
The Next Frontier: Translational Development of Ubiquitination, SUMOylation, and NEDDylation in Cancer

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
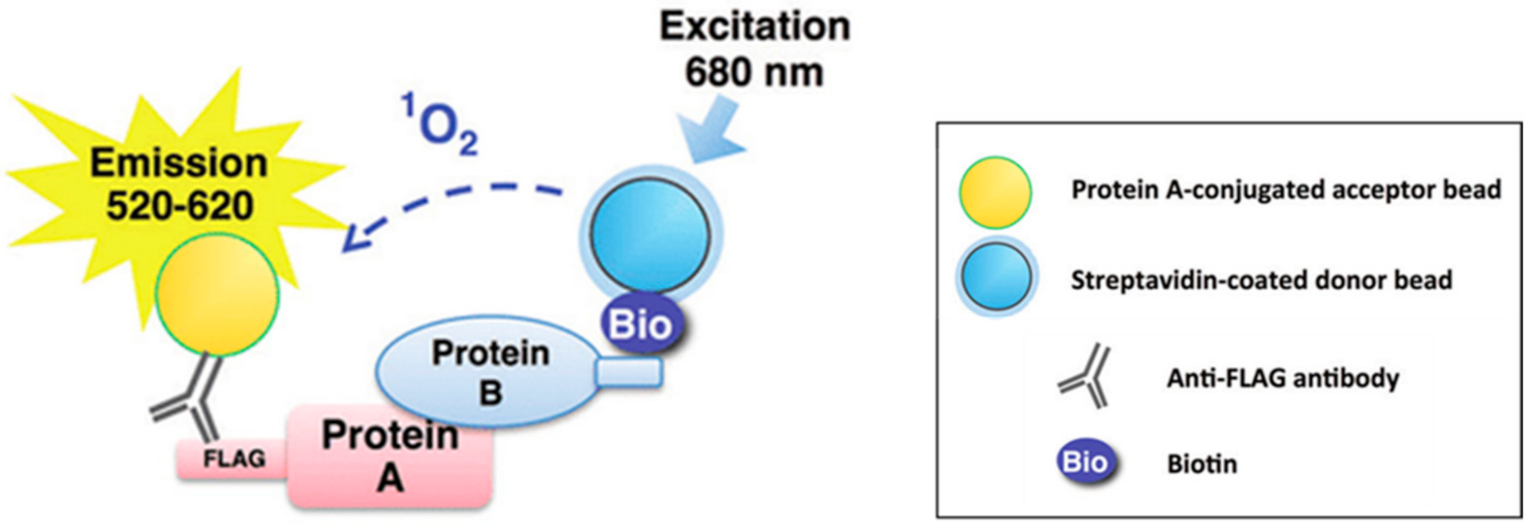{kind=link}
Abstract
1. Introduction
2. Overview of Ubiquitination, SUMOylation, and NEDDylation
2.1. Ubiquitination
2.2. SUMOylation
2.3. NEDDylation
3. Ubiquitination, SUMOylation, and NEDDylation in Cancer
4. Ubiquitination, SUMOylation, and NEDDylation Assay Capabilities
4.1. Ubiquitination
4.2. SUMOylation
4.3. NEDDylation
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Deribe, Y.L.; Pawson, T.; Dikic, I. Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 2010, 17, 666–672. [Google Scholar] [CrossRef]
- El Motiam, A.; Vidal, S.; de la Cruz-Herrera, C.F.; Da Silva-Álvarez, S.; Baz-Martínez, M.; Seoane, R.; Vidal, A.; Rodríguez, M.S.; Xirodimas, D.P.; Carvalho, A.S.; et al. Interplay between SUMOylation and NEDDylation regulates RPL11 localization and function. FASEB J. 2019, 33, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Liu, Z.; Wang, Y.; Cheng, H.; Yang, Q.; Guo, A.; Ren, J.; Xue, Y. UUCD: A family-based database of ubiquitin and ubiquitin-like conjugation. Nucleic Acids Res. 2012, 41, D445–D451. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A. Ubiquitin: Roles in protein modification and breakdown. Cell 1983, 34, 11–12. [Google Scholar] [CrossRef]
- French, M.E.; Koehler, C.F.; Hunter, T. Emerging functions of branched ubiquitin chains. Cell Discov. 2021, 7, 6. [Google Scholar] [CrossRef]
- Husnjak, K.; Dikic, I. Ubiquitin-binding proteins: Decoders of ubiquitin-mediated cellular functions. Annu. Rev. Biochem. 2012, 81, 291–322. [Google Scholar] [CrossRef]
- Chen, Z.J.; Sun, L.J. Nonproteolytic functions of ubiquitin in cell signaling. Mol. Cell 2009, 33, 275–286. [Google Scholar] [CrossRef]
- Grabbe, C.; Husnjak, K.; Dikic, I. The spatial and temporal organization of ubiquitin networks. Nat. Rev. Mol. Cell Biol. 2011, 12, 295–307. [Google Scholar] [CrossRef]
- Wang, D.; Ma, L.; Wang, B.; Liu, J.; Wei, W. E3 ubiquitin ligases in cancer and implications for therapies. Cancer Metastasis Rev. 2017, 36, 683–702. [Google Scholar] [CrossRef]
- Deshaies, R.J.; Joazeiro, C.A. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef]
- Rotin, D.; Kumar, S. Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2009, 10, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Pickart, C.M. Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 2001, 70, 503–533. [Google Scholar] [CrossRef]
- Matunis, M.J.; Rodriguez, M.S. Concepts and Methodologies to Study Protein SUMOylation: An Overview. Methods Mol. Biol. 2016, 1475, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Geiss-Friedlander, R.; Melchior, F. Concepts in sumoylation: A decade on. Nat. Rev. Mol. Cell Biol. 2007, 8, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, K.A.; Henley, J.M. Mechanisms, regulation and consequences of protein SUMOylation. Biochem. J. 2010, 428, 133–145. [Google Scholar] [CrossRef]
- Saitoh, H.; Hinchey, J. Functional heterogeneity of small ubiquitin-related protein modifiers SUMO-1 versus SUMO-2/3. J. Biol. Chem. 2000, 275, 6252–6258. [Google Scholar] [CrossRef]
- Keiten-Schmitz, J.; Schunck, K.; Müller, S. SUMO Chains Rule on Chromatin Occupancy. Front. Cell Dev. Biol. 2019, 7, 343. [Google Scholar] [CrossRef]
- Xu, J.; He, Y.; Qiang, B.; Yuan, J.; Peng, X.; Pan, X.M. A novel method for high accuracy sumoylation site prediction from protein sequences. BMC Bioinform. 2008, 9, 8. [Google Scholar] [CrossRef]
- Hilgarth, R.S.; Sarge, K.D. Detection of sumoylated proteins. Methods Mol. Biol. 2005, 301, 329–338. [Google Scholar] [CrossRef]
- Garvin, A.J.; Morris, J.R. SUMO, a small, but powerful, regulator of double-strand break repair. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160281. [Google Scholar] [CrossRef] [PubMed]
- Kamitani, T.; Kito, K.; Nguyen, H.P.; Yeh, E.T.H. Characterization of NEDD8, a Developmentally Down-regulated Ubiquitin-like Protein *. J. Biol. Chem. 1997, 272, 28557–28562. [Google Scholar] [CrossRef] [PubMed]
- Wada, H.; Kito, K.; Caskey, L.S.; Yeh, E.T.H.; Kamitani, T. Cleavage of the C-Terminus of NEDD8 by UCH-L3. Biochem. Biophys. Res. Commun. 1998, 251, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Schwechheimer, C.; Mergner, J. The NEDD8 modification pathway in plants. Front. Plant Sci. 2014, 5, 103. [Google Scholar] [CrossRef]
- Shin, Y.-C.; Tang, S.-J.; Chen, J.-H.; Liao, P.-H.; Chang, S.-C. The Molecular Determinants of NEDD8 Specific Recognition by Human SENP8. PLoS ONE 2011, 6, e27742. [Google Scholar] [CrossRef] [PubMed]
- Rabut, G.; Peter, M. Function and regulation of protein neddylation. ‘Protein modifications: Beyond the usual suspects’ review series. EMBO Rep. 2008, 9, 969–976. [Google Scholar] [CrossRef]
- Walden, H.; Podgorski, M.S.; Huang, D.T.; Miller, D.W.; Howard, R.J.; Minor, D.L.; Holton, J.M.; Schulman, B.A. The Structure of the APPBP1-UBA3-NEDD8-ATP Complex Reveals the Basis for Selective Ubiquitin-like Protein Activation by an E1. Mol. Cell 2003, 12, 1427–1437. [Google Scholar] [CrossRef]
- Soucy, T.A.; Dick, L.R.; Smith, P.G.; Milhollen, M.A.; Brownell, J.E. The NEDD8 Conjugation Pathway and Its Relevance in Cancer Biology and Therapy. Genes Cancer 2010, 1, 708–716. [Google Scholar] [CrossRef]
- Gong, L.; Yeh, E.T. Identification of the activating and conjugating enzymes of the NEDD8 conjugation pathway. J. Biol. Chem. 1999, 274, 12036–12042. [Google Scholar] [CrossRef]
- Zhou, L.; Jiang, Y.; Luo, Q.; Li, L.; Jia, L. Neddylation: A novel modulator of the tumor microenvironment. Mol. Cancer 2019, 18, 77. [Google Scholar] [CrossRef]
- Leidecker, O.; Matic, I.; Mahata, B.; Pion, E.; Xirodimas, D.P. The ubiquitin E1 enzyme Ube1 mediates NEDD8 activation under diverse stress conditions. Cell Cycle 2012, 11, 1142–1150. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.; Wu, K.; Yang, Y.; Guerrero, C.; Nillegoda, N.; Pan, Z.Q.; Huang, L. A targeted proteomic analysis of the ubiquitin-like modifier nedd8 and associated proteins. J. Proteome Res. 2008, 7, 1274–1287. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Zerath, S.; Kleifeld, O.; Scheffner, M.; Glickman, M.H.; Fushman, D. Recognition and cleavage of related to ubiquitin 1 (Rub1) and Rub1-ubiquitin chains by components of the ubiquitin-proteasome system. Mol. Cell Proteom. 2012, 11, 1595–1611. [Google Scholar] [CrossRef] [PubMed]
- Boh, B.K.; Smith, P.G.; Hagen, T. Neddylation-induced conformational control regulates cullin RING ligase activity in vivo. J. Mol. Biol. 2011, 409, 136–145. [Google Scholar] [CrossRef]
- Huang, D.T.; Ayrault, O.; Hunt, H.W.; Taherbhoy, A.M.; Duda, D.M.; Scott, D.C.; Borg, L.A.; Neale, G.; Murray, P.J.; Roussel, M.F.; et al. E2-RING expansion of the NEDD8 cascade confers specificity to cullin modification. Mol. Cell 2009, 33, 483–495. [Google Scholar] [CrossRef]
- Hammill, J.T.; Scott, D.C.; Min, J.; Connelly, M.C.; Holbrook, G.; Zhu, F.; Matheny, A.; Yang, L.; Singh, B.; Schulman, B.A.; et al. Piperidinyl Ureas Chemically Control Defective in Cullin Neddylation 1 (DCN1)-Mediated Cullin Neddylation. J. Med. Chem. 2018, 61, 2680–2693. [Google Scholar] [CrossRef]
- Xirodimas, D.P.; Saville, M.K.; Bourdon, J.-C.; Hay, R.T.; Lane, D.P. Mdm2-Mediated NEDD8 Conjugation of p53 Inhibits Its Transcriptional Activity. Cell 2004, 118, 83–97. [Google Scholar] [CrossRef]
- Abidi, N.; Xirodimas, D.P. Regulation of cancer-related pathways by protein NEDDylation and strategies for the use of NEDD8 inhibitors in the clinic. Endocr. -Relat. Cancer 2015, 22, T55–T70. [Google Scholar] [CrossRef]
- Zhou, Q.; Zheng, Y.; Sun, Y. Neddylation regulation of mitochondrial structure and functions. Cell Biosci. 2021, 11, 55. [Google Scholar] [CrossRef]
- Seeler, J.S.; Dejean, A. SUMO and the robustness of cancer. Nat. Rev. Cancer 2017, 17, 184–197. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, W.; Sun, Y.; Jia, L. Protein neddylation and its alterations in human cancers for targeted therapy. Cell Signal 2018, 44, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Cadena, C.; Ahmad, S.; Xavier, A.; Willemsen, J.; Park, S.; Park, J.W.; Oh, S.W.; Fujita, T.; Hou, F.; Binder, M.; et al. Ubiquitin-Dependent and -Independent Roles of E3 Ligase RIPLET in Innate Immunity. Cell 2019, 177, 1187–1200.e1116. [Google Scholar] [CrossRef] [PubMed]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Lipkowitz, S.; Weissman, A.M. RINGs of good and evil: RING finger ubiquitin ligases at the crossroads of tumour suppression and oncogenesis. Nat. Rev. Cancer 2011, 11, 629–643. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Pan, C.; Bei, J.X.; Li, B.; Liang, C.; Xu, Y.; Fu, X. Mutant p53 in Cancer Progression and Targeted Therapies. Front. Oncol. 2020, 10, 595187. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Bayle, J.H.; Olson, D.; Levine, A.J. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993, 7, 1126–1132. [Google Scholar] [CrossRef]
- Freedman, D.A.; Wu, L.; Levine, A.J. Functions of the MDM2 oncoprotein. Cell Mol. Life Sci. 1999, 55, 96–107. [Google Scholar] [CrossRef]
- Oliner, J.D.; Saiki, A.Y.; Caenepeel, S. The Role of MDM2 Amplification and Overexpression in Tumorigenesis. Cold Spring Harb. Perspect. Med. 2016, 6, a026336. [Google Scholar] [CrossRef]
- Sheikh, M.S.; Shao, Z.M.; Hussain, A.; Fontana, J.A. The p53-binding protein MDM2 gene is differentially expressed in human breast carcinoma. Cancer Res. 1993, 53, 3226–3228. [Google Scholar]
- Reifenberger, G.; Liu, L.; Ichimura, K.; Schmidt, E.E.; Collins, V.P. Amplification and overexpression of the MDM2 gene in a subset of human malignant gliomas without p53 mutations. Cancer Res. 1993, 53, 2736–2739. [Google Scholar]
- Watanabe, T.; Hotta, T.; Ichikawa, A.; Kinoshita, T.; Nagai, H.; Uchida, T.; Murate, T.; Saito, H. The MDM2 oncogene overexpression in chronic lymphocytic leukemia and low-grade lymphoma of B-cell origin. Blood 1994, 84, 3158–3165. [Google Scholar] [CrossRef] [PubMed]
- Bueso-Ramos, C.E.; Yang, Y.; deLeon, E.; McCown, P.; Stass, S.A.; Albitar, M. The human MDM-2 oncogene is overexpressed in leukemias. Blood 1993, 82, 2617–2623. [Google Scholar] [CrossRef] [PubMed]
- Capoulade, C.; Bressac-de Paillerets, B.; Lefrère, I.; Ronsin, M.; Feunteun, J.; Tursz, T.; Wiels, J. Overexpression of MDM2, due to enhanced translation, results in inactivation of wild-type p53 in Burkitt’s lymphoma cells. Oncogene 1998, 16, 1603–1610. [Google Scholar] [CrossRef]
- Bond, G.L.; Hu, W.; Levine, A.J. MDM2 is a central node in the p53 pathway: 12 years and counting. Curr. Cancer Drug Targets 2005, 5, 3–8. [Google Scholar] [CrossRef]
- Bond, G.L.; Hu, W.; Bond, E.E.; Robins, H.; Lutzker, S.G.; Arva, N.C.; Bargonetti, J.; Bartel, F.; Taubert, H.; Wuerl, P.; et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell 2004, 119, 591–602. [Google Scholar] [CrossRef]
- Hashizume, R.; Fukuda, M.; Maeda, I.; Nishikawa, H.; Oyake, D.; Yabuki, Y.; Ogata, H.; Ohta, T. The RING Heterodimer BRCA1-BARD1 Is a Ubiquitin Ligase Inactivated by a Breast Cancer-derived Mutation *. J. Biol. Chem. 2001, 276, 14537–14540. [Google Scholar] [CrossRef]
- Tarsounas, M.; Sung, P. The antitumorigenic roles of BRCA1-BARD1 in DNA repair and replication. Nat. Rev. Mol. Cell Biol. 2020, 21, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Drost, R.; Bouwman, P.; Rottenberg, S.; Boon, U.; Schut, E.; Klarenbeek, S.; Klijn, C.; van der Heijden, I.; van der Gulden, H.; Wientjens, E.; et al. BRCA1 RING function is essential for tumor suppression but dispensable for therapy resistance. Cancer Cell 2011, 20, 797–809. [Google Scholar] [CrossRef]
- Metzger, M.B.; Pruneda, J.N.; Klevit, R.E.; Weissman, A.M. RING-type E3 ligases: Master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2014, 1843, 47–60. [Google Scholar] [CrossRef]
- Mallery, D.L.; Vandenberg, C.J.; Hiom, K. Activation of the E3 ligase function of the BRCA1/BARD1 complex by polyubiquitin chains. EMBO J. 2002, 21, 6755–6762. [Google Scholar] [CrossRef]
- Martini, E.M.; Keeney, S.; Osley, M.A. A role for histone H2B during repair of UV-induced DNA damage in Saccharomyces cerevisiae. Genetics 2002, 160, 1375–1387. [Google Scholar] [CrossRef] [PubMed]
- Welchman, R.L.; Gordon, C.; Mayer, R.J. Ubiquitin and ubiquitin-like proteins as multifunctional signals. Nat. Rev. Mol. Cell Biol. 2005, 6, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Benavente, B.; Nasresfahani, A.F.; Farràs, R. Ubiquitin-Regulated Cell Proliferation and Cancer. Adv. Exp. Med. Biol. 2020, 1233, 3–28. [Google Scholar] [CrossRef]
- Huangfu, W.C.; Fuchs, S.Y. Ubiquitination-dependent regulation of signaling receptors in cancer. Genes Cancer 2010, 1, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.D.; Ritterhoff, T.; Klevit, R.E.; Brzovic, P.S. E2 enzymes: More than just middle men. Cell Res. 2016, 26, 423–440. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Zhang, R.; Liu, S.; Duan, T.; Zhai, L.; Zhang, M.; Han, X.; Xiang, Y.; Huang, X.; Lin, H.; et al. RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front Pharm. 2018, 9, 1371. [Google Scholar] [CrossRef]
- Zhao, Z.K.; Wu, W.G.; Chen, L.; Dong, P.; Gu, J.; Mu, J.S.; Yang, J.H.; Liu, Y.B. Expression of UbcH10 in pancreatic ductal adenocarcinoma and its correlation with prognosis. Tumour Biol. 2013, 34, 1473–1477. [Google Scholar] [CrossRef]
- Kim, Y.H.; Kim, C.Y. Current surgical management of insular gliomas. Neurosurg. Clin. N. Am. 2012, 23, 199–206. [Google Scholar] [CrossRef]
- Fristrup, N.; Birkenkamp-Demtröder, K.; Reinert, T.; Sanchez-Carbayo, M.; Segersten, U.; Malmström, P.U.; Palou, J.; Alvarez-Múgica, M.; Pan, C.C.; Ulhøi, B.P.; et al. Multicenter validation of cyclin D1, MCM7, TRIM29, and UBE2C as prognostic protein markers in non-muscle-invasive bladder cancer. Am. J. Pathol. 2013, 182, 339–349. [Google Scholar] [CrossRef]
- Hussain, S.; Zhang, Y.; Galardy, P.J. DUBs and cancer: The role of deubiquitinating enzymes as oncogenes, non-oncogenes and tumor suppressors. Cell Cycle 2009, 8, 1688–1697. [Google Scholar] [CrossRef] [PubMed]
- Vucic, D.; Dixit, V.M.; Wertz, I.E. Ubiquitylation in apoptosis: A post-translational modification at the edge of life and death. Nat. Rev. Mol. Cell Biol. 2011, 12, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Venuto, S.; Merla, G. E3 Ubiquitin Ligase TRIM Proteins, Cell Cycle and Mitosis. Cells 2019, 8, 510. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Zhou, Z.; Chen, W.; Wang, C.; Zhang, H.; Ge, G.; Shao, M.; You, D.; Fan, Z.; Xia, H.; et al. BAP1 promotes breast cancer cell proliferation and metastasis by deubiquitinating KLF5. Nat. Commun. 2015, 6, 8471. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Sun, T.; Liu, Z.; Yang, Q. The role of ubiquitination and deubiquitination in cancer metabolism. Mol. Cancer 2020, 19, 146. [Google Scholar] [CrossRef]
- Steinberg, G.R.; Carling, D. AMP-activated protein kinase: The current landscape for drug development. Nat. Rev. Drug Discov. 2019, 18, 527–551. [Google Scholar] [CrossRef]
- Araya, J.; Tsubouchi, K.; Sato, N.; Ito, S.; Minagawa, S.; Hara, H.; Hosaka, Y.; Ichikawa, A.; Saito, N.; Kadota, T.; et al. PRKN-regulated mitophagy and cellular senescence during COPD pathogenesis. Autophagy 2019, 15, 510–526. [Google Scholar] [CrossRef]
- Hu, H.; Sun, S.C. Ubiquitin signaling in immune responses. Cell Res. 2016, 26, 457–483. [Google Scholar] [CrossRef]
- Chang, S.C.; Ding, J.L. Ubiquitination and SUMOylation in the chronic inflammatory tumor microenvironment. Biochim. Biophys Acta Rev. Cancer 2018, 1870, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Tognon, C.E.; Rafn, B.; Cetinbas, N.M.; Kamura, T.; Trigo, G.; Rotblat, B.; Okumura, F.; Matsumoto, M.; Chow, C.; Davare, M.; et al. Insulin-like growth factor 1 receptor stabilizes the ETV6-NTRK3 chimeric oncoprotein by blocking its KPC1/Rnf123-mediated proteasomal degradation. J. Biol. Chem. 2018, 293, 12502–12515. [Google Scholar] [CrossRef] [PubMed]
- Rabl, J.; Bunker, R.D.; Schenk, A.D.; Cavadini, S.; Gill, M.E.; Abdulrahman, W.; Andrés-Pons, A.; Luijsterburg, M.S.; Ibrahim, A.F.M.; Branigan, E.; et al. Structural Basis of BRCC36 Function in DNA Repair and Immune Regulation. Mol. Cell 2019, 75, 483–497.e489. [Google Scholar] [CrossRef] [PubMed]
- Hoeller, D.; Hecker, C.M.; Dikic, I. Ubiquitin and ubiquitin-like proteins in cancer pathogenesis. Nat. Rev. Cancer 2006, 6, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Ben-Neriah, Y. Phosphorylation meets ubiquitination: The control of NF-[kappa]B activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef]
- Potu, H.; Kandarpa, M.; Peterson, L.F.; Donato, N.J.; Talpaz, M. Tumor necrosis factor related apoptosis inducing ligand (TRAIL) regulates deubiquitinase USP5 in tumor cells. Oncotarget 2019, 10, 5745–5754. [Google Scholar] [CrossRef][Green Version]
- Wang, S.; Juan, J.; Zhang, Z.; Du, Y.; Xu, Y.; Tong, J.; Cao, B.; Moran, M.F.; Zeng, Y.; Mao, X. Inhibition of the deubiquitinase USP5 leads to c-Maf protein degradation and myeloma cell apoptosis. Cell Death Dis. 2017, 8, e3058. [Google Scholar] [CrossRef]
- Kim, D.; Hong, A.; Park, H.I.; Shin, W.H.; Yoo, L.; Jeon, S.J.; Chung, K.C. Deubiquitinating enzyme USP22 positively regulates c-Myc stability and tumorigenic activity in mammalian and breast cancer cells. J. Cell Physiol 2017, 232, 3664–3676. [Google Scholar] [CrossRef]
- Popovic, D.; Vucic, D.; Dikic, I. Ubiquitination in disease pathogenesis and treatment. Nat. Med. 2014, 20, 1242–1253. [Google Scholar] [CrossRef]
- da Silva, S.R.; Paiva, S.L.; Lukkarila, J.L.; Gunning, P.T. Exploring a new frontier in cancer treatment: Targeting the ubiquitin and ubiquitin-like activating enzymes. J. Med. Chem. 2013, 56, 2165–2177. [Google Scholar] [CrossRef]
- Micel, L.N.; Tentler, J.J.; Smith, P.G.; Eckhardt, G.S. Role of ubiquitin ligases and the proteasome in oncogenesis: Novel targets for anticancer therapies. J. Clin. Oncol. 2013, 31, 1231–1238. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R.J. Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy. BMC Biol. 2014, 12, 94. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.K.; Lin, H.K.; Sun, S.C.; Zhang, S. Targeting ubiquitination for cancer therapies. Future Med. Chem. 2015, 7, 2333–2350. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Zhou, Q.; He, J.; Jiang, Z.; Peng, C.; Tong, R.; Shi, J. Recent advances in the development of protein-protein interactions modulators: Mechanisms and clinical trials. Signal Transduct. Target 2020, 5, 213. [Google Scholar] [CrossRef]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef]
- Meng, L.; Mohan, R.; Kwok, B.H.; Elofsson, M.; Sin, N.; Crews, C.M. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc. Natl. Acad. Sci. USA 1999, 96, 10403–10408. [Google Scholar] [CrossRef]
- Richardson, P.G.; Barlogie, B.; Berenson, J.; Singhal, S.; Jagannath, S.; Irwin, D.; Rajkumar, S.V.; Srkalovic, G.; Alsina, M.; Alexanian, R.; et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N. Engl. J. Med. 2003, 348, 2609–2617. [Google Scholar] [CrossRef]
- Antao, A.M.; Tyagi, A.; Kim, K.S.; Ramakrishna, S. Advances in Deubiquitinating Enzyme Inhibition and Applications in Cancer Therapeutics. Cancers 2020, 12, 1579. [Google Scholar] [CrossRef]
- Yuan, T.; Yan, F.; Ying, M.; Cao, J.; He, Q.; Zhu, H.; Yang, B. Inhibition of Ubiquitin-Specific Proteases as a Novel Anticancer Therapeutic Strategy. Front Pharm. 2018, 9, 1080. [Google Scholar] [CrossRef]
- Ivanschitz, L.; Takahashi, Y.; Jollivet, F.; Ayrault, O.; Le Bras, M.; de Thé, H. PML IV/ARF interaction enhances p53 SUMO-1 conjugation, activation, and senescence. Proc. Natl. Acad. Sci. USA 2015, 112, 14278. [Google Scholar] [CrossRef]
- Li, R.; Wei, J.; Jiang, C.; Liu, D.; Deng, L.; Zhang, K.; Wang, P. Akt SUMOylation Regulates Cell Proliferation and Tumorigenesis. Cancer Res. 2013, 73, 5742–5753. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yang, C.; Hong, Y.; Bi, H.; Zhao, F.; Liu, Y.; Ao, X.; Pang, P.; Xing, X.; Chang, A.K.; et al. The transcriptional activity of co-activator AIB1 is regulated by the SUMO E3 Ligase PIAS1. Biol. Cell 2012, 104, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Bawa-Khalfe, T.; Yeh, E.T. SUMO Losing Balance: SUMO Proteases Disrupt SUMO Homeostasis to Facilitate Cancer Development and Progression. Genes Cancer 2010, 1, 748–752. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.; Chung, K.C. Small Ubiquitin-like Modifier (SUMO) Modification of Zinc Finger Protein 131 Potentiates Its Negative Effect on Estrogen Signaling *. J. Biol. Chem. 2012, 287, 17517–17529. [Google Scholar] [CrossRef]
- Morris, J.R.; Boutell, C.; Keppler, M.; Densham, R.; Weekes, D.; Alamshah, A.; Butler, L.; Galanty, Y.; Pangon, L.; Kiuchi, T.; et al. The SUMO modification pathway is involved in the BRCA1 response to genotoxic stress. Nature 2009, 462, 886–890. [Google Scholar] [CrossRef]
- Wang, Z.; Jin, J.; Zhang, J.; Wang, L.; Cao, J. Depletion of SENP1 suppresses the proliferation and invasion of triple-negative breast cancer cells. Oncol. Rep. 2016, 36, 2071–2078. [Google Scholar] [CrossRef]
- Chen, C.-H.; Chang, C.-C.; Lee, T.H.; Luo, M.; Huang, P.; Liao, P.-H.; Wei, S.; Li, F.-A.; Chen, R.-H.; Zhou, X.Z.; et al. SENP1 deSUMOylates and Regulates Pin1 Protein Activity and Cellular Function. Cancer Res. 2013, 73, 3951–3962. [Google Scholar] [CrossRef]
- Bawa-Khalfe, T.; Lu, L.-S.; Zuo, Y.; Huang, C.; Dere, R.; Lin, F.-M.; Yeh, E.T.H. Differential expression of SUMO-specific protease 7 variants regulates epithelial–mesenchymal transition. Proc. Natl. Acad. Sci. USA 2012, 109, 17466–17471. [Google Scholar] [CrossRef]
- Mooney, S.M.; Grande, J.P.; Salisbury, J.L.; Janknecht, R. Sumoylation of p68 and p72 RNA Helicases Affects Protein Stability and Transactivation Potential. Biochemistry 2010, 49, 1–10. [Google Scholar] [CrossRef]
- Kessler, J.D.; Kahle, K.T.; Sun, T.; Meerbrey, K.L.; Schlabach, M.R.; Schmitt, E.M.; Skinner, S.O.; Xu, Q.; Li, M.Z.; Hartman, Z.C.; et al. A SUMOylation-Dependent Transcriptional Subprogram Is Required for Myc-Driven Tumorigenesis. Science 2012, 335, 348–353. [Google Scholar] [CrossRef]
- Zhu, S.; Sachdeva, M.; Wu, F.; Lu, Z.; Mo, Y.Y. Ubc9 promotes breast cell invasion and metastasis in a sumoylation-independent manner. Oncogene 2010, 29, 1763–1772. [Google Scholar] [CrossRef] [PubMed]
- Wen, D.; Xu, Z.; Xia, L.; Liu, X.; Tu, Y.; Lei, H.; Wang, W.; Wang, T.; Song, L.; Ma, C.; et al. Important Role of SUMOylation of Spliceosome Factors in Prostate Cancer Cells. J. Proteome Res. 2014, 13, 3571–3582. [Google Scholar] [CrossRef]
- Hu, L.; Yang, F.; Lu, L.; Dai, W. Arsenic-induced sumoylation of Mus81 is involved in regulating genomic stability. Cell Cycle 2017, 16, 802–811. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xia, N.; Li, T.; Xu, Y.; Zou, Y.; Zuo, Y.; Fan, Q.; Bawa-Khalfe, T.; Yeh, E.T.H.; Cheng, J. SUMO-specific protease 1 promotes prostate cancer progression and metastasis. Oncogene 2013, 32, 2493–2498. [Google Scholar] [CrossRef] [PubMed]
- Toropainen, S.; Malinen, M.; Kaikkonen, S.; Rytinki, M.; Jääskeläinen, T.; Sahu, B.; Jänne, O.A.; Palvimo, J.J. SUMO ligase PIAS1 functions as a target gene selective androgen receptor coregulator on prostate cancer cell chromatin. Nucleic Acids Res. 2015, 43, 848–861. [Google Scholar] [CrossRef] [PubMed]
- Núñez-O’Mara, A.; Gerpe-Pita, A.; Pozo, S.; Carlevaris, O.; Urzelai, B.; Lopitz-Otsoa, F.; Rodríguez, M.S.; Berra, E. PHD3–SUMO conjugation represses HIF1 transcriptional activity independently of PHD3 catalytic activity. J. Cell Sci. 2015, 128, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.-Y.; Chen, T.-T.; Xia, L.; Guo, M.; Xu, Y.; Yue, F.; Jiang, Y.; Chen, G.-Q.; Zhao, K.-W. Hypoxia inducible factor-1 mediates expression of galectin-1: The potential role in migration/invasion of colorectal cancer cells. Carcinogenesis 2010, 31, 1367–1375. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhu, J.; Dong, M.; Yu, H.; Dai, X.; Li, K. Knockdown of tripartite motif containing 24 by lentivirus suppresses cell growth and induces apoptosis in human colorectal cancer cells. Oncol. Res. 2014, 22, 39–45. [Google Scholar] [CrossRef]
- Huang, H.J.; Zhou, L.L.; Fu, W.J.; Zhang, C.Y.; Jiang, H.; Du, J.; Hou, J. β-catenin SUMOylation is involved in the dysregulated proliferation of myeloma cells. Am. J. Cancer Res. 2015, 5, 309–320. [Google Scholar]
- Xu, J.; Sun, H.Y.; Xiao, F.J.; Wang, H.; Yang, Y.; Wang, L.; Gao, C.J.; Guo, Z.K.; Wu, C.T.; Wang, L.S. SENP1 inhibition induces apoptosis and growth arrest of multiple myeloma cells through modulation of NF-κB signaling. Biochem Biophys Res. Commun 2015, 460, 409–415. [Google Scholar] [CrossRef]
- Zhao, Z.; Tan, X.; Zhao, A.; Zhu, L.; Yin, B.; Yuan, J.; Qiang, B.; Peng, X. microRNA-214-mediated UBC9 expression in glioma. BMB Rep. 2012, 45, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Bellail, A.C.; Olson, J.J.; Hao, C. SUMO1 modification stabilizes CDK6 protein and drives the cell cycle and glioblastoma progression. Nat. Commun. 2014, 5, 4234. [Google Scholar] [CrossRef]
- Li, H.; Niu, H.; Peng, Y.; Wang, J.; He, P. Ubc9 promotes invasion and metastasis of lung cancer cells. Oncol. Rep. 2013, 29, 1588–1594. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Huang, F.-F.; Wu, D.-S.; Li, W.-J.; Zhan, H.-E.; Peng, M.-Y.; Fang, P.; Cao, P.-F.; Zhang, M.-M.; Zeng, H.; et al. SUMOylation of insulin-like growth factor 1 receptor, promotes proliferation in acute myeloid leukemia. Cancer Lett. 2015, 357, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Moschos, S.J.; Smith, A.P.; Mandic, M.; Athanassiou, C.; Watson-Hurst, K.; Jukic, D.M.; Edington, H.D.; Kirkwood, J.M.; Becker, D. SAGE and antibody array analysis of melanoma-infiltrated lymph nodes: Identification of Ubc9 as an important molecule in advanced-stage melanomas. Oncogene 2007, 26, 4216–4225. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Shen, K.; Chen, T.; Yu, W.; Zhang, H. SUMO-1 Gene Silencing Inhibits Proliferation and Promotes Apoptosis of Human Gastric Cancer SGC-7901 Cells. Cell. Physiol. Biochem. 2017, 41, 987–998. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.Y.; Mu, X.Y.; Liu, B.; Wang, Y.; Bao, E.D.; Qiu, J.X.; Fan, Y. SUMO-Specific Protease 2 Suppresses Cell Migration and Invasion through Inhibiting the Expression of MMP13 in Bladder Cancer Cells. Cell. Physiol. Biochem. 2013, 32, 542–548. [Google Scholar] [CrossRef]
- Wang, K.; Zhang, X.C. Inhibition of SENP5 suppresses cell growth and promotes apoptosis in osteosarcoma cells. Exp. Med. 2014, 7, 1691–1695. [Google Scholar] [CrossRef]
- Jin, Z.L.; Pei, H.; Xu, Y.H.; Yu, J.; Deng, T. The SUMO-specific protease SENP5 controls DNA damage response and promotes tumorigenesis in hepatocellular carcinoma. Eur. Rev. Med. Pharm. Sci. 2016, 20, 3566–3573. [Google Scholar]
- Ding, X.; Sun, J.; Wang, L.; Li, G.; Shen, Y.; Zhou, X.; Chen, W. Overexpression of SENP5 in oral squamous cell carcinoma and its association with differentiation. Oncol. Rep. 2008, 20, 1041–1045. [Google Scholar]
- Ma, C.; Wu, B.; Huang, X.; Yuan, Z.; Nong, K.; Dong, B.; Bai, Y.; Zhu, H.; Wang, W.; Ai, K. SUMO-specific protease 1 regulates pancreatic cancer cell proliferation and invasion by targeting MMP-9. Tumor Biol. 2014, 35, 12729–12735. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.J.; Feng, Y.H.; Gu, B.H.; Li, Y.M.; Chen, H. The post-translational modification, SUMOylation, and cancer (Review). Int J. Oncol. 2018, 52, 1081–1094. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, I.; Ito, A.; Hirai, G.; Nishimura, S.; Kawasaki, H.; Saitoh, H.; Kimura, K.; Sodeoka, M.; Yoshida, M. Ginkgolic acid inhibits protein SUMOylation by blocking formation of the E1-SUMO intermediate. Chem. Biol. 2009, 16, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Kroonen, J.S.; Vertegaal, A.C.O. Targeting SUMO Signaling to Wrestle Cancer. Trends Cancer 2021, 7, 496–510. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, M.; Yu, G.; Chen, P.; Li, H.; Wei, D.; Zhu, J.; Xie, L.; Jia, H.; Shi, J.; et al. Overactivated neddylation pathway as a therapeutic target in lung cancer. J. Natl. Cancer Inst. 2014, 106, dju083. [Google Scholar] [CrossRef]
- Barbier-Torres, L.; Delgado, T.C.; García-Rodríguez, J.L.; Zubiete-Franco, I.; Fernández-Ramos, D.; Buqué, X.; Cano, A.; Gutiérrez-de Juan, V.; Fernández-Domínguez, I.; Lopitz-Otsoa, F.; et al. Stabilization of LKB1 and Akt by neddylation regulates energy metabolism in liver cancer. Oncotarget 2015, 6, 2509–2523. [Google Scholar] [CrossRef]
- Gao, Q.; Guangyang, Y.; Shi, J.-Y.; Li, L.-H.; Zhang, W.-J.; Wang, Z.; Yang, L.-X.; Duan, M.; Zhao, H.; Wang, X.-Y.; et al. Neddylation pathway is up-regulated in human intrahepatic cholangiocarcinoma and serves as a potential therapeutic target. Oncotarget 2014, 5, 7820. [Google Scholar] [CrossRef]
- Xie, P.; Yang, J.P.; Cao, Y.; Peng, L.X.; Zheng, L.S.; Sun, R.; Meng, D.F.; Wang, M.Y.; Mei, Y.; Qiang, Y.Y.; et al. Promoting tumorigenesis in nasopharyngeal carcinoma, NEDD8 serves as a potential theranostic target. Cell Death Dis. 2017, 8, e2834. [Google Scholar] [CrossRef]
- Chen, P.; Hu, T.; Liang, Y.; Li, P.; Chen, X.; Zhang, J.; Ma, Y.; Hao, Q.; Wang, J.; Zhang, P.; et al. Neddylation Inhibition Activates the Extrinsic Apoptosis Pathway through ATF4–CHOP–DR5 Axis in Human Esophageal Cancer Cells. Clin. Cancer Res. 2016, 22, 4145. [Google Scholar] [CrossRef]
- Hua, W.; Li, C.; Yang, Z.; Li, L.; Jiang, Y.; Yu, G.; Zhu, W.; Liu, Z.; Duan, S.; Chu, Y.; et al. Suppression of glioblastoma by targeting the overactivated protein neddylation pathway. Neuro Oncol. 2015, 17, 1333–1343. [Google Scholar] [CrossRef]
- Haagenson, K.K.; Tait, L.; Wang, J.; Shekhar, M.P.; Polin, L.; Chen, W.; Wu, G.S. Cullin-3 protein expression levels correlate with breast cancer progression. Cancer Biol. 2012, 13, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Welcker, M.; Clurman, B.E. FBW7 ubiquitin ligase: A tumour suppressor at the crossroads of cell division, growth and differentiation. Nat. Rev. Cancer 2008, 8, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.; Nagahama, H.; Minamishima, Y.A.; Matsumoto, M.; Nakamichi, I.; Kitagawa, K.; Shirane, M.; Tsunematsu, R.; Tsukiyama, T.; Ishida, N.; et al. Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication. EMBO J. 2000, 19, 2069–2081. [Google Scholar] [CrossRef]
- Margottin-Goguet, F.; Hsu, J.Y.; Loktev, A.; Hsieh, H.-M.; Reimann, J.D.R.; Jackson, P.K. Prophase Destruction of Emi1 by the SCFβTrCP/Slimb Ubiquitin Ligase Activates the Anaphase Promoting Complex to Allow Progression beyond Prometaphase. Dev. Cell 2003, 4, 813–826. [Google Scholar] [CrossRef]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef]
- Ryu, J.-H.; Li, S.-H.; Park, H.-S.; Park, J.-W.; Lee, B.; Chun, Y.-S. Hypoxia-inducible Factor α Subunit Stabilization by NEDD8 Conjugation Is Reactive Oxygen Species-dependent. J. Biol. Chem. 2011, 286, 6963–6970. [Google Scholar] [CrossRef]
- Read, M.A.; Brownell, J.E.; Gladysheva, T.B.; Hottele, M.; Parent, L.A.; Coggins, M.B.; Pierce, J.W.; Podust, V.N.; Luo, R.-S.; Chau, V.; et al. Nedd8 Modification of Cul-1 Activates SCFβTrCP-Dependent Ubiquitination of IκBα. Mol. Cell. Biol. 2000, 20, 2326–2333. [Google Scholar] [CrossRef]
- Lan, H.; Tang, Z.; Jin, H.; Sun, Y. Neddylation inhibitor MLN4924 suppresses growth and migration of human gastric cancer cells. Sci. Rep. 2016, 6, 24218. [Google Scholar] [CrossRef]
- Xie, P.; Zhang, M.; He, S.; Lu, K.; Chen, Y.; Xing, G.; Lu, Y.; Liu, P.; Li, Y.; Wang, S.; et al. The covalent modifier Nedd8 is critical for the activation of Smurf1 ubiquitin ligase in tumorigenesis. Nat. Commun. 2014, 5, 3733. [Google Scholar] [CrossRef]
- Kitching, R.; Li, H.; Wong, M.J.; Kanaganayakam, S.; Kahn, H.; Seth, A. Characterization of a novel human breast cancer associated gene (BCA3) encoding an alternatively spliced proline-rich protein. Biochim. Biophys Acta 2003, 1625, 116–121. [Google Scholar] [CrossRef]
- Gao, F.; Cheng, J.; Shi, T.; Yeh, E.T.H. Neddylation of a breast cancer-associated protein recruits a class III histone deacetylase that represses NFκB-dependent transcription. Nat. Cell Biol. 2006, 8, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Zuo, W.; Huang, F.; Chiang, Y.J.; Li, M.; Du, J.; Ding, Y.; Zhang, T.; Lee, H.W.; Jeong, L.S.; Chen, Y.; et al. c-Cbl-mediated neddylation antagonizes ubiquitination and degradation of the TGF-β type II receptor. Mol. Cell 2013, 49, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Wang, Q.; Cao, H.; Qiu, G.; Cao, J.; Li, X.; Wang, J.; Shen, B.; Zhang, J. The NEDD8-activating enzyme inhibitor MLN4924 induces G2 arrest and apoptosis in T-cell acute lymphoblastic leukemia. Oncotarget 2016, 7, 23812–23824. [Google Scholar] [CrossRef]
- Nawrocki, S.T.; Griffin, P.; Kelly, K.R.; Carew, J.S. MLN4924: A novel first-in-class inhibitor of NEDD8-activating enzyme for cancer therapy. Expert Opin. Investig. Drugs 2012, 21, 1563–1573. [Google Scholar] [CrossRef]
- Gai, W.; Peng, Z.; Liu, C.H.; Zhang, L.; Jiang, H. Advances in Cancer Treatment by Targeting the Neddylation Pathway. Front. Cell Dev. Biol. 2021, 9, 653882. [Google Scholar] [CrossRef]
- Zhou, H.; Lu, J.; Liu, L.; Bernard, D.; Yang, C.Y.; Fernandez-Salas, E.; Chinnaswamy, K.; Layton, S.; Stuckey, J.; Yu, Q.; et al. A potent small-molecule inhibitor of the DCN1-UBC12 interaction that selectively blocks cullin 3 neddylation. Nat. Commun. 2017, 8, 1150. [Google Scholar] [CrossRef]
- Hammill, J.T.; Bhasin, D.; Scott, D.C.; Min, J.; Chen, Y.; Lu, Y.; Yang, L.; Kim, H.S.; Connelly, M.C.; Hammill, C.; et al. Discovery of an Orally Bioavailable Inhibitor of Defective in Cullin Neddylation 1 (DCN1)-Mediated Cullin Neddylation. J. Med. Chem. 2018, 61, 2694–2706. [Google Scholar] [CrossRef]
- Sims, J.J.; Scavone, F.; Cooper, E.M.; Kane, L.A.; Youle, R.J.; Boeke, J.D.; Cohen, R.E. Polyubiquitin-sensor proteins reveal localization and linkage-type dependence of cellular ubiquitin signaling. Nat. Methods 2012, 9, 303–309. [Google Scholar] [CrossRef]
- Hospenthal, M.K.; Mevissen, T.E.T.; Komander, D. Deubiquitinase-based analysis of ubiquitin chain architecture using Ubiquitin Chain Restriction (UbiCRest). Nat. Protoc 2015, 10, 349–361. [Google Scholar] [CrossRef]
- Newton, K.; Matsumoto, M.L.; Wertz, I.E.; Kirkpatrick, D.S.; Lill, J.R.; Tan, J.; Dugger, D.; Gordon, N.; Sidhu, S.S.; Fellouse, F.A.; et al. Ubiquitin Chain Editing Revealed by Polyubiquitin Linkage-Specific Antibodies. Cell 2008, 134, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Guven, A.; Wu, W.S.; Patil, S.; Gokul, K.; Tekumalla, P.; Sharma, S.; Diers, A.; Gesta, S.; Vishnudas, V.; Sarangarajan, R.; et al. Diablo ubiquitination analysis by sandwich immunoassay. J. Pharm. Biomed. Anal. 2019, 173, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Hjerpe, R.; Aillet, F.; Lopitz-Otsoa, F.; Lang, V.; England, P.; Rodriguez, M.S. Efficient protection and isolation of ubiquitylated proteins using tandem ubiquitin-binding entities. EMBO Rep. 2009, 10, 1250–1258. [Google Scholar] [CrossRef] [PubMed]
- Kenten, J.H.; Davydov, I.V.; Safiran, Y.J.; Stewart, D.H.; Oberoi, P.; Biebuyck, H.A. Assays for high-throughput screening of E2 AND E3 ubiquitin ligases. Methods Enzym. 2005, 399, 682–701. [Google Scholar] [CrossRef]
- Herman, A.G.; Hayano, M.; Poyurovsky, M.V.; Shimada, K.; Skouta, R.; Prives, C.; Stockwell, B.R. Discovery of Mdm2-MdmX E3 ligase inhibitors using a cell-based ubiquitination assay. Cancer Discov. 2011, 1, 312–325. [Google Scholar] [CrossRef]
- Tian, X.; Isamiddinova, N.S.; Peroutka, R.J.; Goldenberg, S.J.; Mattern, M.R.; Nicholson, B.; Leach, C. Characterization of Selective Ubiquitin and Ubiquitin-Like Protease Inhibitors Using a Fluorescence-Based Multiplex Assay Format. ASSAY Drug Dev. Technol. 2010, 9, 165–173. [Google Scholar] [CrossRef]
- Eglen, R.M.; Reisine, T.; Roby, P.; Rouleau, N.; Illy, C.; Bossé, R.; Bielefeld, M. The use of AlphaScreen technology in HTS: Current status. Curr Chem. Genom. 2008, 1, 2–10. [Google Scholar] [CrossRef]
- Franklin, T.G.; Pruneda, J.N. A High-Throughput Assay for Monitoring Ubiquitination in Real Time. Front Chem. 2019, 7, 816. [Google Scholar] [CrossRef]
- Peng, J.; Schwartz, D.; Elias, J.E.; Thoreen, C.C.; Cheng, D.; Marsischky, G.; Roelofs, J.; Finley, D.; Gygi, S.P. A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol. 2003, 21, 921–926. [Google Scholar] [CrossRef]
- Xiao, Y.; Pollack, D.; Nieves, E.; Winchell, A.; Callaway, M.; Vigodner, M. Can your protein be sumoylated? A quick summary and important tips to study SUMO-modified proteins. Anal. Biochem. 2015, 477, 95–97. [Google Scholar] [CrossRef]
- Hansen, F.M.; Tanzer, M.C.; Brüning, F.; Bludau, I.; Stafford, C.; Schulman, B.A.; Robles, M.S.; Karayel, O.; Mann, M. Data-independent acquisition method for ubiquitinome analysis reveals regulation of circadian biology. Nat. Commun. 2021, 12, 254. [Google Scholar] [CrossRef] [PubMed]
- Sidoli, S.; Lu, C.; Coradin, M.; Wang, X.; Karch, K.R.; Ruminowicz, C.; Garcia, B.A. Metabolic labeling in middle-down proteomics allows for investigation of the dynamics of the histone code. Epigenetics Chromatin 2017, 10, 34. [Google Scholar] [CrossRef] [PubMed]
- Valkevich, E.M.; Sanchez, N.A.; Ge, Y.; Strieter, E.R. Middle-down mass spectrometry enables characterization of branched ubiquitin chains. Biochemistry 2014, 53, 4979–4989. [Google Scholar] [CrossRef]
- Swatek, K.N.; Usher, J.L.; Kueck, A.F.; Gladkova, C.; Mevissen, T.E.T.; Pruneda, J.N.; Skern, T.; Komander, D. Insights into ubiquitin chain architecture using Ub-clipping. Nature 2019, 572, 533–537. [Google Scholar] [CrossRef]
- Sun, H.; Leverson, J.D.; Hunter, T. Conserved function of RNF4 family proteins in eukaryotes: Targeting a ubiquitin ligase to SUMOylated proteins. EMBO J. 2007, 26, 4102–4112. [Google Scholar] [CrossRef] [PubMed]
- Eifler, K.; Vertegaal, A.C. Mapping the SUMOylated landscape. FEBS J. 2015, 282, 3669–3680. [Google Scholar] [CrossRef]
- Pirone, L.; Xolalpa, W.; Sigurðsson, J.O.; Ramirez, J.; Pérez, C.; González, M.; de Sabando, A.R.; Elortza, F.; Rodriguez, M.S.; Mayor, U.; et al. A comprehensive platform for the analysis of ubiquitin-like protein modifications using in vivo biotinylation. Sci. Rep. 2017, 7, 40756. [Google Scholar] [CrossRef]
- Tripathi, V.; Das, R. A Fluorescence-Based Assay to Monitor SUMOylation in Real-Time. Curr. Protoc. Protein Sci. 2020, 101, e111. [Google Scholar] [CrossRef] [PubMed]
- Obata, S.; Yuasa, E.; Seki, D.; Kitano, T.; Saitoh, H. Molecular Cloning and Bacterial Expression of the Catalytic Domain of the SENP1 Gene of Oryzias latipes. Biosci. Biotechnol. Biochem. 2013, 77, 1788–1791. [Google Scholar] [CrossRef]
- Lumpkin, R.J.; Gu, H.; Zhu, Y.; Leonard, M.; Ahmad, A.S.; Clauser, K.R.; Meyer, J.G.; Bennett, E.J.; Komives, E.A. Site-specific identification and quantitation of endogenous SUMO modifications under native conditions. Nat. Commun. 2017, 8, 1171. [Google Scholar] [CrossRef]
- Hendriks, I.A.; Lyon, D.; Su, D.; Skotte, N.H.; Daniel, J.A.; Jensen, L.J.; Nielsen, M.L. Site-specific characterization of endogenous SUMOylation across species and organs. Nat. Commun. 2018, 9, 2456. [Google Scholar] [CrossRef] [PubMed]
- Impens, F.; Radoshevich, L.; Cossart, P.; Ribet, D. Mapping of SUMO sites and analysis of SUMOylation changes induced by external stimuli. Proc. Natl. Acad. Sci. USA 2014, 111, 12432. [Google Scholar] [CrossRef] [PubMed]
- Schwinn, M.K.; Hoang, T.; Yang, X.; Zhao, X.; Ma, J.; Li, P.; Wood, K.V.; Mallender, W.D.; Bembenek, M.E.; Yan, Z.H. Antibody-free detection of cellular neddylation dynamics of Cullin1. Anal. Biochem. 2018, 555, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Y.L.; Qiu, G.; Pian, L.; Guo, L.; Cao, H.; Liu, J.; Zhao, Y.; Li, X.; Xu, Z.; et al. Hepatic neddylation targets and stabilizes electron transfer flavoproteins to facilitate fatty acid β-oxidation. Proc. Natl. Acad. Sci. USA 2020, 117, 2473–2483. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, B.; Lai, G.; Xu, R.; Chu, G.; Zhao, Y. 20-Hydroxyeicosatetraenoic acid regulates the expression of Nedd4-2 in kidney and liver through a neddylation modification pathway. Mol. Med. Rep. 2017, 16, 9671–9677. [Google Scholar] [CrossRef][Green Version]
- Li, X.; Yokoyama, N.N.; Zhang, S.; Ding, L.; Liu, H.M.; Lilly, M.B.; Mercola, D.; Zi, X. Flavokawain A induces deNEDDylation and Skp2 degradation leading to inhibition of tumorigenesis and cancer progression in the TRAMP transgenic mouse model. Oncotarget 2015, 6, 41809–41824. [Google Scholar] [CrossRef]
- An, H.; Statsyuk, A.V. Development of Activity-Based Probes for Ubiquitin and Ubiquitin-like Protein Signaling Pathways. J. Am. Chem. Soc. 2013, 135, 16948–16962. [Google Scholar] [CrossRef]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458, 732–736. [Google Scholar] [CrossRef]
- Hyer, M.L.; Milhollen, M.A.; Ciavarri, J.; Fleming, P.; Traore, T.; Sappal, D.; Huck, J.; Shi, J.; Gavin, J.; Brownell, J.; et al. A small-molecule inhibitor of the ubiquitin activating enzyme for cancer treatment. Nat. Med. 2018, 24, 186–193. [Google Scholar] [CrossRef]
- Zhong, H.J.; Wang, W.; Kang, T.S.; Yan, H.; Yang, Y.; Xu, L.; Wang, Y.; Ma, D.L.; Leung, C.H. A Rhodium(III) Complex as an Inhibitor of Neural Precursor Cell Expressed, Developmentally Down-Regulated 8-Activating Enzyme with in Vivo Activity against Inflammatory Bowel Disease. J. Med. Chem. 2017, 60, 497–503. [Google Scholar] [CrossRef]
- Zhou, H.; Zhou, W.; Zhou, B.; Liu, L.; Chern, T.R.; Chinnaswamy, K.; Lu, J.; Bernard, D.; Yang, C.Y.; Li, S.; et al. High-Affinity Peptidomimetic Inhibitors of the DCN1-UBC12 Protein-Protein Interaction. J. Med. Chem. 2018, 61, 1934–1950. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.J.; Ma, V.P.; Cheng, Z.; Chan, D.S.; He, H.Z.; Leung, K.H.; Ma, D.L.; Leung, C.H. Discovery of a natural product inhibitor targeting protein neddylation by structure-based virtual screening. Biochimie 2012, 94, 2457–2460. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Jiang, Y.; Sun, Y. Anticancer drug discovery by targeting cullin neddylation. Acta Pharm Sin. B 2020, 10, 746–765. [Google Scholar] [CrossRef] [PubMed]
- Lobato-Gil, S.; Heidelberger, J.B.; Maghames, C.; Bailly, A.; Brunello, L.; Rodriguez, M.S.; Beli, P.; Xirodimas, D.P. Proteome-wide identification of NEDD8 modification sites reveals distinct proteomes for canonical and atypical NEDDylation. Cell Rep. 2021, 34, 108635. [Google Scholar] [CrossRef]
- Vogl, A.M.; Phu, L.; Becerra, R.; Giusti, S.A.; Verschueren, E.; Hinkle, T.B.; Bordenave, M.D.; Adrian, M.; Heidersbach, A.; Yankilevich, P.; et al. Global site-specific neddylation profiling reveals that NEDDylated cofilin regulates actin dynamics. Nat. Struct. Mol. Biol. 2020, 27, 210–220. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pellegrino, N.E.; Guven, A.; Gray, K.; Shah, P.; Kasture, G.; Nastke, M.-D.; Thakurta, A.; Gesta, S.; Vishnudas, V.K.; Narain, N.R.; et al. The Next Frontier: Translational Development of Ubiquitination, SUMOylation, and NEDDylation in Cancer. Int. J. Mol. Sci. 2022, 23, 3480. https://doi.org/10.3390/ijms23073480
Pellegrino NE, Guven A, Gray K, Shah P, Kasture G, Nastke M-D, Thakurta A, Gesta S, Vishnudas VK, Narain NR, et al. The Next Frontier: Translational Development of Ubiquitination, SUMOylation, and NEDDylation in Cancer. International Journal of Molecular Sciences. 2022; 23(7):3480. https://doi.org/10.3390/ijms23073480
Chicago/Turabian StylePellegrino, Nicole E., Arcan Guven, Kayleigh Gray, Punit Shah, Gargi Kasture, Maria-Dorothea Nastke, Anjan Thakurta, Stephane Gesta, Vivek K. Vishnudas, Niven R. Narain, and et al. 2022. "The Next Frontier: Translational Development of Ubiquitination, SUMOylation, and NEDDylation in Cancer" International Journal of Molecular Sciences 23, no. 7: 3480. https://doi.org/10.3390/ijms23073480
APA StylePellegrino, N. E., Guven, A., Gray, K., Shah, P., Kasture, G., Nastke, M.-D., Thakurta, A., Gesta, S., Vishnudas, V. K., Narain, N. R., & Kiebish, M. A. (2022). The Next Frontier: Translational Development of Ubiquitination, SUMOylation, and NEDDylation in Cancer. International Journal of Molecular Sciences, 23(7), 3480. https://doi.org/10.3390/ijms23073480