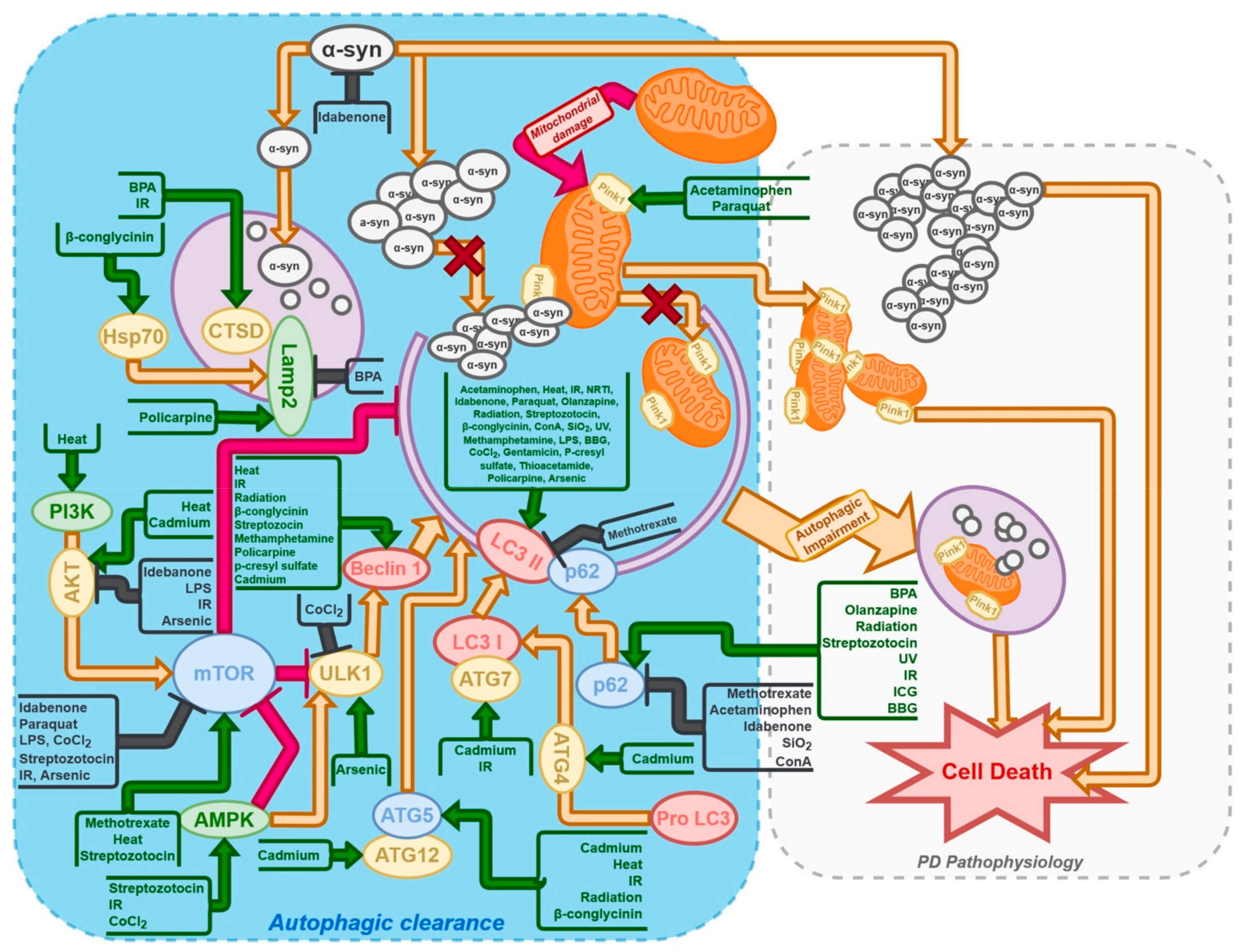

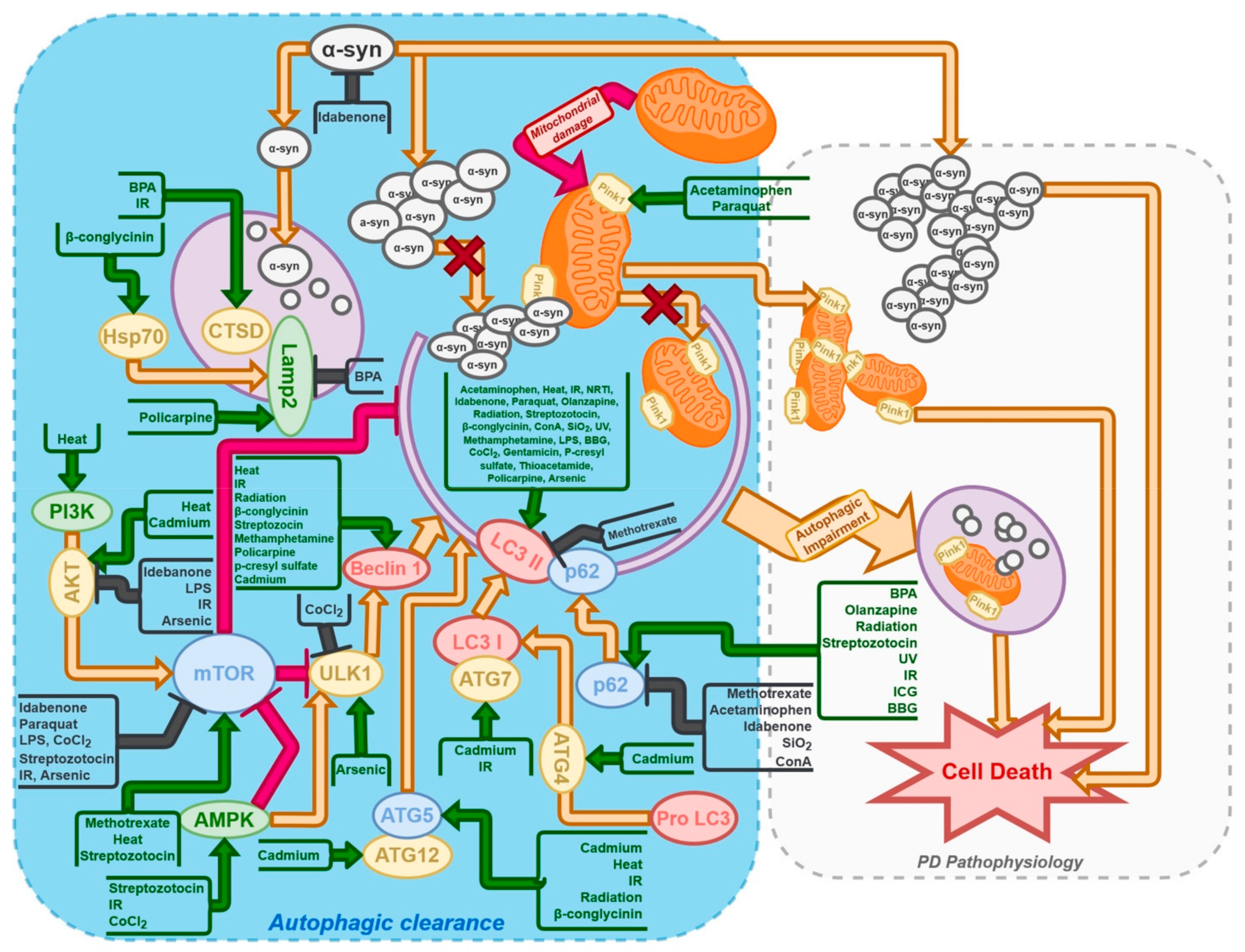{kind=link}
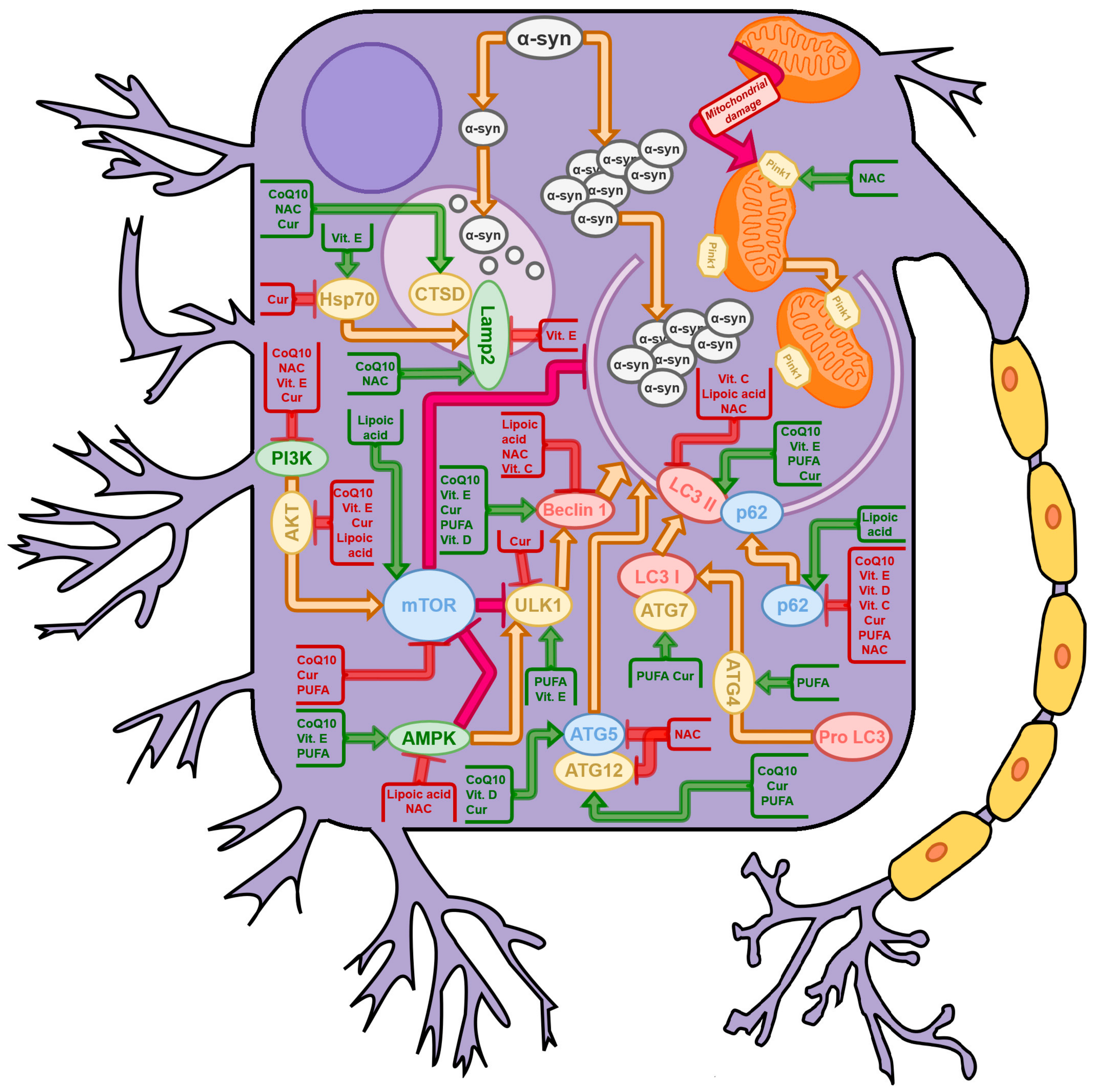
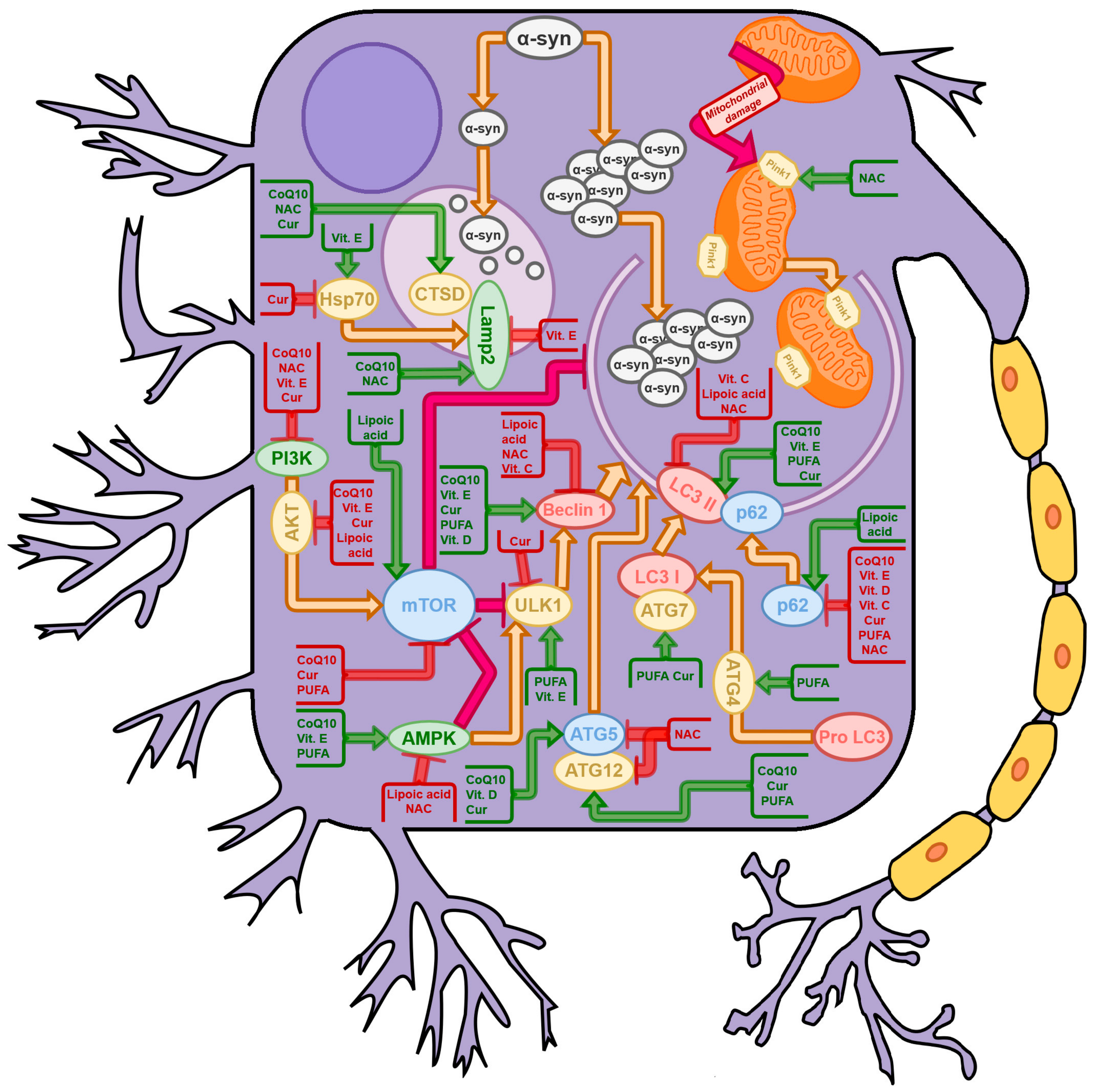{kind=link}
| Compound Used | Experimental Model | Signaling Mediators | Results | Ref. |
|---|---|---|---|---|
| ATRA | SKBR3 and MDA-MB453 breast cancer cell lines | ↑LC3-II/I (SKBR3) | ATRA-induced autophagy was mediated by RARα in SKBR3 cells. | [49] |
| Crocin | Middle cerebral artery occlusion rat model | ↑p62 ↑p-mTOR/mTOR ↓LC3-II/I ↓p-AMPK/AMPK ↓ULK1 | Crocin reduced the level of autophagy following cerebral ischemia by activating mTOR. | [50] |
| β-carotene | Rats with LPS-induced intestinal inflammation | ↑p-AKT/AKT ↓LC3-II/I | β-carotene protected rat intestinal cells, most probably via the JAK2/STAT3 and JNK/p38 MAPK signaling pathways. | [51] |
| Lycopene (Lyc) | Cadmium-induced hippocampal dysfunction mice | ↓Beclin-1 ↓AKT1 ↓MAPK ↓Atg * mRNA | Lyc reversed Cd-induced dysfunctions and neurotoxicity. | [52] |
| Endothelial progenitor cells isolated from T2DM rats | ↓Beclin-1 ↓LC3-II/I | Lyc promoted EPCs survival and protected EPCs from apoptosis and autophagy induced by AGEs. | [53] | |
| Gentamicin-induced renal cortical oxidative stress rat model | ↓LC3-II/I | Lyc decreased the level of the LC3-II/I autophagy marker. | [54] | |
| 4HPR | hiPSC-derived motoneurons from ALS patients’ keratinocytes | ↑LC3-II/I ↑SQSTM1 ↓ATG10 | 4HPR induced autophagy and downregulated Atg10 expression via modulation of TBK1. | [55] |
| Astaxanthin | H. pylori-infected AGS cells | ↑LC3-II/I ↑p-AMPK/AMPK ↑p-ULK1/ULK1 ↓p62 ↓p-mTOR/mTOR | Astaxanthin increased autophagy through activation of the AMPK pathway. | [56] |
| Lutein | Cobalt (II) Chloride-Induced Hypoxia in Rat-derived Müller Cells | ↓LC3-II/I | Lutein suppressed autophagosome formation after hypoxic insult and inhibited autophagy after rapamycin treatment. | [57] |
| IEC-6 rat intestinal epithelial cells | ↑LC3-II/I ↑Beclin-1 ↑p-AMPK ↑p-JNK ↑p-p38 ↑p-mTOR | Lutein induced autophagy via the upregulation of Beclin-1 in IEC-6 cells. | [58] | |
| ICG and BBG-treated ARPE-19 and 661W cell lines | ↑LC3-II/I | Lutein induced autophagy and diminished the cytotoxic effects of ICG and BBG in ocular cells. | [59] | |
| Fucoxanthin | Nasopharyngeal carcinoma C666-1 cell line | ↑LC3-II/I ↑p62 ↑ATG7 ↓ATG4B | Fucoxanthin induced autophagy and apoptosis in C666-1 cells. | [60] |
| Traumatic brain injury (TBI) mice model | ↑LC3-II/I ↑Beclin-1 | Fucoxanthin exerted protective effects, potentially via regulation of the Nrf2-autophagy pathway. | [61] |
| Compound Used | Experimental Model | Signaling Mediators | Results | Ref. |
|---|---|---|---|---|
| AA | Pilocarpine-induced rat model of seizures | ↓Beclin-1 | AA partially inhibited the pilocarpine-mediated induction of oxidative stress and autophagy. | [73] |
| Murine bone marrow stromal cells (BMSCs) | ↓LC3 ↓p62 | VitC significantly rescued BMSCs from oxidative stress by regulating autophagy. | [74] | |
| SA | Methamphetamine-treated primary rat cortical neuron-glia cells | ↓LC3-II/I ↓Beclin-1 | VitC partially attenuated the induction of autophagy, most probably via an ROS-dependent mechanism. | [75] |
| Compound Used | Experimental Model | Signaling Mediators | Results | Ref. |
|---|---|---|---|---|
| 1,25(OH)2D3 | SiO2-mediated lung injury in vitro model (THP-1 and BEAS-2B cells) | ↑LC3 ↓p62 | VitD protected against particle-induced cell damage via the induction of autophagy in an Nrf2-dependent manner. | [79] |
| P. gingivalis-infected U937-derived macrophages | ↑LC3-II/I ↑Atg5 ↓p62 | VitD induced autophagy to degrade live P. gingivalis. | [80] | |
| STZ-induced T2DM mouse model | ↑Beclin-1 mRNA ↑LC3 mRNA | VitD induced autophagy and suppressed apoptosis of pancreatic β cells. | [81] | |
| MCF-7 cell line (with or without VDR knockout) | ↑LC3 | VitD increased the level of autophagy in MCF-7 cells. VDR gene knockout caused even higher than vitD upregulation of autophagy, suggesting a possible VDR-dependent mechanism of autophagy modulation. | [82] | |
| 25(OH)D3 | UV-mediated acute skin injury mouse model | ↑LC3-II/I ↓p62 | VitD resolved skin injury via inhibition of inflammatory cytokines associated with enhanced autophagy in myeloid anti-inflammatory M2 macrophages. | [83] |
| VitD3 | Aspergillus fumigatus-infected mice model | ↓LC3-II | VitD delayed the formation of lysosomes against A. fumigatus through autophagy. | [84] |
| VitD deficiency | Two groups of patients: HCV HCV + HCC | ↓LC3 | Downregulation of autophagy was observed in vitD3-deficient patients. | [85] |
| Compound Used | Experimental Model | Signaling Mediators | Results | Ref. |
|---|---|---|---|---|
| VitE deficiency | Hippocampal neurons isolated from vitE-deficient mice | ↑LC3-II | VitE deficiency led to axonal degeneration. LC3-II expression is higher in short-term rather than long-term deficiency. | [94] |
| α-tocph | Rats with pilocarpine-induced status epilepticus | ↓LC3-II/I ↓Beclin-1 | α-tocph inhibited autophagy in the hippocampus of the animals. | [95] |
| ↓Lamp2a | α-tocph inhibited CMA in the hippocampus of rats. | [96] | ||
| Proximal tubules isolated from a DN patients/rat model | ↓LC3-II ↓SQSTM1 | High dose of α-tocph downregulated the autophagy markers. | [97] | |
| Chronic unpredictable mild stress mice | ↑LC3-II ↑p-AMPK/AMPK ↑ULK1Ser317 ↓p62 ↓p-mTOR/mTOR ↓p-P70S6K1 | α-tocph induced antidepressive responses via the promotion of autophagy in chronic unpredictable mild stress mice. | [98] | |
| Primary rat hepatocytes | ↑LC3-II/I | Both compounds increased autophagy by accelerating LC3 conversion. | [99] | |
| α-toctr | H-4-II-E cells | [85] | ||
| γ-toctr | Ischemia/reperfusion rat model | ↑p-Akt/Akt ↑LC3-II/I ↑Beclin-1 ↓p-mTOR/mTOR | γ-toctr-mediated cardioprotection was achieved by its ability to induce autophagy. | [100] |
| Human prostate cancer cell lines, PC-3 and LNCaP cells | ↑LC3-II/I | γ-toctr promoted autophagy in PC-3 and LNCaP cells. | [101] | |
| HCT-116 cells | ↑LC3-II/I | γ-toctr induced autophagy. | [102] | |
| Mouse (+SA) and human (MCF-7) mammary cancer cells | ↑LC3-II/I ↑Beclin-1 ↓PI3K ↓p-Akt ↓p-mTOR | γ-toctr induced autophagy in cancer (+SA, MCF-7) cell lines but did not in normal mammary cell lines (CL-S1, MCF-10A). | [103] | |
| Palm oil TRF | Rat pancreatic stellate cells | ↑LC3-II/I | TRF reduced the viability of activated PSCs by targeting the mitochondrial permeability transition pore. | [104] |
| α-TEA | 4T1 and 3LL cells | ↑LC3-II/I | Autophagy and apoptosis signaling pathways are activated during α-TEA-induced death of cells. | [105] |
| Compound Used | Experimental Model | Signaling Mediators | Results | Ref. |
|---|---|---|---|---|
| CoQ10 | Methotrexate-induced lung and liver fibrosis rat model | ↑LC3 ↑p62 ↓mTOR | CoQ protected against lung and liver fibrosis via induction of autophagy. | [113] |
| BPA-treated C2C12 cells | ↑LC3-II ↑Lamp2 ↓p62 | CoQ promoted autophagy by improving lysosomal function. | [114] | |
| Fibroblasts derived from an MERRF patient | ↑phospho-AMPK ↓LC3-II ↓p62 | CoQ restored the autophagic flux in MERRF fibroblasts. | [115] | |
| Acetaminophen-induced liver injury mice model | ↑LC3-II ↑Parkin ↑mito-p62 ↓p62 | CoQ activated mitophagy and protected against acetaminophen-induced liver injury. | [116] | |
| Heat-stressed chicken primary myocardial cells | ↑LC3 ↑Beclin-1 ↑Atg5 ↓p-Akt/Akt ↓p-mTOR/mTOR ↓p-PI3K/PI3K | CoQ protected the cells during heat stress by upregulation of autophagy via the PI3K/Akt/mTOR pathway. | [117] | |
| Fibroblasts from patients with MELAS | ↓LC3-II ↓Beclin-1 ↓Atg12 | CoQ partially alleviated MELAS-induced activation of autophagy. | [119] | |
| NRTI-treated HUVEC cells | ↓LC3-II | CoQ prevented an NRTI-mediated increase in LC3-II. | [120] | |
| Acute myocardial ischemia-reperfusion rat model | ↑LC3-II ↑Beclin-1 ↑Atg5 ↓p62 | CoQ protected against acute myocardial ischemia-reperfusion injury via the autophagy pathway. | [118] | |
| Primary pancreatic stellate cells isolated from a mice model of pancreatic fibrosis | ↑p-Akt ↑p-mTOR ↑p-PI3K ↓LC3-II/I ↓Atg5 ↓Beclin-1 ↓p62 | CoQ alleviated pancreatic fibrosis by the ROS-triggered PI3K/Akt/mTOR pathway. | [121,122] | |
| MitoQ | Sepsis-induced acute lung injury rat model | ↑p-Akt/Akt ↑p-mTOR/mTOR ↑p-GSK-3β/GSK-3β ↓Beclin-1 ↓LC3-II/I | MitoQ protected sepsis-induced acute lung injury by activating the PI3K/Akt/GSK-3β/mTOR pathway. | [123] |
| CoQ deficiency | Fibroblasts from patients with a CoQ deficiency | ↑Atg12 ↑Beclin ↑LC3 ↑cathepsin D | Authors suggested a protective role of autophagy in CoQ deficiency. | [124] |
| Idabenone | SH-SY5Y-A53T cells | ↑LC3-II | Idabenone enhanced the autophagy-mediated clearance of α-syn. | [125] |
| Antroquinonol | PANC-1 and AsPC-1 cells | ↑LC3-II ↓p-AKT ↓p-mTOR | Antroquinonol induced anticancer activity through an inhibitory effect on PI3K/Akt/mTOR pathways. | [126] |
| Compound Used | Experimental Model | Signaling Mediators | Results | Ref. |
|---|---|---|---|---|
| Cur | SH-SY5Y neuroblastoma cells | ↑LAMP1 ↑LC3-II ↓p62 | Cur regulated autophagy by controlling TFEB through the inhibition of GSK-3β. | [14] |
| NTERA2 stem cells | ↑LC3 ↑LAMP1 ↑Atg12 ↑Atg5 | Cur induced neurogenesis of NTERA2 cells via the activation of ROS-mediated autophagy. | [132] | |
| Passive Heymann nephritis rat model | ↑LC3-II/I ↑Beclin-1 ↓PI3K ↓p-mTOR | Cur induced autophagy through the PI3K/AKT/mTOR and Nrf2/HO-1 pathways. | [128] | |
| A172 glioblastoma cells | ↑LC3-II ↑Atg5 ↑Atg7 ↑Atg12 ↑Beclin-1 | Cur induced autophagy and led to the death of cells. | [133] | |
| CRPC cells (DU145 and PC3 cell lines) | ↑LC3-II | Cur induced apoptosis and protective autophagy in CRPC cells. | [134] | |
| Arsenic-treated PC12 cells | ↑mTOR ↑Akt ↑ERK ↑Nrf2 ↓ULK ↓LC3-II | Cur alleviated arsenic-triggered toxicity in PC12 cells by regulating autophagy and apoptosis. | [135] | |
| Rat neural stem cells differentiated into GFAP+ astrocytes or dcX+ immature neurons | ↓Atg7 ↓p62 ↓ULK | Cur inhibited NSC differentiation into GFAP+ astrocytes or dcX+ immature neurons. | [136] | |
| Renal tissue derived from an STZ-induced diabetic nephropathy rat model | ↑LC3-II/I ↓p62 ↓p-mTOR ↓p-Akt ↓PI3K | Cur protected podocytes by alleviating EMT via the PI3K/Akt/mTOR pathway. | [129] | |
| SH-SY5Y cells treated with paraquat | ↓LC3-II | Cur reversed the paraquat-mediated induction of autophagy in SH-SY5Y cells. | [137] | |
| Rat model of sciatic nerve injury | ↑LC3-II/I ↑Beclin-1 ↑p-Erk1/2 ↓p62 ↓p-Akt | Cur promoted injury-induced cell autophagy, remyelination, and axon regeneration in the sciatic nerve of rats. | [138] | |
| Double-transgenic mice (hAPP and mhPS1) | ↑LC3-II/I ↑Beclin-1 ↓PI3K ↓p-Akt ↓p-mTOR | Cur inhibited Aβ generation and induced autophagy by downregulation of the PI3K/Akt/mTOR pathway. | [139] | |
| OGD/R model of the PC12 cell line | ↑p62 ↓LC3-II/I | Cur exerted neuroprotection via regulation of the reciprocal function between autophagy and HIF-1α. | [140] | |
| SH-SY5Y neuroblastoma cells with A53T mutation in the SNCA gene | ↑LC3-II/I ↓p-mTOR ↓α-syn | Cur efficiently reduced the accumulation of A53T α-synuclein through downregulation of the mTOR/p70S6K signaling and recovery of macroautophagy. | [141] | |
| SK-OV-3, A2780 cell lines | ↑LC3-II/I ↓p-Akt ↓p-mTOR ↓p-p70S6K | Cur induced protective autophagy via inhibition of the AKT/mTOR/p70S6K pathway. | [142] | |
| Hepatic fibrosis rat model | ↑Atg-7 ↑Beclin-1 ↑LC3-II/I ↓PI3K mRNA ↓mTOR mRNA ↓SQSTM1 mRNA | Cur effectively reduced the occurrence of EMT via the activation of autophagy. | [131] | |
| Cur and SLCP | U-87MG, GL261, F98 cell lines | ↑Atg5 (U-87MG) ↑Atg7 (U-87MG and GL261) ↑Beclin-1 ↑LC3-II/I ↑p62 ↓mTOR ↓PI3K ↓Akt | Increased levels of autophagy and decreased levels of mitophagy markers, along with inhibition of the PI3K-Akt/mTOR pathway were noted. The effects were greater in the SLCP-treated group when compared to Cur. | [143] |
| C6-glioma and N2a cell lines | ↑Beclin-1 (C6-glioma) ↑p-Akt (C6-glioma) ↓Atg7 | |||
| “E4” Cur derivative [144] | N2a cell line | ↑LC3-II ↑LAMP1 ↑CSTD ↓p-Akt ↓p-mTOR | E4 induced TFEB activation mainly through Akt-mTORC1 inhibition, promoting the degradation of α-syn, and protected against the cytotoxicity of MPP+. | [15] |
| THCu | Traumatic brain injury (TBI) rat model | ↑LC3-II/I ↑Beclin-1 | Treatment with THCu improved neurological function via the activation of autophagy and attenuation of oxidative stress. | [145] |
| ↑LC3-II/I ↑Beclin-1 ↓p62 | THCu protected neurons from TBI-induced apoptotic neuronal death. | [144] | ||
| AO-2 | OGD/R model of primary culture of rat cortical neurons | ↑p-Akt ↑p-mTOR ↓LC3-II/I | AO-2 increased the resistance of cortical neurons to OGD/R by decreasing autophagy and cell apoptosis, which involves an mTOR-dependent mechanism. | [146] |
| Compound Used | Experimental Model | Signaling Mediators | Results | Ref. |
|---|---|---|---|---|
| α-LA | p-Cresyl sulfate-induced renal tubular injury HK-2 cells | ↓LC3-II/I ↓Beclin-1 ↓p-ERK ↓p-p38 ↓p-JNK | LA treatment reduced apoptosis and autophagy by modulating the ER stress and MAPK/NF-κB signaling pathways. | [179] |
| TAA-induced liver fibrosis rat model | ↓LC3-II/I | LA inhibited autophagy and induced apoptotic clearance of activated HSCs. | [180] | |
| Colorectal cancer cell lines: HCT116, RKO | ↑LC3B ↓p-Akt (HCT116) | LA inhibited MGMT and induced autophagy. | [181] | |
| H9c2 cardiomyocytes derived from rat myocardium under H/RI | ↓Beclin-1 ↓LC3-II/I | Pretreatment with LA inhibited the degree of autophagy and increased the viability of cells. | [182] | |
| Vascular smooth muscle cells isolated from rats with STZ-induced T2DM | ↑p62 ↑p-mTOR ↓Beclin-1 ↓p-AMPK | LA treatment reduced the autophagy-related index and activation of the AMPK/mTOR pathway in an H2S-dependent manner. | [183] | |
| Heart, kidney, and small intestine cells isolated from rats with sepsis | ↑LC3-II/I ↑Atg5 ↑Atg7 ↑Beclin-1 ↓p62 | LA upregulated autophagy in the myocardium, kidney, and small intestine of septic rats and reduced apoptosis. | [184] | |
| 3T3-L1 preadipocytes during adipogenesis | ↑p-mTOR ↑p62 ↓p-AMPK ↓LC3-II/I | LA significantly attenuated adipocyte differentiation and consequently decreased the intracellular fat deposit of adipocytes. | [185] |
| Compound Used | Experimental Model | Signaling Mediators | Results | Ref. |
|---|---|---|---|---|
| NAC | PQ-treated primary murine neural progenitor cells | ↑mTOR ↓LC3B ↓Pink1/Parkin | NAC alleviated PQ-induced cytotoxicity and reversed the induction of autophagy. | [189] |
| Abdominal aortic constriction rat model | ↓LC3B ↓Beclin-1 ↓Atg12 ↓p-PI3K/PI3K | NAC reversed the AAC-induced activation of autophagy. | [191] | |
| Olanzapine-treated mHypoA-59 cells | ↓LC3-II | NAC mitigated the olanzapine-induced upregulation of LC3-II. | [190] | |
| Piglets challenged with β-conglycinin | ↑Beclin-1 ↑LC3B-I ↓Atg5 | NAC supplementation improved intestinal function and attenuated intestinal autophagy in β-CG-challenged piglets. | [192] | |
| Radiation-treated HaCaT cells | ↓Beclin-1 ↓p62 ↓LC3 ↓Atg5 | NAC treatment significantly inhibited radiation-induced autophagy in keratinocytes. | [193] | |
| Primary microglia cells isolated from cART-treated rats with HIV | ↑Lamp2 ↑CTSD ↓LC3-II ↓SQSTM1 | NAC reversed the damaging effects of cART. | [194] | |
| STZ-induced rats with T2DM subjected to myocardial I/RI | ↓AMPKα ↓LC3-II/I ↓p62 ↓mTOR | NAC exerted cardioprotective effects primarily through inhibition of excessive autophagy. | [195] |
| Compound Used | Experimental Model | Signaling Mediators | Results | Ref. |
|---|---|---|---|---|
| ω-3 PUFA | Rats with TBI | ↑LC3 ↑Beclin-1 ↑Atg3 ↑Atg7 ↑p62 | ω-3 PUFA supplementation attenuated TBI-induced apoptosis by inducing autophagy through upregulation of the SIRT1-mediated deacetylation of Beclin-1. | [198] |
| ω-6 PUFA (linoleic acid) | Larimichthys crocea (in vivo), Larimichthys crocea hepatocytes in vitro | ↑Beclin-1 ↑ULK1 ↑Atg101 ↑Atg12 ↑Atg4b ↑LC3 ↑p62 | Linoleic acid induced autophagy through the AMPK/mTOR signaling pathway. | [199] |
| DHA, EPA | L02 cell line | ↑LC3-II/I | DHA and EPA protected hepatocytes during lipotoxicity through the induction of autophagy. | [200] |
| STZ-treated Fat-1 transgenic mice | ↑LC3 ↓p62 | Fat-1 modification protected against STZ-induced β cell death by the activation of autophagy. | [201] | |
| Fat-1 transgenic mice | ↑LC3-II/I ↑Atg7 ↓p62 | Fat-1 modification caused a reduction in the body weight and activation of autophagy in the hypothalamus. | [202] | |
| Purkinje cells of fat-1 transgenic mice with STZ-induced diabetes | ↑LC3-II/I ↑Beclin-1 ↑p-Akt ↓p62 | STZ-treated fat-1 mice were protected from Purkinje cell loss and exhibited increased BDNF signaling, which enhanced autophagy. | [203] | |
| Fat-1 transgenic mice with ConA-induced T cell-mediated hepatitis | ↑LC3-II/I ↓p62 | n-3 PUFAs limited ConA-induced hepatitis via an autophagy-dependent mechanism. | [204] | |
| Fat-1 transgenic mice with I/R-mediated renal injury | ↑Beclin-1 ↑Atg7 ↑LC3-II/I ↑p-AMPK/AMPK ↓p62 (baseline lower than wt) ↓p-mTOR/mTOR | ω3-PUFAs in fat-1 mice contributed to AMPK-mediated autophagy activation, leading to a renoprotective response. | [205] | |
| DHA | Bone marrow-derived macrophages from fat-1 transgenic mice | ↑LC3-II/I ↑p-AMPKα | ω3-PUFAs and DHA-mediated control of T. gondii infection suggested that ω3-PUFAs might serve as a therapeutic candidate to prevent toxoplasmosis. | [206] |
| D54MG, U87MG, U251MG, GL261 cell lines | ↑LC3-II/I ↓p-Akt/Akt ↓p-mTOR/mTOR ↓p-AMPK/AMPK | DHA induced cell death through apoptosis and autophagy in glioblastoma cells. | [207] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rakowski, M.; Porębski, S.; Grzelak, A. Nutraceuticals as Modulators of Autophagy: Relevance in Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 3625. https://doi.org/10.3390/ijms23073625
Rakowski M, Porębski S, Grzelak A. Nutraceuticals as Modulators of Autophagy: Relevance in Parkinson’s Disease. International Journal of Molecular Sciences. 2022; 23(7):3625. https://doi.org/10.3390/ijms23073625
Chicago/Turabian StyleRakowski, Michał, Szymon Porębski, and Agnieszka Grzelak. 2022. "Nutraceuticals as Modulators of Autophagy: Relevance in Parkinson’s Disease" International Journal of Molecular Sciences 23, no. 7: 3625. https://doi.org/10.3390/ijms23073625
APA StyleRakowski, M., Porębski, S., & Grzelak, A. (2022). Nutraceuticals as Modulators of Autophagy: Relevance in Parkinson’s Disease. International Journal of Molecular Sciences, 23(7), 3625. https://doi.org/10.3390/ijms23073625