
Iron Metabolism in Aging and Age-Related Diseases

 , ,
, ,
Abstract
1. Introduction
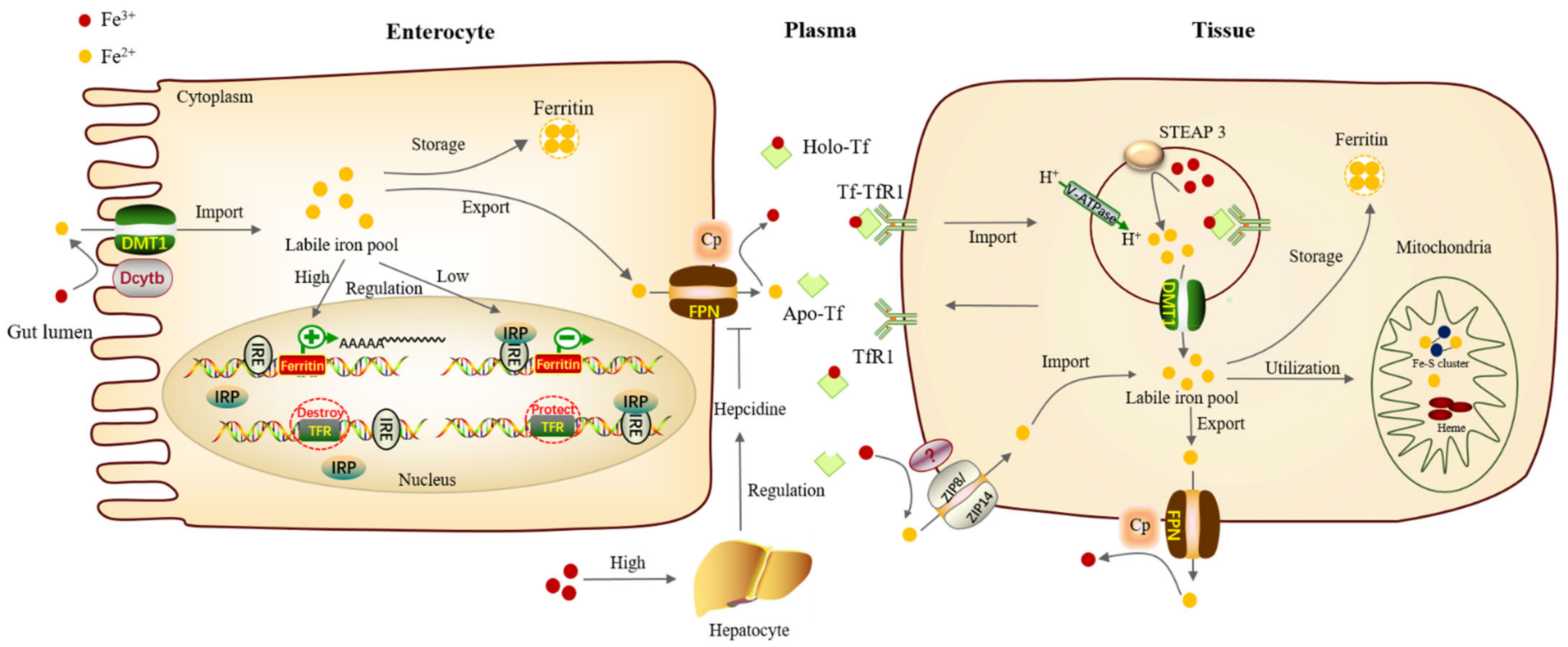
2. Iron Metabolism
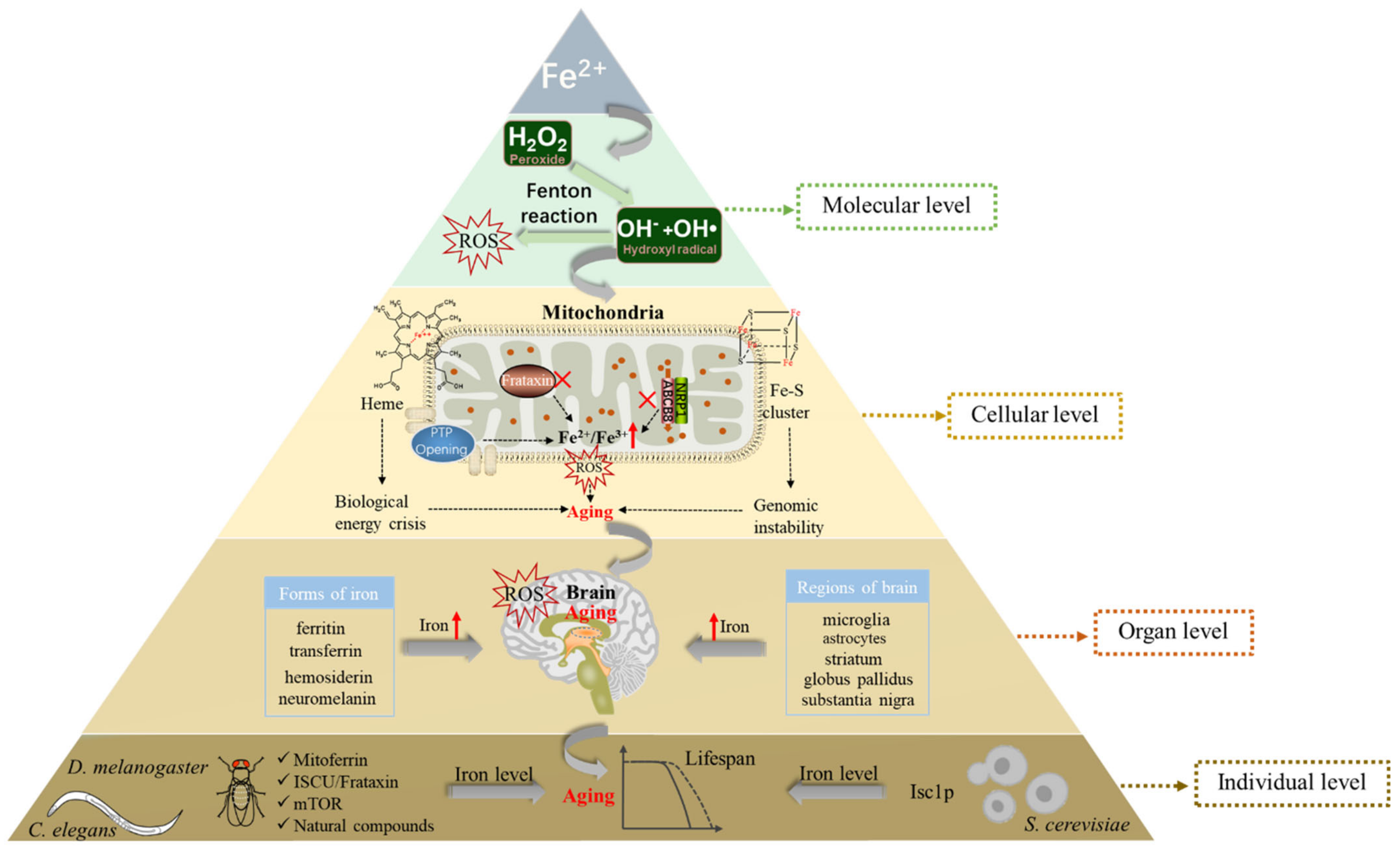
3. Iron Metabolism and Aging
3.1. Iron and the Production of ROS
3.2. Iron Accumulation in the Mitochondria and Aging
3.3. Iron Accumulation and Brain Aging
3.4. Iron Metabolism and Life Expectancy
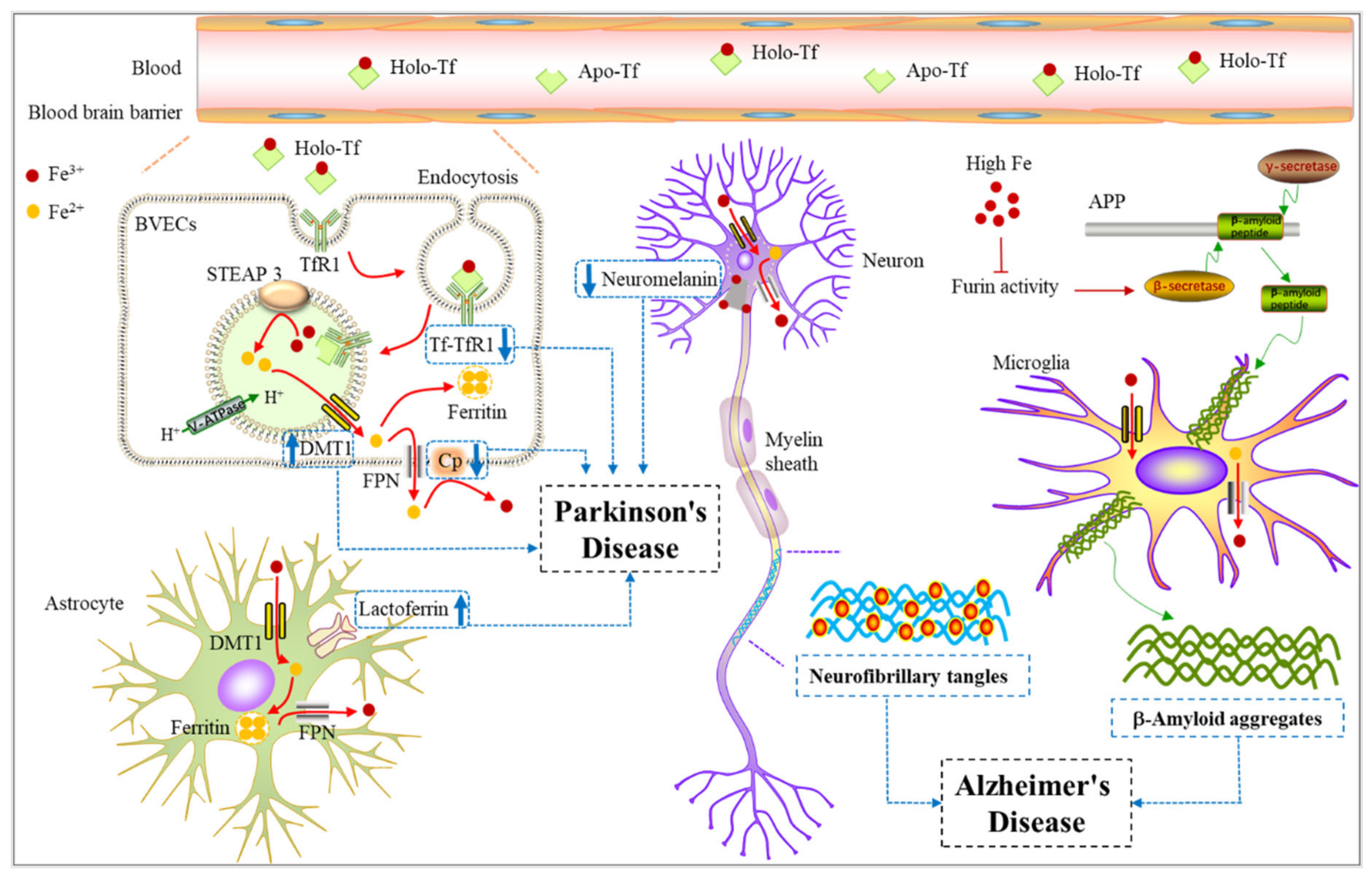
4. Iron Metabolism and Neurodegenerative Diseases
4.1. Iron Metabolism and Alzheimer’s Disease
4.2. Iron Metabolism and Parkinson’s Disease
5. Other Iron-Related Neurological Diseases
6. Treatment Strategies Using Iron Chelators
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, S.; Uliana, A.; Taylor, M.K.; Chakarawet, K.; Bandaru, S.R.S.; Gul, S.; Xu, J.; Ackerman, C.M.; Chatterjee, R.; Furukawa, H.; et al. Iron detection and remediation with a functionalized porous polymer applied to environmental water samples. Chem. Sci. 2019, 10, 6651–6660. [Google Scholar] [CrossRef] [PubMed]
- Sheykhansari, S.; Kozielski, K.; Bill, J.; Sitti, M.; Gemmati, D.; Zamboni, P.; Singh, A.V. Redox metals homeostasis in multiple sclerosis and amyotrophic lateral sclerosis: A review. Cell Death Dis. 2018, 9, 348. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Muckenthaler, M.U.; Andrews, N.C. Balancing acts: Molecular control of mammalian iron metabolism. Cell 2004, 117, 285–297. [Google Scholar] [CrossRef]
- McLean, E.; Cogswell, M.; Egli, I.; Wojdyla, D.; de Benoist, B. Worldwide prevalence of anaemia, WHO Vitamin and Mineral Nutrition Information System, 1993–2005. Public Health Nutr. 2009, 12, 444–454. [Google Scholar] [CrossRef]
- Dev, S.; Babitt, J.L. Overview of iron metabolism in health and disease. Hemodial. Int. 2017, 21, S6–S20. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Tan, Y.; Ouyang, S.; He, J.; Liu, L. Resveratrol protects against myocardial ischemia-reperfusion injury via attenuating ferroptosis. Gene 2022, 808, 145968. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Buffenstein, R.; Edrey, Y.H.; Yang, T.; Mele, J. The oxidative stress theory of aging: Embattled or invincible? Insights from non-traditional model organisms. Age 2008, 30, 99–109. [Google Scholar] [CrossRef]
- Levi, S.; Rovida, E. The role of iron in mitochondrial function. Biochim. Biophys. Acta 2009, 1790, 629–636. [Google Scholar] [CrossRef]
- Harman, D. Free radical theory of aging: An update: Increasing the functional life span. Ann. N. Y. Acad. Sci. 2006, 1067, 10–21. [Google Scholar] [CrossRef]
- Chen, H.; Lukas, T.J.; Du, N.; Suyeoka, G.; Neufeld, A.H. Dysfunction of the retinal pigment epithelium with age: Increased iron decreases phagocytosis and lysosomal activity. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1895–1902. [Google Scholar] [CrossRef] [PubMed]
- Rouault, T.A. Iron on the brain. Nat. Genet. 2001, 28, 299–300. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Youdim, M.B.; Riederer, P.; Connor, J.R.; Crichton, R.R. Iron, brain ageing and neurodegenerative disorders. Nat. Rev. Neurosci. 2004, 5, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, M.; Ben-Shachar, D.; Riederer, P.; Youdim, M.B. Altered brain metabolism of iron as a cause of neurodegenerative diseases? J. Neurochem. 1994, 63, 793–807. [Google Scholar] [CrossRef]
- Chiang, S.; Huang, M.L.H.; Park, K.C.; Richardson, D.R. Antioxidant defense mechanisms and its dysfunctional regulation in the mitochondrial disease, Friedreich’s ataxia. Free Radic. Biol. Med. 2020, 159, 177–188. [Google Scholar] [CrossRef]
- Svetel, M.; Dragašević, N.; Petrović, I.; Novaković, I.; Tomić, A.; Kresojević, N.; Stanković, I.; Kostić, V. NBIA Syndromes: A Step Forward from the Previous Knowledge. Neurol. India 2021, 69, 1380–1388. [Google Scholar] [CrossRef]
- Fan, X.; McLaughlin, C.; Robinson, C.; Ravasini, J.; Schelch, K.; Johnson, T.; van Zandwijk, N.; Reid, G.; George, A.M. Zeolites ameliorate asbestos toxicity in a transgenic model of malignant mesothelioma. FASEB Bioadv. 2019, 1, 550–560. [Google Scholar] [CrossRef]
- Tang, X.; Zhou, B. Iron homeostasis in insects: Insights from Drosophila studies. IUBMB Life 2013, 65, 863–872. [Google Scholar] [CrossRef]
- Liuzzi, J.P.; Aydemir, F.; Nam, H.; Knutson, M.D.; Cousins, R.J. Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13612–13617. [Google Scholar] [CrossRef]
- Wang, D.; Li, W.B.; Wei, X.E.; Li, Y.H.; Dai, Y.M. An investigation of age-related iron deposition using susceptibility weighted imaging. PLoS ONE 2012, 7, e50706. [Google Scholar] [CrossRef]
- Zeidan, R.S.; Han, S.M.; Leeuwenburgh, C.; Xiao, R. Iron homeostasis and organismal aging. Ageing Res. Rev. 2021, 72, 101510. [Google Scholar] [CrossRef] [PubMed]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [PubMed]
- Sobh, A.; Loguinov, A.; Zhou, J.; Jenkitkasemwong, S.; Zeidan, R.; El Ahmadie, N.; Tagmount, A.; Knutson, M.; Fraenkel, P.G.; Vulpe, C.D. Genetic screens reveal CCDC115 as a modulator of erythroid iron and heme trafficking. Am. J. Hematol. 2020, 95, 1085–1098. [Google Scholar] [CrossRef]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to tango: Regulation of Mammalian iron metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Ingrassia, R.; Cavadini, P. Ferritins: A family of molecules for iron storage, antioxidation and more. Biochim. Biophys. Acta 2009, 1790, 589–599. [Google Scholar] [CrossRef]
- Mumbauer, S.; Pascual, J.; Kolotuev, I.; Hamaratoglu, F. Ferritin heavy chain protects the developing wing from reactive oxygen species and ferroptosis. PLoS Genet. 2019, 15, e1008396. [Google Scholar] [CrossRef]
- Baldi, A.; Lombardi, D.; Russo, P.; Palescandolo, E.; De Luca, A.; Santini, D.; Baldi, F.; Rossiello, L.; Dell’Anna, M.L.; Mastrofrancesco, A.; et al. Ferritin contributes to melanoma progression by modulating cell growth and sensitivity to oxidative stress. Clin. Cancer Res. 2005, 11, 3175–3183. [Google Scholar] [CrossRef][Green Version]
- Qiao, B.; Sugianto, P.; Fung, E.; Del-Castillo-Rueda, A.; Moran-Jimenez, M.J.; Ganz, T.; Nemeth, E. Hepcidin-induced endocytosis of ferroportin is dependent on ferroportin ubiquitination. Cell Metab. 2012, 15, 918–924. [Google Scholar] [CrossRef]
- Rishi, G.; Subramaniam, V.N. Signaling pathways regulating hepcidin. Vitam. Horm. 2019, 110, 47–70. [Google Scholar] [CrossRef]
- Gao, G.; Li, J.; Zhang, Y.; Chang, Y.Z. Cellular Iron Metabolism and Regulation. Adv. Exp. Med. Biol. 2019, 1173, 21–32. [Google Scholar]
- Torti, S.V.; Torti, F.M. Iron: The cancer connection. Mol. Asp. Med. 2020, 75, 100860. [Google Scholar] [CrossRef] [PubMed]
- Sfera, A.; Bullock, K.; Price, A.; Inderias, L.; Osorio, C. Ferrosenescence: The iron age of neurodegeneration? Mech. Ageing Dev. 2018, 174, 63–75. [Google Scholar] [CrossRef]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Bhutia, Y.D.; Ogura, J.; Grippo, P.J.; Torres, C.; Sato, T.; Wachtel, M.; Ramachandran, S.; Babu, E.; Sivaprakasam, S.; Rajasekaran, D.; et al. Chronic exposure to excess iron promotes EMT and cancer via p53 loss in pancreatic cancer. Asian J. Pharm. Sci. 2020, 15, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Poprac, P.; Jomova, K.; Simunkova, M.; Kollar, V.; Rhodes, C.J.; Valko, M. Targeting Free Radicals in Oxidative Stress-Related Human Diseases. Trends Pharmacol. Sci. 2017, 38, 592–607. [Google Scholar] [CrossRef] [PubMed]
- Ghosh-Choudhary, S.K.; Liu, J.; Finkel, T. The role of mitochondria in cellular senescence. FASEB J. 2021, 35, e21991. [Google Scholar] [CrossRef]
- Hughes, C.E.; Coody, T.K.; Jeong, M.Y.; Berg, J.A.; Winge, D.R.; Hughes, A.L. Cysteine Toxicity Drives Age-Related Mitochondrial Decline by Altering Iron Homeostasis. Cell 2020, 180, 296–310.e18. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, Q.; Chen, L.; Jiang, J. Iron-mediated lysosomal-mitochondrial crosstalk: A new direction in the treatment of aging and aging-related diseases. Acta Biochim. Biophys. Sin. 2020, 52, 1293–1295. [Google Scholar] [CrossRef]
- Braymer, J.J.; Lill, R. Iron-sulfur cluster biogenesis and trafficking in mitochondria. J. Biol. Chem. 2017, 292, 12754–12763. [Google Scholar] [CrossRef]
- Chiang, S.; Braidy, N.; Maleki, S.; Lal, S.; Richardson, D.R.; Huang, M.L. Mechanisms of impaired mitochondrial homeostasis and NAD (+) metabolism in a model of mitochondrial heart disease exhibiting redox active iron accumulation. Redox Biol. 2021, 46, 102038. [Google Scholar] [CrossRef]
- Mollet, I.G.; Patel, D.; Govani, F.S.; Giess, A.; Paschalaki, K.; Periyasamy, M.; Lidington, E.C.; Mason, J.C.; Jones, M.D.; Game, L.; et al. Low Dose Iron Treatments Induce a DNA Damage Response in Human Endothelial Cells within Minutes. PLoS ONE 2016, 11, e0147990. [Google Scholar] [CrossRef] [PubMed]
- Bolinches-Amorós, A.; Mollá, B.; Pla-Martín, D.; Palau, F.; González-Cabo, P. Mitochondrial dysfunction induced by frataxin deficiency is associated with cellular senescence and abnormal calcium metabolism. Front. Cell. Neurosci. 2014, 8, 124. [Google Scholar] [CrossRef] [PubMed]
- Navarro, J.A.; Botella, J.A.; Metzendorf, C.; Lind, M.I.; Schneuwly, S. Mitoferrin modulates iron toxicity in a Drosophila model of Friedreich’s ataxia. Free Radic. Biol. Med. 2015, 85, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Chen, R.; Liu, L.; Zhou, Y.; Chen, Z. Lactoferrin ameliorates pathological cardiac hypertrophy related to mitochondrial quality control in aged mice. Food Funct. 2021, 12, 7514–7526. [Google Scholar] [CrossRef] [PubMed]
- Issitt, T.; Bosseboeuf, E.; De Winter, N.; Dufton, N.; Gestri, G.; Senatore, V.; Chikh, A.; Randi, A.M.; Raimondi, C. Neuropilin-1 Controls Endothelial Homeostasis by Regulating Mitochondrial Function and Iron-Dependent Oxidative Stress. iScience 2019, 11, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Seo, A.Y.; Xu, J.; Servais, S.; Hofer, T.; Marzetti, E.; Wohlgemuth, S.E.; Knutson, M.D.; Chung, H.Y.; Leeuwenburgh, C. Mitochondrial iron accumulation with age and functional consequences. Aging Cell 2008, 7, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.L.; Ven, T.N.; Crane, M.M.; Brunner, M.L.C.; Pun, A.K.; Helget, K.L.; Brower, K.; Chen, D.E.; Doan, H.; Dillard-Telm, J.D.; et al. Loss of vacuolar acidity results in iron-sulfur cluster defects and divergent homeostatic responses during aging in Saccharomyces cerevisiae. Geroscience 2020, 42, 749–764. [Google Scholar] [CrossRef]
- Veatch, J.R.; McMurray, M.A.; Nelson, Z.W.; Gottschling, D.E. Mitochondrial dysfunction leads to nuclear genome instability via an iron-sulfur cluster defect. Cell 2009, 137, 1247–1258. [Google Scholar] [CrossRef]
- Rouault, T.A.; Tong, W.H. Iron-sulphur cluster biogenesis and mitochondrial iron homeostasis. Nat. Rev. Mol. Cell Biol. 2005, 6, 345–351. [Google Scholar] [CrossRef]
- Atamna, H.; Liu, J.; Ames, B.N. Heme deficiency selectively interrupts assembly of mitochondrial complex IV in human fibroblasts: Revelance to aging. J. Biol. Chem. 2001, 276, 48410–48416. [Google Scholar] [CrossRef]
- Picca, A.; Saini, S.K.; Mankowski, R.T.; Kamenov, G.; Anton, S.D.; Manini, T.M.; Buford, T.W.; Wohlgemuth, S.E.; Xiao, R.; Calvani, R.; et al. Altered Expression of Mitoferrin and Frataxin, Larger Labile Iron Pool and Greater Mitochondrial DNA Damage in the Skeletal Muscle of Older Adults. Cells 2020, 9, 2579. [Google Scholar] [CrossRef] [PubMed]
- Molz, P.; de Freitas, B.S.; Uberti, V.H.; da Costa, K.M.; Kist, L.W.; Bogo, M.R.; Schröder, N. Effects of lipoic acid supplementation on age- and iron-induced memory impairment, mitochondrial DNA damage and antioxidant responses. Eur. J. Nutr. 2021, 60, 3679–3690. [Google Scholar] [CrossRef] [PubMed]
- Walker, T.; Michaelides, C.; Ekonomou, A.; Geraki, K.; Parkes, H.G.; Suessmilch, M.; Herlihy, A.H.; Crum, W.R.; So, P.W. Dissociation between iron accumulation and ferritin upregulation in the aged substantia nigra: Attenuation by dietary restriction. Aging 2016, 8, 2488–2508. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, A.; Michaelides, C.; Walker, T.A.; Ekonomou, A.; Suessmilch, M.; Sriskanthanathan, A.; Abraha, S.; Parkes, A.; Parkes, H.G.; Geraki, K.; et al. Regional Distributions of Iron, Copper and Zinc and Their Relationships with Glia in a Normal Aging Mouse Model. Front. Aging Neurosci. 2019, 11, 351. [Google Scholar] [CrossRef] [PubMed]
- Connor, J.R.; Menzies, S.L.; St Martin, S.M.; Mufson, E.J. Cellular distribution of transferrin, ferritin, and iron in normal and aged human brains. J. Neurosci. Res. 1990, 27, 595–611. [Google Scholar] [CrossRef] [PubMed]
- Connor, J.R.; Snyder, B.S.; Arosio, P.; Loeffler, D.A.; LeWitt, P. A quantitative analysis of isoferritins in select regions of aged, parkinsonian, and Alzheimer’s diseased brains. J. Neurochem. 1995, 65, 717–724. [Google Scholar] [CrossRef]
- Zecca, L.; Gallorini, M.; Schünemann, V.; Trautwein, A.X.; Gerlach, M.; Riederer, P.; Vezzoni, P.; Tampellini, D. Iron, neuromelanin and ferritin content in the substantia nigra of normal subjects at different ages: Consequences for iron storage and neurodegenerative processes. J. Neurochem. 2001, 76, 1766–1773. [Google Scholar] [CrossRef]
- Lee, D.W.; Andersen, J.K. Iron elevations in the aging Parkinsonian brain: A consequence of impaired iron homeostasis? J. Neurochem. 2010, 112, 332–339. [Google Scholar] [CrossRef]
- Levenson, C.W. Trace metal regulation of neuronal apoptosis: From genes to behavior. Physiol. Behav. 2005, 86, 399–406. [Google Scholar] [CrossRef]
- Weinreb, O.; Amit, T.; Youdim, M.B.H. Targeting dysregulation of brain iron homeostasis in ageing. Nutr. Aging 2012, 1, 27–39. [Google Scholar] [CrossRef]
- Han, K.; Jin, X.; Guo, X.; Cao, G.; Tian, S.; Song, Y.; Zuo, Y.; Yu, P.; Gao, G.; Chang, Y.Z. Nrf2 knockout altered brain iron deposition and mitigated age-related motor dysfunction in aging mice. Free Radic. Biol. Med. 2021, 162, 592–602. [Google Scholar] [CrossRef] [PubMed]
- Uberti, V.H.; de Freitas, B.S.; Molz, P.; Bromberg, E.; Schröder, N. Iron Overload Impairs Autophagy: Effects of Rapamycin in Ameliorating Iron-Related Memory Deficits. Mol. Neurobiol. 2020, 57, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, E.; Medina-Leendertz, S.; Díaz, S. Extension of life span and stress resistance of Drosophila melanogaster by long-term supplementation with melatonin. Exp. Gerontol. 2002, 37, 629–638. [Google Scholar] [CrossRef]
- Massie, H.R.; Aiello, V.R.; Williams, T.R. Inhibition of iron absorption prolongs the life span of Drosophila. Mech. Ageing Dev. 1993, 67, 227–237. [Google Scholar] [CrossRef]
- Kennedy, T.P.; Rao, N.V.; Noah, W.; Michael, J.R.; Jafri, M.H., Jr.; Gurtner, G.H.; Hoidal, J.R. Ibuprofen prevents oxidant lung injury and in vitro lipid peroxidation by chelating iron. J. Clin. Investig. 1990, 86, 1565–1573. [Google Scholar] [CrossRef] [PubMed]
- Lopez, T.E.; Pham, H.M.; Nguyen, B.V.; Tahmasian, Y.; Ramsden, S.; Coskun, V.; Schriner, S.E.; Jafari, M. Green tea polyphenols require the mitochondrial iron transporter, mitoferrin, for lifespan extension in Drosophila melanogaster. Arch. Insect Biochem. Physiol. 2016, 93, 210–221. [Google Scholar] [CrossRef]
- Ren, Y.; Yang, S.; Tan, G.; Ye, W.; Liu, D.; Qian, X.; Ding, Z.; Zhong, Y.; Zhang, J.; Jiang, D.; et al. Reduction of mitoferrin results in abnormal development and extended lifespan in Caenorhabditis elegans. PLoS ONE 2012, 7, e29666. [Google Scholar] [CrossRef]
- Schiavi, A.; Maglioni, S.; Palikaras, K.; Shaik, A.; Strappazzon, F.; Brinkmann, V.; Torgovnick, A.; Castelein, N.; De Henau, S.; Braeckman, B.P.; et al. Iron-Starvation-Induced Mitophagy Mediates Lifespan Extension upon Mitochondrial Stress in C. elegans. Curr. Biol. 2015, 25, 1810–1822. [Google Scholar] [CrossRef]
- Klang, I.M.; Schilling, B.; Sorensen, D.J.; Sahu, A.K.; Kapahi, P.; Andersen, J.K.; Swoboda, P.; Killilea, D.W.; Gibson, B.W.; Lithgow, G.J. Iron promotes protein insolubility and aging in C. elegans. Aging 2014, 6, 975–991. [Google Scholar] [CrossRef]
- Jiao, Y.; Wilkinson, J., IV; Christine Pietsch, E.; Buss, J.L.; Wang, W.; Planalp, R.; Torti, F.M.; Torti, S.V. Iron chelation in the biological activity of curcumin. Free Radic. Biol. Med. 2006, 40, 1152–1160. [Google Scholar] [CrossRef]
- Chin, D.; Huebbe, P.; Frank, J.; Rimbach, G.; Pallauf, K. Curcumin may impair iron status when fed to mice for six months. Redox Biol. 2014, 2, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.R.; Parnell, L.D.; Ordovas, J.M.; Lai, C.Q. Curcumin and aging. Biofactors 2013, 39, 133–140. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Tsuchiyama, S.K.; Nguyen, Q.T.; Plyusnina, E.N.; Terrill, S.R.; Sahibzada, S.; Patel, B.; Faulkner, A.R.; Shaposhnikov, M.V.; Tian, R.; et al. Enhanced longevity by ibuprofen, conserved in multiple species, occurs in yeast through inhibition of tryptophan import. PLoS Genet. 2014, 10, e1004860. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.L.; Hatef, A.; Imran ul-Haq, M.; Nair, N.; Unniappan, S.; Kizhakkedathu, J.N. Clinically approved iron chelators influence zebrafish mortality, hatching morphology and cardiac function. PLoS ONE 2014, 9, e109880. [Google Scholar] [CrossRef]
- Almeida, T.; Marques, M.; Mojzita, D.; Amorim, M.A.; Silva, R.D.; Almeida, B.; Rodrigues, P.; Ludovico, P.; Hohmann, S.; Moradas-Ferreira, P.; et al. Isc1p plays a key role in hydrogen peroxide resistance and chronological lifespan through modulation of iron levels and apoptosis. Mol. Biol. Cell 2008, 19, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Q.; Liu, X.G.; Zhao, W.; Cui, H.; Ruan, J.; Yuan, Y.; Tu, Z. MET18 Deficiency Increases the Sensitivity of Yeast to Oxidative Stress and Shortens Replicative Lifespan by Inhibiting Catalase Activity. BioMed Res. Int. 2017, 2017, 7587395. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.K.; Mittal, N.; Deswal, S.; Roy, N. Calorie restriction up-regulates iron and copper transport genes in Saccharomyces cerevisiae. Mol. Biosyst. 2011, 7, 394–402. [Google Scholar] [CrossRef]
- Sheng, Y.; Yang, G.; Casey, K.; Curry, S.; Oliver, M.; Han, S.M.; Leeuwenburgh, C.; Xiao, R. A novel role of the mitochondrial iron-sulfur cluster assembly protein ISCU-1/ISCU in longevity and stress response. Geroscience 2021, 43, 691–707. [Google Scholar] [CrossRef]
- Prischi, F.; Konarev, P.V.; Iannuzzi, C.; Pastore, C.; Adinolfi, S.; Martin, S.R.; Svergun, D.I.; Pastore, A. Structural bases for the interaction of frataxin with the central components of iron-sulphur cluster assembly. Nat. Commun. 2010, 1, 95. [Google Scholar] [CrossRef]
- Britti, E.; Delaspre, F.; Feldman, A.; Osborne, M.; Greif, H.; Tamarit, J.; Ros, J. Frataxin-deficient neurons and mice models of Friedreich ataxia are improved by TAT-MTScs-FXN treatment. J. Cell. Mol. Med. 2018, 22, 834–848. [Google Scholar] [CrossRef]
- Zarse, K.; Schulz, T.J.; Birringer, M.; Ristow, M. Impaired respiration is positively correlated with decreased life span in Caenorhabditis elegans models of Friedreich Ataxia. FASEB J. 2007, 21, 1271–1275. [Google Scholar] [CrossRef] [PubMed]
- Runko, A.P.; Griswold, A.J.; Min, K.T. Overexpression of frataxin in the mitochondria increases resistance to oxidative stress and extends lifespan in Drosophila. FEBS Lett. 2008, 582, 715–719. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, N.L.; James, S.A.; Salim, A.; Sumardy, F.; Speed, T.P.; Conrad, M.; Richardson, D.R.; Bush, A.I.; McColl, G. Changes in ferrous iron and glutathione promote ferroptosis and frailty in aging Caenorhabditis elegans. Elife 2020, 9, e56580. [Google Scholar] [CrossRef] [PubMed]
- Mangan, D. Iron: An underrated factor in aging. Aging 2021, 13, 23407–23415. [Google Scholar] [CrossRef] [PubMed]
- Go, S.; Kang, M.; Kwon, S.P.; Jung, M.; Jeon, O.H.; Kim, B.S. The Senolytic Drug JQ1 Removes Senescent Cells via Ferroptosis. Tissue Eng. Regen. Med. 2021, 18, 841–850. [Google Scholar] [CrossRef]
- Beydoun, R.; Hamood, M.A.; Gomez Zubieta, D.M.; Kondapalli, K.C. Na+/H+ Exchanger 9 Regulates Iron Mobilization at the Blood-Brain Barrier in Response to Iron Starvation. J. Biol. Chem. 2017, 292, 4293–4301. [Google Scholar] [CrossRef]
- Austin, R.N.; Freeman, J.L.; Guilarte, T.R. Neurochemistry of lead and manganese. Metallomics 2016, 8, 561–562. [Google Scholar] [CrossRef][Green Version]
- Ju, Y.; Tam, K.Y. Pathological mechanisms and therapeutic strategies for Alzheimer’s disease. Neural Regen. Res. 2022, 17, 543–549. [Google Scholar] [CrossRef]
- Khattar, N.; Triebswetter, C.; Kiely, M.; Ferrucci, L.; Resnick, S.M.; Spencer, R.G.; Bouhrara, M. Investigation of the association between cerebral iron content and myelin content in normative aging using quantitative magnetic resonance neuroimaging. Neuroimage 2021, 239, 118267. [Google Scholar] [CrossRef]
- Faux, N.G.; Rembach, A.; Wiley, J.; Ellis, K.A.; Ames, D.; Fowler, C.J.; Martins, R.N.; Pertile, K.K.; Rumble, R.L.; Trounson, B.; et al. An anemia of Alzheimer’s disease. Mol. Psychiatry 2014, 19, 1227–1234. [Google Scholar] [CrossRef]
- Nobili, A.; Krashia, P.; D’Amelio, M. Cisd2: A promising new target in Alzheimer’s disease†. J. Pathol. 2020, 251, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Sayre, L.M.; Perry, G.; Harris, P.L.; Liu, Y.; Schubert, K.A.; Smith, M.A. In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer’s disease: A central role for bound transition metals. J. Neurochem. 2000, 74, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Chang, X.; Lang, M. Iron Homeostasis Disorder and Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 12442. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Song, C.; Du, Y.; Gaur, U.; Yang, M. Pharmacological Treatment of Alzheimer’s Disease: Insights from Drosophila melanogaster. Int. J. Mol. Sci. 2020, 21, 4621. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.H.; Kwon, K.C.; Hwang, D.J.; Koo, J.H.; Um, H.S.; Song, H.S.; Kim, J.S.; Jang, Y.; Cho, J.Y. Treadmill Exercise Alleviates Brain Iron Dyshomeostasis Accelerating Neuronal Amyloid-β Production, Neuronal Cell Death, and Cognitive Impairment in Transgenic Mice Model of Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 3208–3223. [Google Scholar] [CrossRef] [PubMed]
- Spotorno, N.; Acosta-Cabronero, J.; Stomrud, E.; Lampinen, B.; Strandberg, O.T.; van Westen, D.; Hansson, O. Relationship between cortical iron and tau aggregation in Alzheimer’s disease. Brain 2020, 143, 1341–1349. [Google Scholar] [CrossRef]
- Nairz, M.; Metzendorf, C.; Vujic-Spasic, M.; Mitterstiller, A.M.; Schroll, A.; Haschka, D.; Hoffmann, A.; Von Raffay, L.; Sparla, R.; Huck, C.W.; et al. Cell-specific expression of Hfe determines the outcome of Salmonella enterica serovar Typhimurium infection in mice. Haematologica 2021, 106, 3149–3161. [Google Scholar] [CrossRef]
- Lee, S.Y.; Connor, J.R. Regulation of Hfe by stress factors in BV-2 cells. Neurobiol. Aging 2005, 26, 803–812. [Google Scholar] [CrossRef]
- Tisato, V.; Zuliani, G.; Vigliano, M.; Longo, G.; Franchini, E.; Secchiero, P.; Zauli, G.; Paraboschi, E.M.; Vikram Singh, A.; Serino, M.L.; et al. Gene-gene interactions among coding genes of iron-homeostasis proteins and APOE-alleles in cognitive impairment diseases. PLoS ONE 2018, 13, e0193867. [Google Scholar] [CrossRef]
- Lehmann, D.J.; Schuur, M.; Warden, D.R.; Hammond, N.; Belbin, O.; Kölsch, H.; Lehmann, M.G.; Wilcock, G.K.; Brown, K.; Kehoe, P.G.; et al. Transferrin and HFE genes interact in Alzheimer’s disease risk: The Epistasis Project. Neurobiol. Aging 2012, 33, e1–e13. [Google Scholar] [CrossRef]
- Namekata, K.; Imagawa, M.; Terashi, A.; Ohta, S.; Oyama, F.; Ihara, Y. Association of transferrin C2 allele with late-onset Alzheimer’s disease. Hum. Genet. 1997, 101, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Robson, K.J.; Lehmann, D.J.; Wimhurst, V.L.; Livesey, K.J.; Combrinck, M.; Merryweather-Clarke, A.T.; Warden, D.R.; Smith, A.D. Synergy between the C2 allele of transferrin and the C282Y allele of the haemochromatosis gene (HFE) as risk factors for developing Alzheimer’s disease. J. Med. Genet. 2004, 41, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.C.; Domingues, H.S.; Salgado, A.J.; Teixeira, F.G. From regenerative strategies to pharmacological approaches: Can we fine-tune treatment for Parkinson’s disease? Neural Regen. Res. 2022, 17, 933–936. [Google Scholar] [CrossRef] [PubMed]
- Mudd, A.T.; Waworuntu, R.V.; Berg, B.M.; Dilger, R.N. Dietary Alpha-Lipoic Acid Alters Piglet Neurodevelopment. Front. Pediatr. 2016, 4, 44. [Google Scholar] [CrossRef]
- Lhermitte, J.; Kraus, W.M.; McAlpine, D. Original Papers: On the occurrence of abnormal deposits of iron in the brain in parkinsonism with special reference to its localisation. J. Neurol. Psychopathol. 1924, 5, 195–208. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, B.; Tao, K.; Yang, H.; Wang, Y.; Zhou, T.; Yang, Y.; Yuan, L.; Liu, X.; Duan, Y. Iron accumulation and microglia activation contribute to substantia nigra hyperechogenicity in the 6-OHDA-induced rat model of Parkinson’s disease. Parkinsonism Relat. Disord. 2017, 36, 76–82. [Google Scholar] [CrossRef]
- Kaur, D.; Yantiri, F.; Rajagopalan, S.; Kumar, J.; Mo, J.Q.; Boonplueang, R.; Viswanath, V.; Jacobs, R.; Yang, L.; Beal, M.F.; et al. Genetic or pharmacological iron chelation prevents MPTP-induced neurotoxicity in vivo: A novel therapy for Parkinson’s disease. Neuron 2003, 37, 899–909. [Google Scholar] [CrossRef]
- Kaur, D.; Peng, J.; Chinta, S.J.; Rajagopalan, S.; Di Monte, D.A.; Cherny, R.A.; Andersen, J.K. Increased murine neonatal iron intake results in Parkinson-like neurodegeneration with age. Neurobiol. Aging 2007, 28, 907–913. [Google Scholar] [CrossRef]
- Faucheux, B.A.; Bonnet, A.M.; Agid, Y.; Hirsch, E.C. Blood vessels change in the mesencephalon of patients with Parkinson’s disease. Lancet 1999, 353, 981–982. [Google Scholar] [CrossRef]
- Kortekaas, R.; Leenders, K.L.; van Oostrom, J.C.; Vaalburg, W.; Bart, J.; Willemsen, A.T.; Hendrikse, N.H. Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann. Neurol. 2005, 57, 176–179. [Google Scholar] [CrossRef]
- Faucheux, B.A.; Nillesse, N.; Damier, P.; Spik, G.; Mouatt-Prigent, A.; Pierce, A.; Leveugle, B.; Kubis, N.; Hauw, J.J.; Agid, Y.; et al. Expression of lactoferrin receptors is increased in the mesencephalon of patients with Parkinson disease. Proc. Natl. Acad. Sci. USA 1995, 92, 9603–9607. [Google Scholar] [CrossRef]
- Borie, C.; Gasparini, F.; Verpillat, P.; Bonnet, A.M.; Agid, Y.; Hetet, G.; Brice, A.; Dürr, A.; Grandchamp, B. Association study between iron-related genes polymorphisms and Parkinson’s disease. J. Neurol. 2002, 249, 801–804. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.J.; Bras, J.M.; Santana, I.; Januario, C.; Santiago, B.; Morgadinho, A.S.; Ribeiro, M.H.; Hardy, J.; Singleton, A.; Oliveira, C. Association of HFE common mutations with Parkinson’s disease, Alzheimer’s disease and mild cognitive impairment in a Portuguese cohort. BMC Neurol. 2006, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Salazar, J.; Mena, N.; Hunot, S.; Prigent, A.; Alvarez-Fischer, D.; Arredondo, M.; Duyckaerts, C.; Sazdovitch, V.; Zhao, L.; Garrick, L.M.; et al. Divalent metal transporter 1 (DMT1) contributes to neurodegeneration in animal models of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2008, 105, 18578–18583. [Google Scholar] [CrossRef] [PubMed]
- Kopaeva, M.Y.; Cherepov, A.B.; Nesterenko, M.V.; Zarayskaya, I.Y. Pretreatment with Human Lactoferrin Had a Positive Effect on the Dynamics of Mouse Nigrostriatal System Recovery after Acute MPTP Exposure. Biology 2021, 10, 24. [Google Scholar] [CrossRef]
- Ayton, S.; Lei, P.; McLean, C.; Bush, A.I.; Finkelstein, D.I. Transferrin protects against Parkinsonian neurotoxicity and is deficient in Parkinson’s substantia nigra. Signal Transduct. Target. Ther. 2016, 1, 16015. [Google Scholar] [CrossRef]
- He, Q.; Du, T.; Yu, X.; Xie, A.; Song, N.; Kang, Q.; Yu, J.; Tan, L.; Xie, J.; Jiang, H. DMT1 polymorphism and risk of Parkinson’s disease. Neurosci. Lett. 2011, 501, 128–131. [Google Scholar] [CrossRef]
- Jiang, H.; Song, N.; Xu, H.; Zhang, S.; Wang, J.; Xie, J. Up-regulation of divalent metal transporter 1 in 6-hydroxydopamine intoxication is IRE/IRP dependent. Cell Res. 2010, 20, 345–356. [Google Scholar] [CrossRef]
- Martínez-Hernández, R.; Montes, S.; Higuera-Calleja, J.; Yescas, P.; Boll, M.C.; Diaz-Ruiz, A.; Rios, C. Plasma ceruloplasmin ferroxidase activity correlates with the nigral sonographic area in Parkinson’s disease patients: A pilot study. Neurochem. Res. 2011, 36, 2111–2115. [Google Scholar] [CrossRef]
- Olivieri, S.; Conti, A.; Iannaccone, S.; Cannistraci, C.V.; Campanella, A.; Barbariga, M.; Codazzi, F.; Pelizzoni, I.; Magnani, G.; Pesca, M.; et al. Ceruloplasmin oxidation, a feature of Parkinson’s disease CSF, inhibits ferroxidase activity and promotes cellular iron retention. J. Neurosci. 2011, 31, 18568–18577. [Google Scholar] [CrossRef]
- Hochstrasser, H.; Bauer, P.; Walter, U.; Behnke, S.; Spiegel, J.; Csoti, I.; Zeiler, B.; Bornemann, A.; Pahnke, J.; Becker, G.; et al. Ceruloplasmin gene variations and substantia nigra hyperechogenicity in Parkinson disease. Neurology 2004, 63, 1912–1917. [Google Scholar] [CrossRef]
- Sian-Hülsmann, J.; Mandel, S.; Youdim, M.B.; Riederer, P. The relevance of iron in the pathogenesis of Parkinson’s disease. J. Neurochem. 2011, 118, 939–957. [Google Scholar] [CrossRef] [PubMed]
- Faucheux, B.A.; Martin, M.E.; Beaumont, C.; Hauw, J.J.; Agid, Y.; Hirsch, E.C. Neuromelanin associated redox-active iron is increased in the substantia nigra of patients with Parkinson’s disease. J. Neurochem. 2003, 86, 1142–1148. [Google Scholar] [CrossRef] [PubMed]
- Meade, R.M.; Fairlie, D.P.; Mason, J.M. Alpha-synuclein structure and Parkinson’s disease-lessons and emerging principles. Mol. Neurodegener. 2019, 14, 29. [Google Scholar] [CrossRef] [PubMed]
- Riederer, P.; Monoranu, C.; Strobel, S.; Iordache, T.; Sian-Hülsmann, J. Iron as the concert master in the pathogenic orchestra playing in sporadic Parkinson’s disease. J. Neural Transm. 2021, 128, 1577–1598. [Google Scholar] [CrossRef] [PubMed]
- Jansen van Rensburg, Z.; Abrahams, S.; Bardien, S.; Kenyon, C. Toxic Feedback Loop Involving Iron, Reactive Oxygen Species, α-Synuclein and Neuromelanin in Parkinson’s Disease and Intervention with Turmeric. Mol. Neurobiol. 2021, 58, 5920–5936. [Google Scholar] [CrossRef]
- Fernández-Mendívil, C.; Luengo, E.; Trigo-Alonso, P.; García-Magro, N.; Negredo, P.; López, M.G. Protective role of microglial HO-1 blockade in aging: Implication of iron metabolism. Redox Biol. 2021, 38, 101789. [Google Scholar] [CrossRef]
- Sun, W.; Zheng, J.; Ma, J.; Wang, Z.; Shi, X.; Li, M.; Huang, S.; Hu, S.; Zhao, Z.; Li, D. Increased Plasma Heme Oxygenase-1 Levels in Patients with Early-Stage Parkinson’s Disease. Front. Aging Neurosci. 2021, 13, 621508. [Google Scholar] [CrossRef]
- Xu, J.; Xiao, C.; Song, W.; Cui, X.; Pan, M.; Wang, Q.; Feng, Y.; Xu, Y. Elevated Heme Oxygenase-1 Correlates with Increased Brain Iron Deposition Measured by Quantitative Susceptibility Mapping and Decreased Hemoglobin in Patients with Parkinson’s Disease. Front. Aging Neurosci. 2021, 13, 656626. [Google Scholar] [CrossRef]
- Monda, E.; Lioncino, M.; Rubino, M.; Passantino, S.; Verrillo, F.; Caiazza, M.; Cirillo, A.; Fusco, A.; Di Fraia, F.; Fimiani, F.; et al. Diagnosis and Management of Cardiovascular Involvement in Friedreich Ataxia. Heart Fail. Clin. 2022, 18, 31–37. [Google Scholar] [CrossRef]
- Yang, W.; Thompson, B.; Kwa, F.A.A. Molecular approaches for the treatment and prevention of Friedreich’s ataxia. Drug Discov. Today 2021, 27, 866–880. [Google Scholar] [CrossRef]
- Nittis, T.; Gitlin, J.D. The copper-iron connection: Hereditary aceruloplasminemia. Semin. Hematol. 2002, 39, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, M.; Zhang, C.; Zhou, S.; Ji, G. Molecular Functions of Ceruloplasmin in Metabolic Disease Pathology. Diabetes Metab. Syndr. Obes. 2022, 15, 695–711. [Google Scholar] [CrossRef] [PubMed]
- Morita, H.; Ikeda, S.; Yamamoto, K.; Morita, S.; Yoshida, K.; Nomoto, S.; Kato, M.; Yanagisawa, N. Hereditary ceruloplasmin deficiency with hemosiderosis: A clinicopathological study of a Japanese family. Ann. Neurol. 1995, 37, 646–656. [Google Scholar] [CrossRef]
- Nair, T.; Ram, C.V.S. Normotensive aging: Finally, an ‘iron in the fire’. J. Hypertens. 2021, 39, 1102–1103. [Google Scholar] [CrossRef]
- Crompton, D.E.; Chinnery, P.F.; Fey, C.; Curtis, A.R.; Morris, C.M.; Kierstan, J.; Burt, A.; Young, F.; Coulthard, A.; Curtis, A.; et al. Neuroferritinopathy: A window on the role of iron in neurodegeneration. Blood Cells Mol. Dis. 2002, 29, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Dusek, P.; Tovar Martinez, E.M.; Madai, V.I.; Jech, R.; Sobesky, J.; Paul, F.; Niendorf, T.; Wuerfel, J.; Schneider, S.A. 7-Tesla Magnetic Resonance Imaging for Brain Iron Quantification in Homozygous and Heterozygous PANK2 Mutation Carriers. Mov. Disord. Clin. Pract. 2014, 1, 329–335. [Google Scholar] [CrossRef]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef]
- Hagemeier, J.; Geurts, J.J.; Zivadinov, R. Brain iron accumulation in aging and neurodegenerative disorders. Expert Rev. Neurother. 2012, 12, 1467–1480. [Google Scholar] [CrossRef]
- Prass, K.; Ruscher, K.; Karsch, M.; Isaev, N.; Megow, D.; Priller, J.; Scharff, A.; Dirnagl, U.; Meisel, A. Desferrioxamine induces delayed tolerance against cerebral ischemia in vivo and in vitro. J. Cereb. Blood Flow Metab. 2002, 22, 520–525. [Google Scholar] [CrossRef]
- Jiang, H.; Luan, Z.; Wang, J.; Xie, J. Neuroprotective effects of iron chelator Desferal on dopaminergic neurons in the substantia nigra of rats with iron-overload. Neurochem. Int. 2006, 49, 605–609. [Google Scholar] [CrossRef]
- Crapper McLachlan, D.R.; Dalton, A.J.; Kruck, T.P.; Bell, M.Y.; Smith, W.L.; Kalow, W.; Andrews, D.F. Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet 1991, 337, 1304–1308. [Google Scholar] [CrossRef]
- Lin, G.; Zhu, F.; Kanaan, N.M.; Asano, R.; Shirafuji, N.; Sasaki, H.; Yamaguchi, T.; Enomoto, S.; Endo, Y.; Ueno, A.; et al. Clioquinol Decreases Levels of Phosphorylated, Truncated, and Oligomerized Tau Protein. Int. J. Mol. Sci. 2021, 22, 12063. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Huang, C.; Luo, Q.; Xia, Y.; Liu, W.; Zeng, W.; Cheng, A.; Shi, R.; Zhengli, C. Clioquinol improves motor and non-motor deficits in MPTP-induced monkey model of Parkinson’s disease through AKT/mTOR pathway. Aging 2020, 12, 9515–9533. [Google Scholar] [CrossRef] [PubMed]
- Summers, K.L.; Roseman, G.P.; Sopasis, G.J.; Millhauser, G.L.; Harris, H.H.; Pickering, I.J.; George, G.N. Copper (II) Binding to PBT2 Differs from That of Other 8-Hydroxyquinoline Chelators: Implications for the Treatment of Neurodegenerative Protein Misfolding Diseases. Inorg. Chem. 2020, 59, 17519–17534. [Google Scholar] [CrossRef]
- Lakey-Beitia, J.; González, Y.; Doens, D.; Stephens, D.E.; Santamaría, R.; Murillo, E.; Gutiérrez, M.; Fernández, P.L.; Rao, K.S.; Larionov, O.V.; et al. Assessment of Novel Curcumin Derivatives as Potent Inhibitors of Inflammation and Amyloid-β Aggregation in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 60, s59–s68. [Google Scholar] [CrossRef]
- Elmegeed, G.A.; Ahmed, H.H.; Hashash, M.A.; Abd-Elhalim, M.M.; El-kady, D.S. Synthesis of novel steroidal curcumin derivatives as anti-Alzheimer’s disease candidates: Evidences-based on in vivo study. Steroids 2015, 101, 78–89. [Google Scholar] [CrossRef]
- Bardestani, A.; Ebrahimpour, S.; Esmaeili, A.; Esmaeili, A. Quercetin attenuates neurotoxicity induced by iron oxide nanoparticles. J. Nanobiotechnol. 2021, 19, 327. [Google Scholar] [CrossRef]
- Zheng, H.; Gal, S.; Weiner, L.M.; Bar-Am, O.; Warshawsky, A.; Fridkin, M.; Youdim, M.B. Novel multifunctional neuroprotective iron chelator-monoamine oxidase inhibitor drugs for neurodegenerative diseases: In vitro studies on antioxidant activity, prevention of lipid peroxide formation and monoamine oxidase inhibition. J. Neurochem. 2005, 95, 68–78. [Google Scholar] [CrossRef]
- Wu, Z.; Palanimuthu, D.; Braidy, N.; Salikin, N.H.; Egan, S.; Huang, M.L.H.; Richardson, D.R. Novel multifunctional iron chelators of the aroyl nicotinoyl hydrazone class that markedly enhance cellular NAD+/NADH ratios. Br. J. Pharmacol. 2020, 177, 1967–1987. [Google Scholar] [CrossRef]
- Dexter, D.T.; Statton, S.A.; Whitmore, C.; Freinbichler, W.; Weinberger, P.; Tipton, K.F.; Della Corte, L.; Ward, R.J.; Crichton, R.R. Clinically available iron chelators induce neuroprotection in the 6-OHDA model of Parkinson’s disease after peripheral administration. J. Neural Transm. 2011, 118, 223–231. [Google Scholar] [CrossRef]
- Kenche, V.B.; Barnham, K.J. Alzheimer’s disease & metals: Therapeutic opportunities. Br. J. Pharmacol. 2011, 163, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.B. Therapeutic developments in Friedreich ataxia. J. Child Neurol. 2012, 27, 1212–1216. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Sánchez, D.; Aracil, A.; Montero, R.; Mas, A.; Jiménez, L.; O’Callaghan, M.; Tondo, M.; Capdevila, A.; Blanch, J.; Artuch, R.; et al. Combined therapy with idebenone and deferiprone in patients with Friedreich’s ataxia. Cerebellum 2011, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Leed, M.G.; Wolkow, N.; Pham, D.M.; Daniel, C.L.; Dunaief, J.L.; Franz, K.J. Prochelators triggered by hydrogen peroxide provide hexadentate iron coordination to impede oxidative stress. J. Inorg. Biochem. 2011, 105, 1161–1172. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Iron Chelating Agent | Characteristic | Targeted Diseases |
|---|---|---|
| Desferrioxamine | Induced HIF-1 DNA binding and transcription of erythropoietin in vivo; brain-impermeable; hydrophilic and high molecular weight; low selectivity for metal ions. | PD/AD [140,141,142] |
| Clioquinol | Brain-permeable; lipophilic; high toxicity. | PD/AD [143,144] |
| PBT2 | high selectivity. | AD [145] |
| Natural polyphenols (curcumin/EGCG/quercetin) | Nontoxic; brain-permeable; anti-oxidation/inflammatory. | PD/AD [146,147,148] |
| Novel chelating agents (VK-28/HLA-20/M30/SNH6) | Low-toxicity; lipophilic; brain-permeable. | PD/AD [149,150] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, Y.; Tian, Y.; Yuan, Z.; Zeng, Y.; Wang, S.; Fan, X.; Yang, D.; Yang, M. Iron Metabolism in Aging and Age-Related Diseases. Int. J. Mol. Sci. 2022, 23, 3612. https://doi.org/10.3390/ijms23073612
Tian Y, Tian Y, Yuan Z, Zeng Y, Wang S, Fan X, Yang D, Yang M. Iron Metabolism in Aging and Age-Related Diseases. International Journal of Molecular Sciences. 2022; 23(7):3612. https://doi.org/10.3390/ijms23073612
Chicago/Turabian StyleTian, Yao, Yuanliangzi Tian, Zhixiao Yuan, Yutian Zeng, Shuai Wang, Xiaolan Fan, Deying Yang, and Mingyao Yang. 2022. "Iron Metabolism in Aging and Age-Related Diseases" International Journal of Molecular Sciences 23, no. 7: 3612. https://doi.org/10.3390/ijms23073612
APA StyleTian, Y., Tian, Y., Yuan, Z., Zeng, Y., Wang, S., Fan, X., Yang, D., & Yang, M. (2022). Iron Metabolism in Aging and Age-Related Diseases. International Journal of Molecular Sciences, 23(7), 3612. https://doi.org/10.3390/ijms23073612