
Human MicroRNAs Attenuate the Expression of Immediate Early Proteins and HCMV Replication during Lytic and Latent Infection in Connection with Enhancement of Phosphorylated RelA/p65 (Serine 536) That Binds to MIEP

 , and
, and
Abstract
1. Introduction
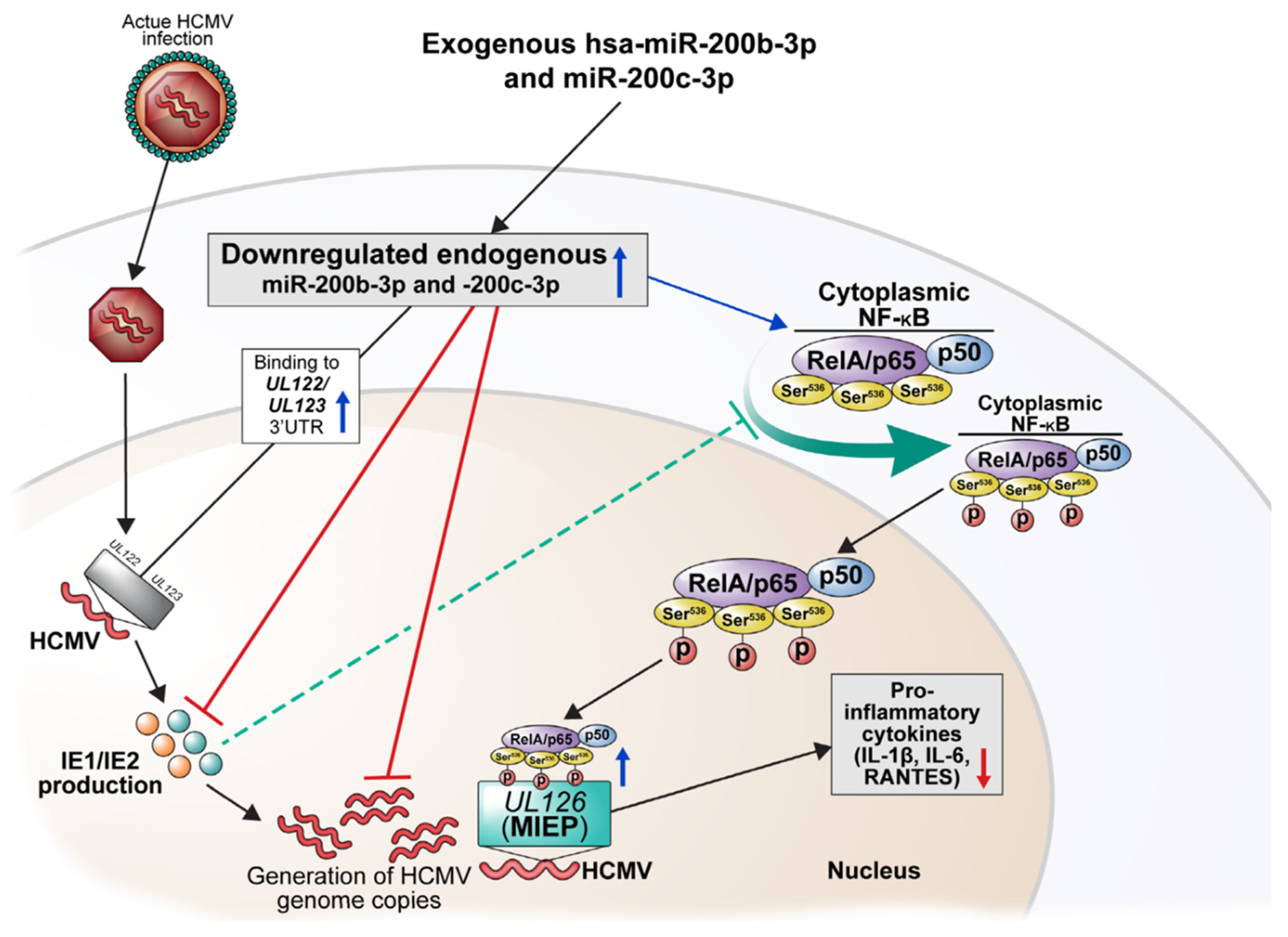
2. Results
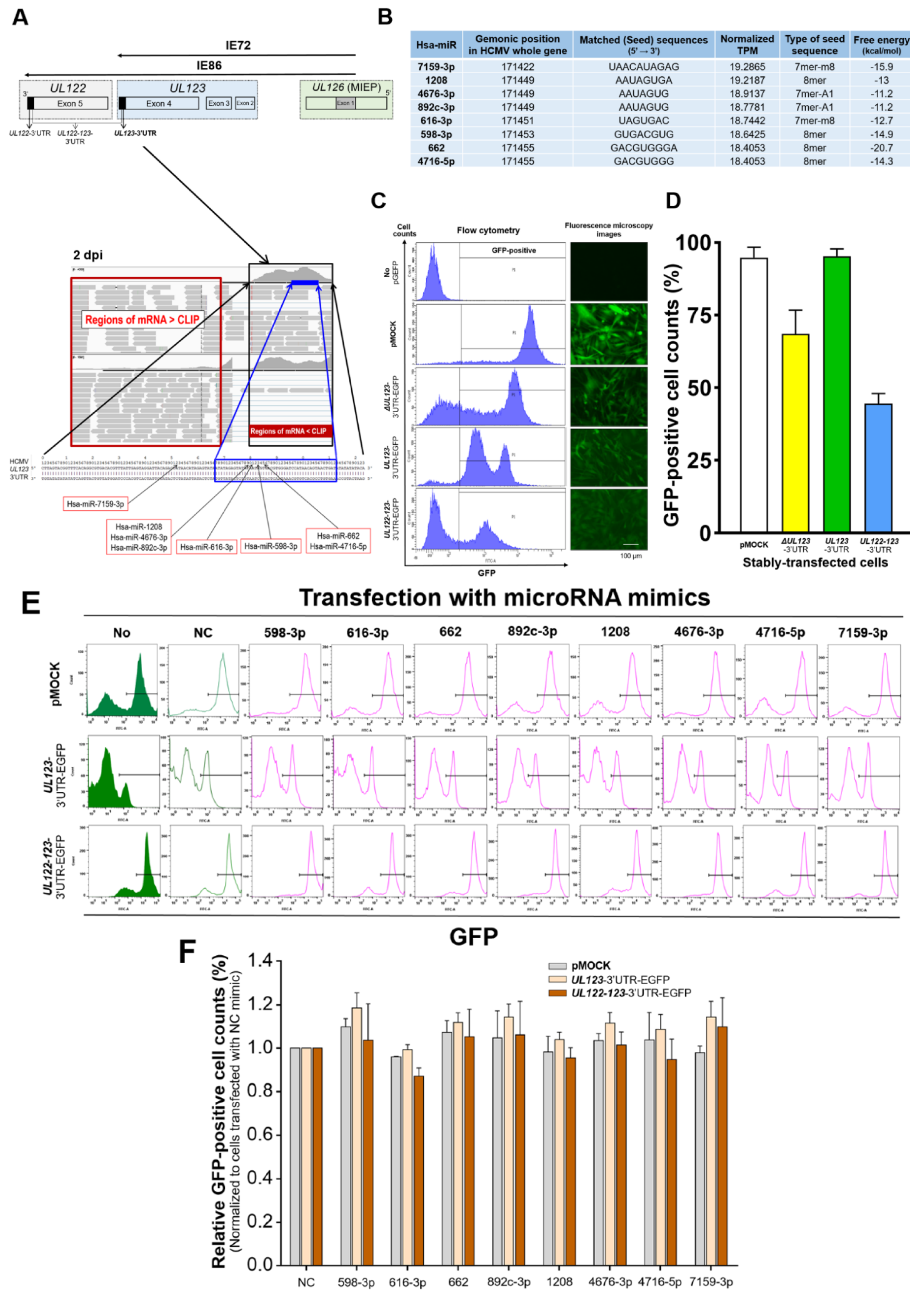
2.1. Identification of miRNAs That Bind to the 3′-UTR of UL123 Based on AGO-CLIP-Seq Data
2.2. Binding of the Predicted miRNAs to the 3′-UTR of UL123
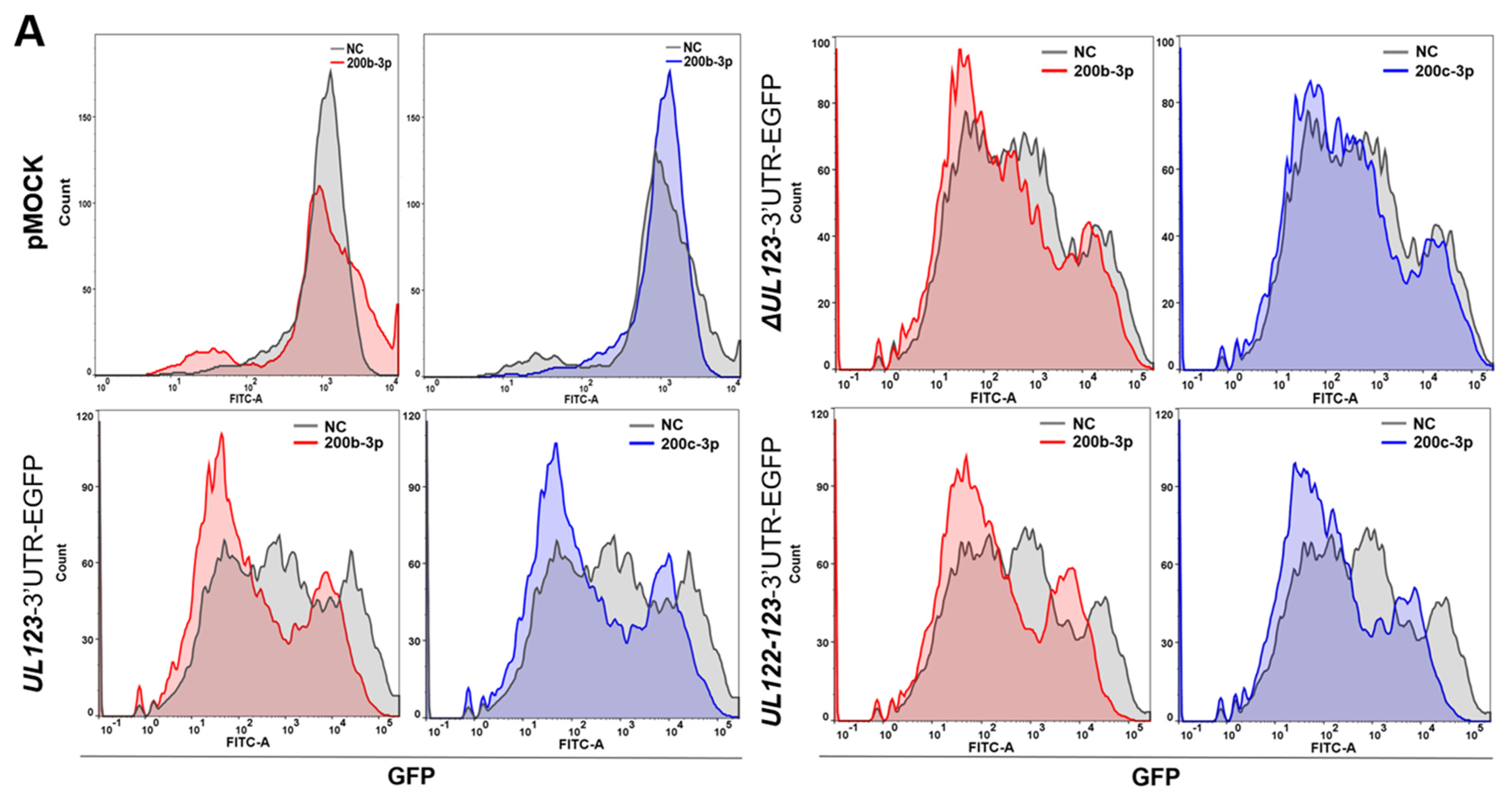
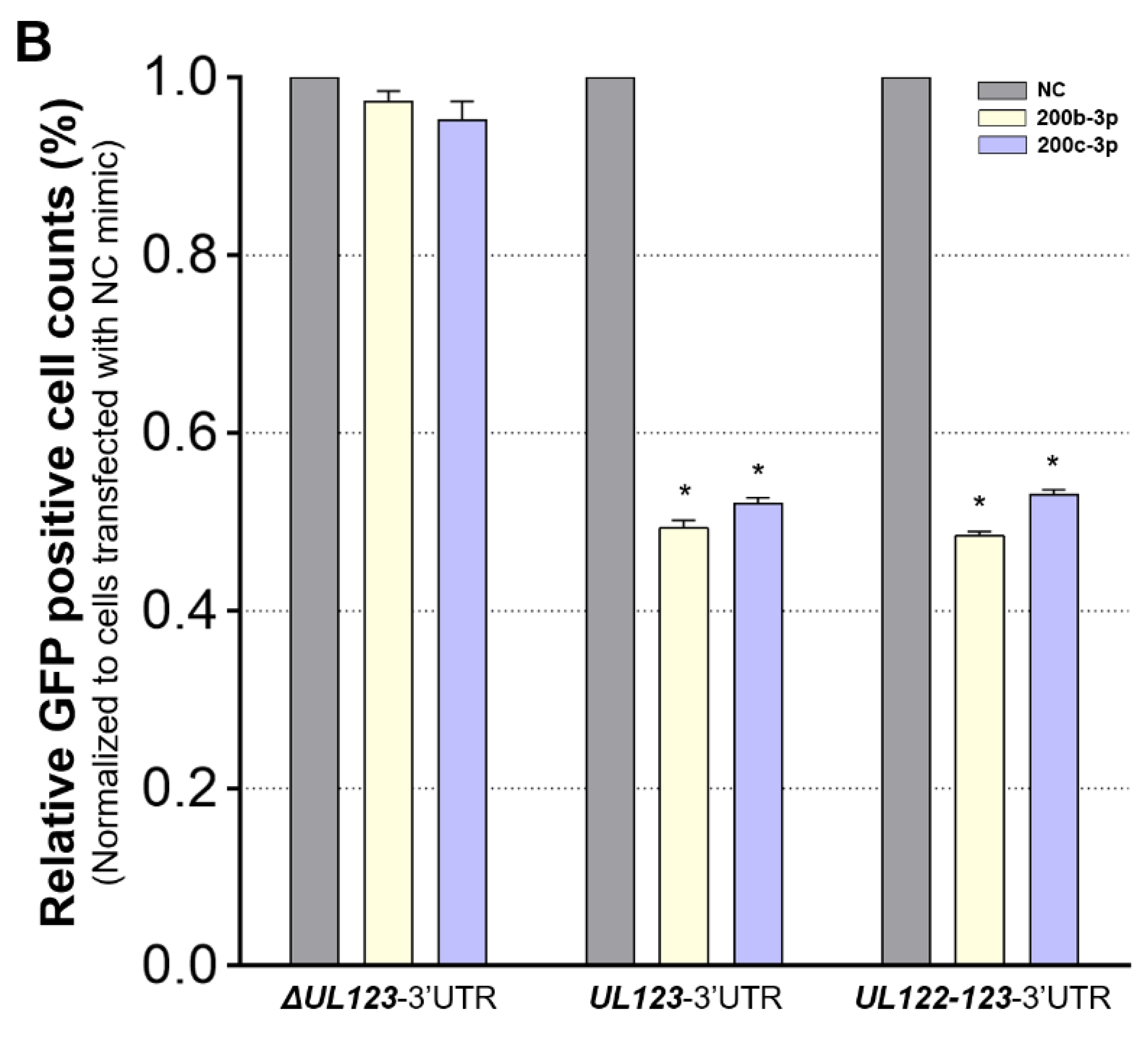
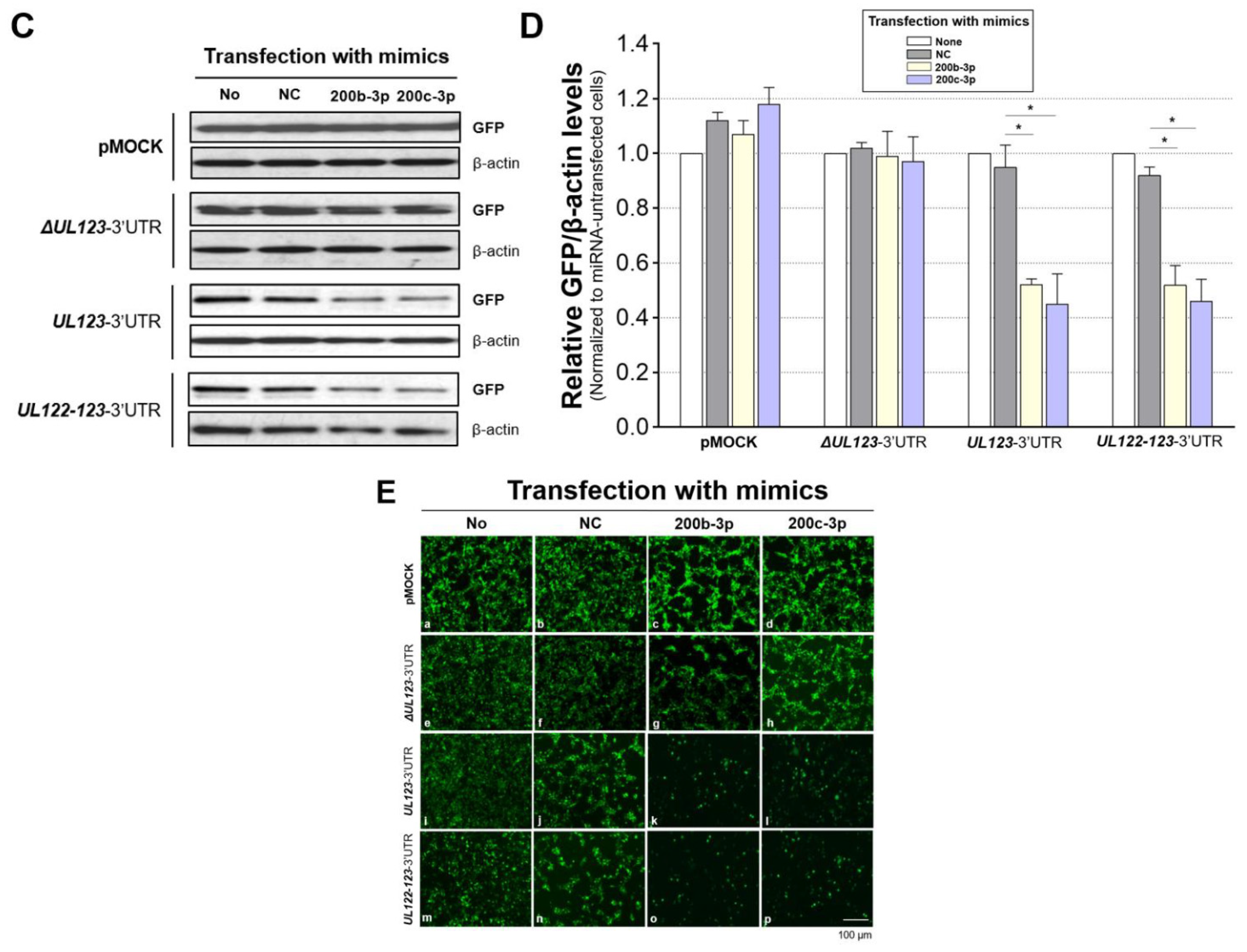
2.3. MiR-200b-3p and miR-200c-3p Bound to the 3-UTR of HCMV UL123
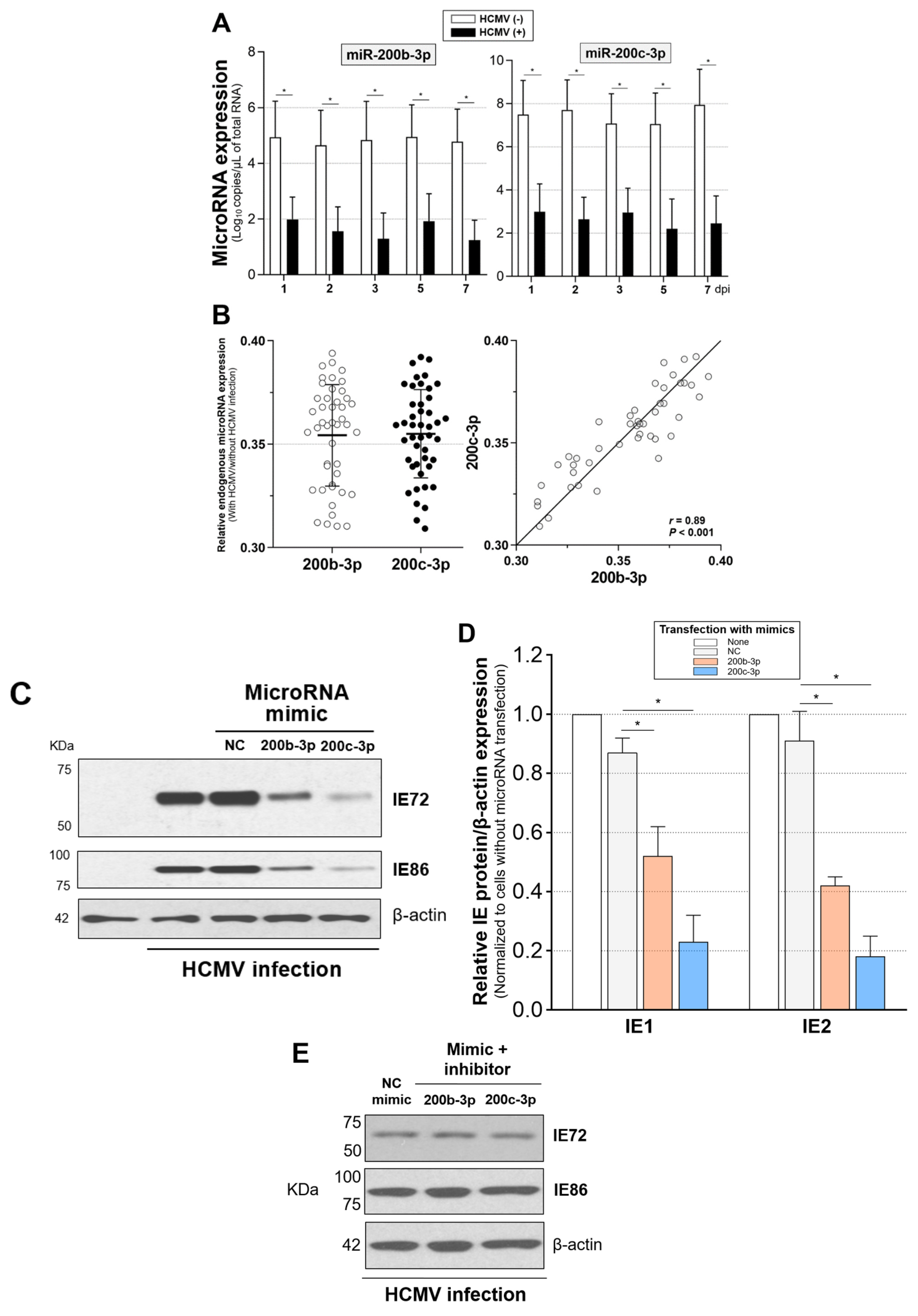
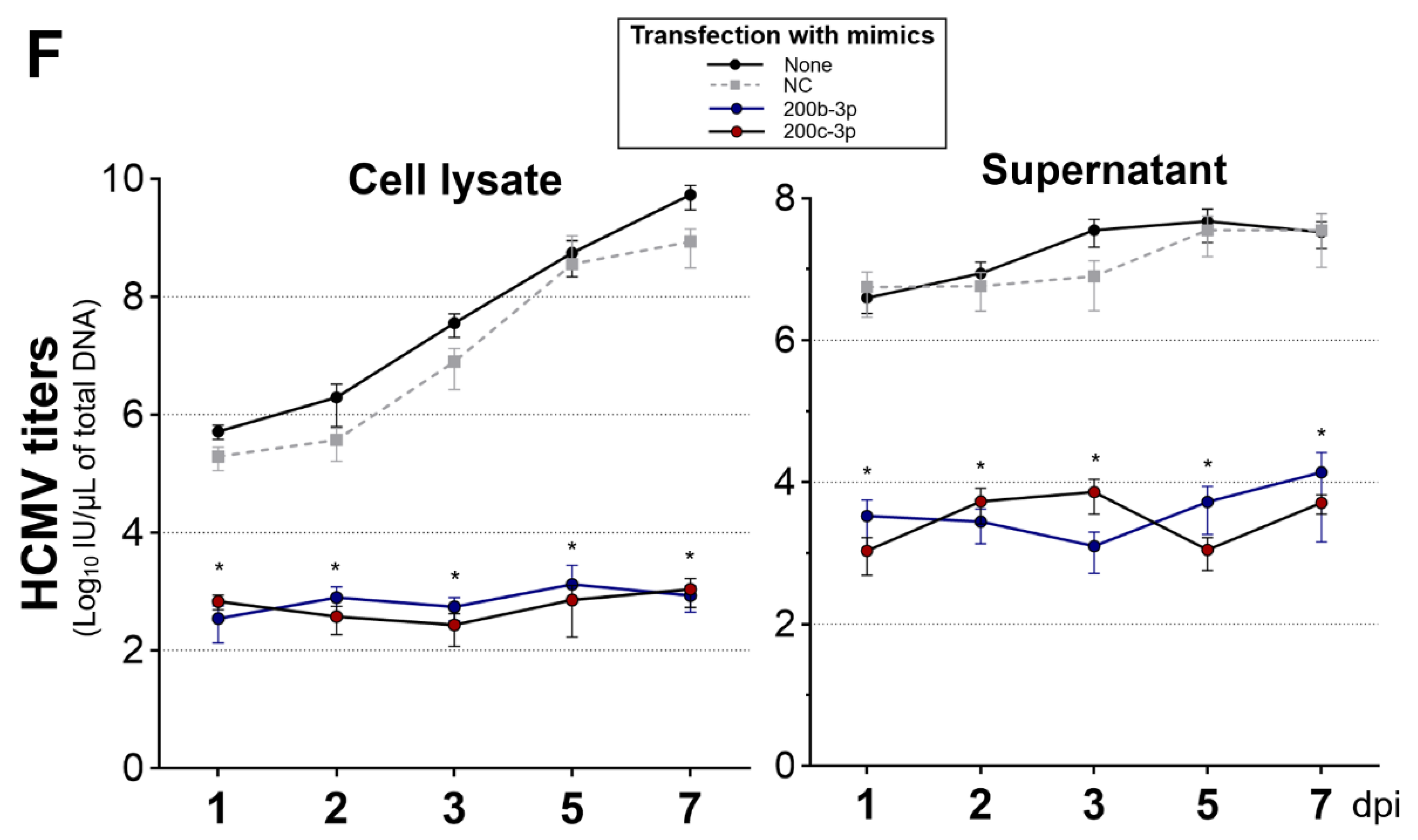
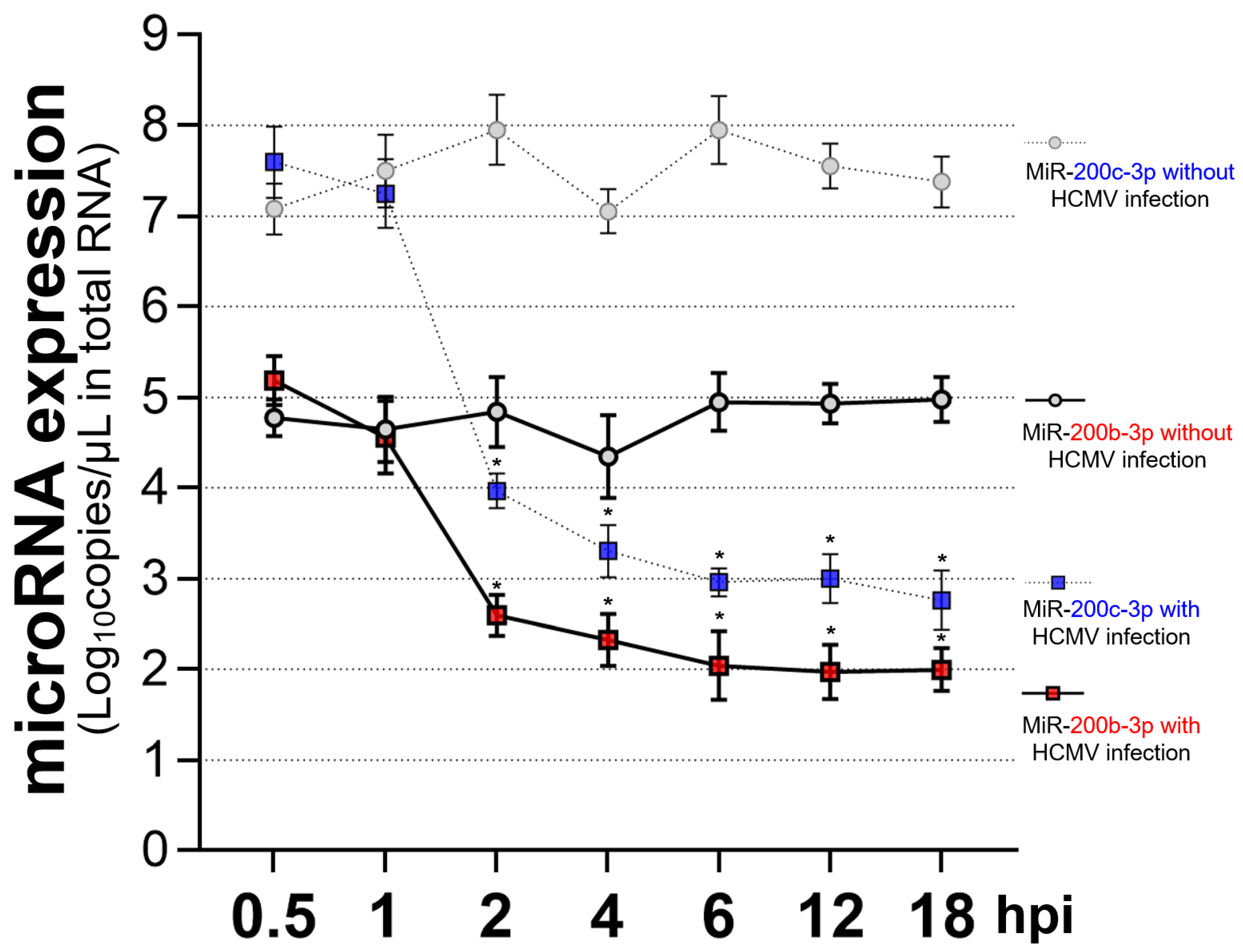
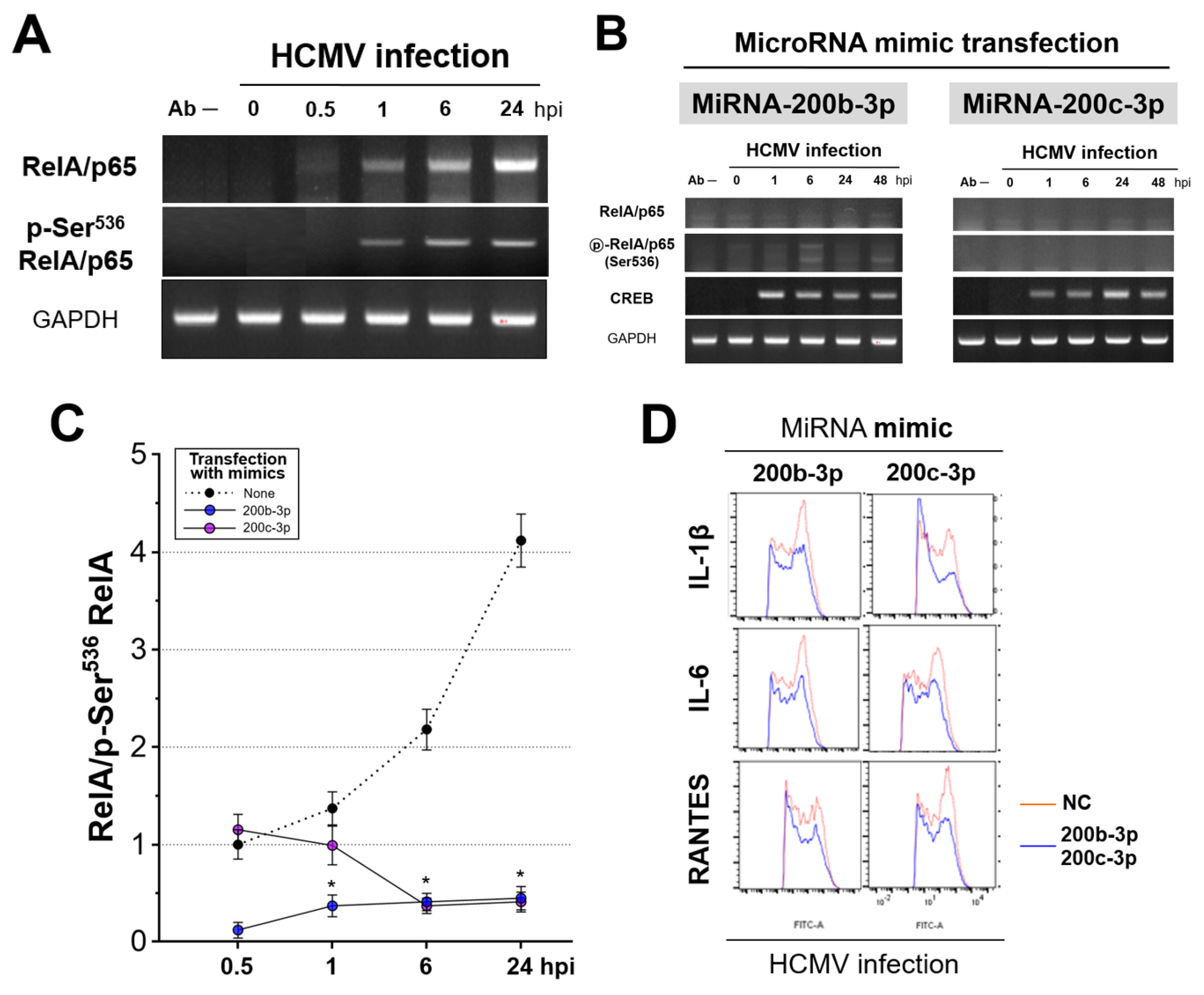
2.4. Restoration of Downregulated Expression of miR-200b-3p and miR-200c-3p after Acute Lytic HCMV Infection Decreased IE72 and IE86 Expression and HCMV Titers
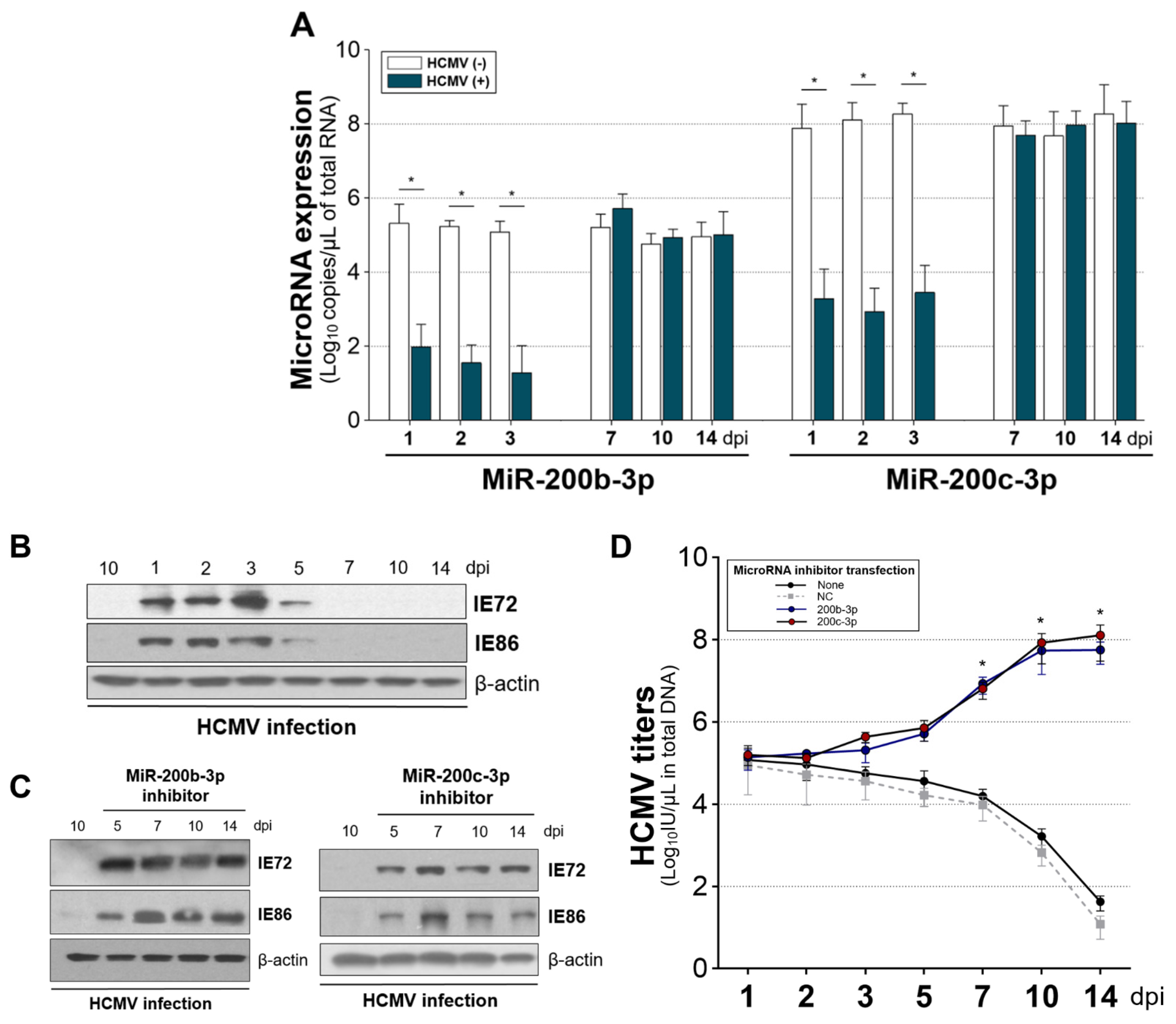
2.5. The Expression of miR-200b-3p and miR-200c-3p Is Not Downregulated during Latent HCMV Infection and Exogenous Inhibition of miRNA Expression Induced IE Expression and Virus Replication during Latency
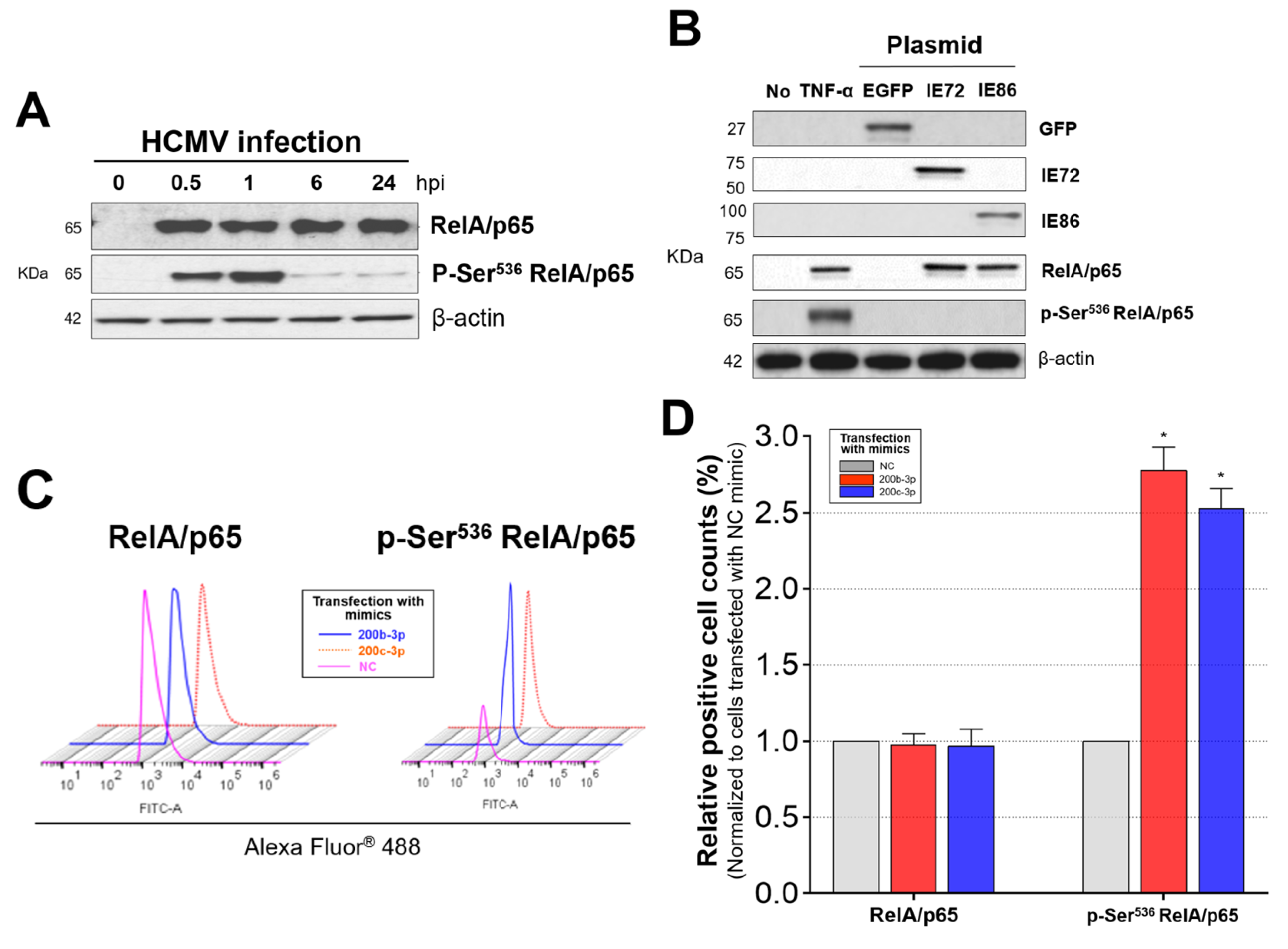
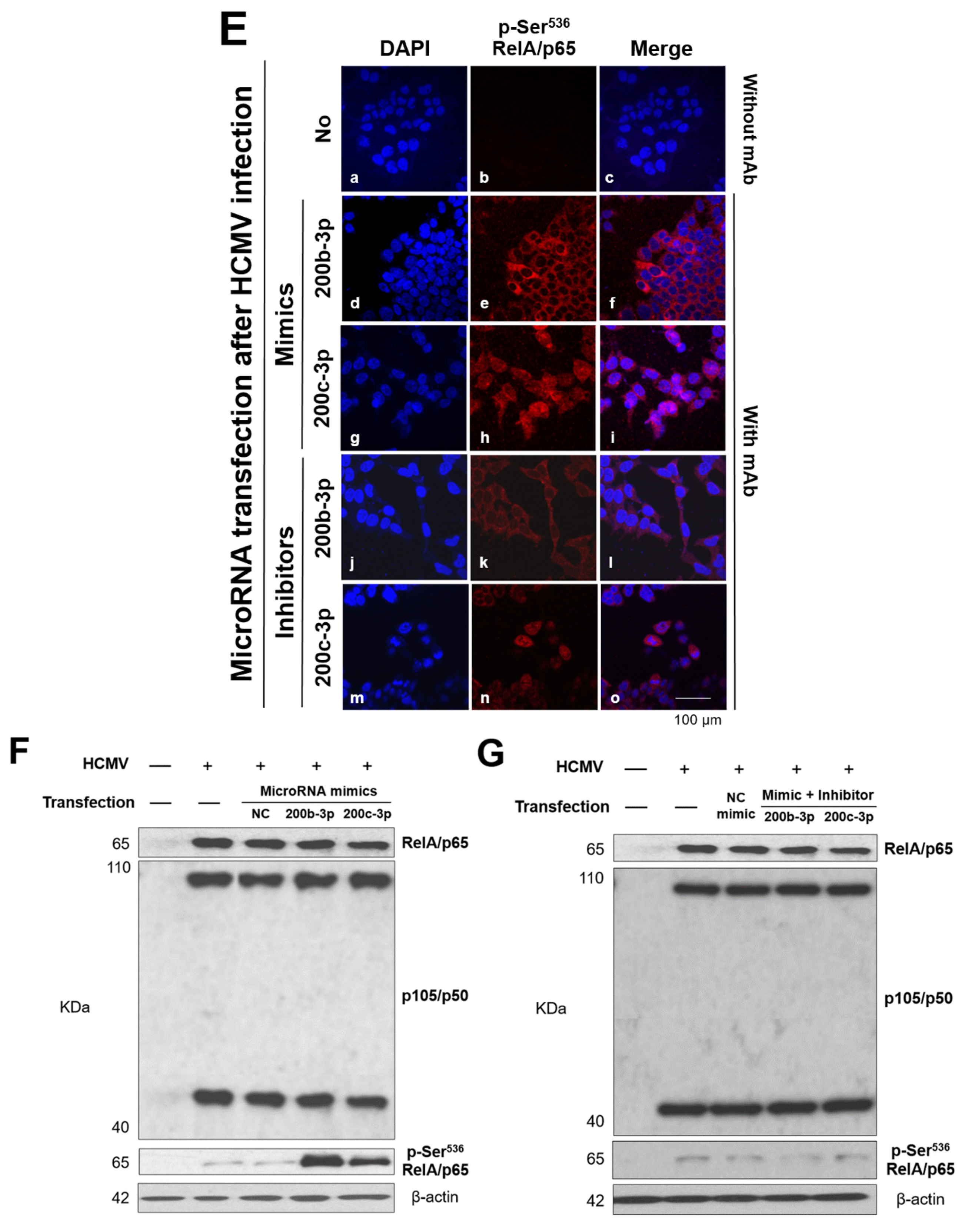
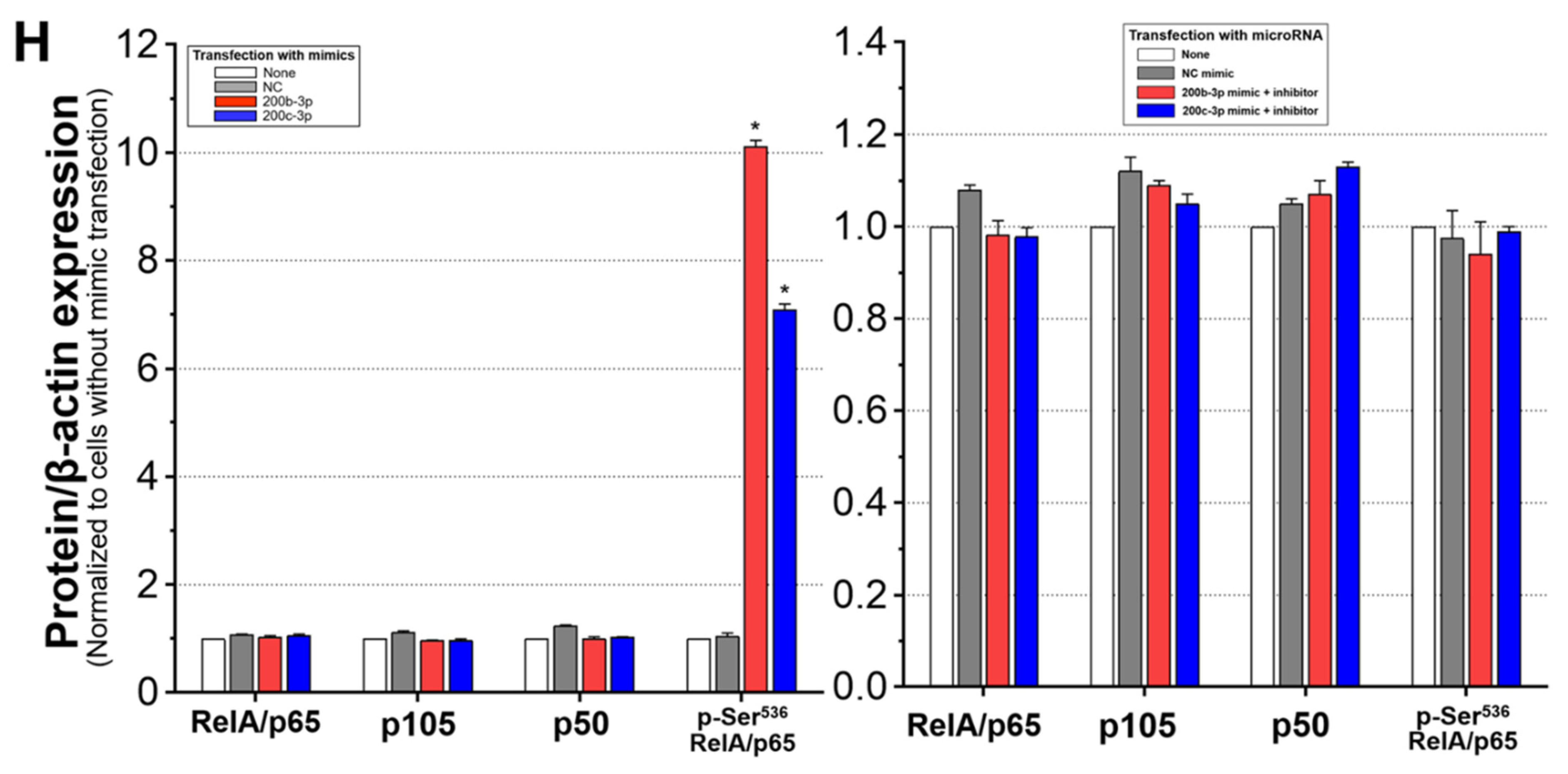
2.6. IE Proteins and MiR-200b-3p or miR-200c-3p Are Associated with Phosphorylation of RelA/p65 at Ser536
2.7. Phosphorylated RelA/p65 (Ser536) Binds to MIEP Region
3. Discussion
4. Materials and Methods
4.1. Cell Culture and HCMV Infection
4.2. Construction of IE Plasmids and Plasmids Containing the 3′-UTR of UL123
4.3. Generation of Cell Lines Stably Transfected with 3′-UTR-EGFP
4.4. Measurement of the Binding of miRNAs to the 3′-UTR Targets
4.5. Evaluation of MicroRNA Expression and HCMV Viral Loads
4.6. Immunoblotting
4.7. Flow Cytometry
4.8. Immunocytochemistry
4.9. Cross-Linking Chromatin Immunoprecipitation (ChiP)
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Klenerman, P.; Oxenius, A. T cell responses to cytomegalovirus. Nat. Rev. Immunol. 2016, 16, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.; Rao, S. Mechanisms Underlying T Cell Immunosenescence: Aging and Cytomegalovirus Infection. Front. Microbiol. 2016, 7, 2111. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Peng, G.; Bai, J.; He, B.; Huang, K.; Hu, X.; Liu, D. Cytomegalovirus Infection and Relative Risk of Cardiovascular Disease (Ischemic Heart Disease, Stroke, and Cardiovascular Death): A Meta-Analysis of Prospective Studies up to 2016. J. Am. Heart Assoc. 2017, 6, e005025. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.G.; Han, K.D.; Lee, K.H.; La, Y.; Kwon, D.E.; Han, S.H. Impact of Cytomegalovirus Disease on New-Onset Type 2 Diabetes Mellitus: Population-Based Matched Case-Control Cohort Study. Diabetes Metab. J. 2019, 43, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Dupont, L.; Reeves, M.B. Cytomegalovirus latency and reactivation: Recent insights into an age old problem. Rev. Med. Virol. 2016, 26, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, B.; Tang, X.; Liu, Y.L.; He, H.Y.; Li, X.Y.; Wang, R.; Guo, F.; Tong, Z.H. Application of metagenomic next-generation sequencing for bronchoalveolar lavage diagnostics in critically ill patients. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Han, Y.; Feng, J. Metagenomic next-generation sequencing for mixed pulmonary infection diagnosis. BMC Pulm. Med. 2019, 19, 252. [Google Scholar] [CrossRef]
- Kotton, C.N.; Kumar, D.; Caliendo, A.M.; Huprikar, S.; Chou, S.; Danziger-Isakov, L.; Humar, A. The Third International Consensus Guidelines on the Management of Cytomegalovirus in Solid-organ Transplantation. Transplantation 2018, 102, 900–931. [Google Scholar] [CrossRef]
- Ljungman, P.; de la Camara, R.; Robin, C.; Crocchiolo, R.; Einsele, H.; Hill, J.A.; Hubacek, P.; Navarro, D.; Cordonnier, C.; Ward, K.N. Guidelines for the management of cytomegalovirus infection in patients with haematological malignancies and after stem cell transplantation from the 2017 European Conference on Infections in Leukaemia (ECIL 7). Lancet Infect. Dis. 2019, 19, e260–e272. [Google Scholar] [CrossRef]
- Griffiths, P. The direct and indirect consequences of cytomegalovirus infection and potential benefits of vaccination. Antivir. Res. 2020, 176, 104732. [Google Scholar] [CrossRef]
- Nicholson, E.; Peggs, K.S. Cytomegalovirus-specific T-cell therapies: Current status and future prospects. Immunotherapy 2015, 7, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Wills, M.R.; Poole, E.; Lau, B.; Krishna, B.; Sinclair, J.H. The immunology of human cytomegalovirus latency: Could latent infection be cleared by novel immunotherapeutic strategies? Cell. Mol. Immunol. 2015, 12, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Adamson, C.S.; Nevels, M.M. Bright and Early: Inhibiting Human Cytomegalovirus by Targeting Major Immediate-Early Gene Expression or Protein Function. Viruses 2020, 12, 110. [Google Scholar] [CrossRef]
- Marchini, A.; Liu, H.; Zhu, H. Human cytomegalovirus with IE-2 (UL122) deleted fails to express early lytic genes. J. Virol. 2001, 75, 1870–1878. [Google Scholar] [CrossRef]
- Collins-McMillen, D.; Kamil, J.; Moorman, N.; Goodrum, F. Control of Immediate Early Gene Expression for Human Cytomegalovirus Reactivation. Front. Cell. Infect. Microbiol. 2020, 10, 476. [Google Scholar] [CrossRef]
- Stinski, M.F.; Petrik, D.T. Functional roles of the human cytomegalovirus essential IE86 protein. Curr. Top. Microbiol. Immunol. 2008, 325, 133–152. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Herbein, G. Epigenetic regulation of human cytomegalovirus latency: An update. Epigenomics 2014, 6, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Dooley, A.L.; O’Connor, C.M. Regulation of the MIE Locus During HCMV Latency and Reactivation. Pathogens 2020, 9, 869. [Google Scholar] [CrossRef] [PubMed]
- Tarrant-Elorza, M.; Rossetto, C.C.; Pari, G.S. Maintenance and replication of the human cytomegalovirus genome during latency. Cell Host Microbe 2014, 16, 43–54. [Google Scholar] [CrossRef]
- Caposio, P.; Luganini, A.; Hahn, G.; Landolfo, S.; Gribaudo, G. Activation of the virus-induced IKK/NF-kappaB signalling axis is critical for the replication of human cytomegalovirus in quiescent cells. Cell. Microbiol. 2007, 9, 2040–2054. [Google Scholar] [CrossRef]
- Collins-McMillen, D.; Buehler, J.; Peppenelli, M.; Goodrum, F. Molecular Determinants and the Regulation of Human Cytomegalovirus Latency and Reactivation. Viruses 2018, 10, 444. [Google Scholar] [CrossRef] [PubMed]
- Elder, E.; Sinclair, J. HCMV latency: What regulates the regulators? Med. Microbiol. Immunol. 2019, 208, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Paulus, C.; Nevels, M. The human cytomegalovirus major immediate-early proteins as antagonists of intrinsic and innate antiviral host responses. Viruses 2009, 1, 760–779. [Google Scholar] [CrossRef] [PubMed]
- Moller, R.; Schwarz, T.M.; Noriega, V.M.; Panis, M.; Sachs, D.; Tortorella, D.; tenOever, B.R. miRNA-mediated targeting of human cytomegalovirus reveals biological host and viral targets of IE2. Proc. Natl. Acad. Sci. USA 2018, 115, 1069–1074. [Google Scholar] [CrossRef]
- McCormick, D.; Lin, Y.T.; Grey, F. Identification of Host Factors Involved in Human Cytomegalovirus Replication, Assembly, and Egress Using a Two-Step Small Interfering RNA Screen. MBio 2018, 9, e00716-18. [Google Scholar] [CrossRef] [PubMed]
- Polachek, W.S.; Moshrif, H.F.; Franti, M.; Coen, D.M.; Sreenu, V.B.; Strang, B.L. High-Throughput Small Interfering RNA Screening Identifies Phosphatidylinositol 3-Kinase Class II Alpha as Important for Production of Human Cytomegalovirus Virions. J. Virol. 2016, 90, 8360–8371. [Google Scholar] [CrossRef]
- Kim, S.; Seo, D.; Kim, D.; Hong, Y.; Chang, H.; Baek, D.; Kim, V.N.; Lee, S.; Ahn, K. Temporal Landscape of MicroRNA-Mediated Host-Virus Crosstalk during Productive Human Cytomegalovirus Infection. Cell Host Microbe 2015, 17, 838–851. [Google Scholar] [CrossRef]
- Fu, M.; Gao, Y.; Zhou, Q.; Zhang, Q.; Peng, Y.; Tian, K.; Wang, J.; Zheng, X. Human cytomegalovirus latent infection alters the expression of cellular and viral microRNA. Gene 2014, 536, 272–278. [Google Scholar] [CrossRef]
- Tuddenham, L.; Pfeffer, S. Roles and regulation of microRNAs in cytomegalovirus infection. Biochim. Biophys. Acta 2011, 1809, 613–622. [Google Scholar] [CrossRef]
- Xiao, Y.; MacRae, I.J. Toward a Comprehensive View of MicroRNA Biology. Mol. Cell 2019, 75, 666–668. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef]
- Woolston, C. How RNA therapies could be used to tackle the world’s biggest killer. Nature 2019, 574, S13–S14. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, C.M.; Vanicek, J.; Murphy, E.A. Host microRNA regulation of human cytomegalovirus immediate early protein translation promotes viral latency. J. Virol. 2014, 88, 5524–5532. [Google Scholar] [CrossRef]
- Han, S.H.; Kumar, D.; Ferreira, V.H.; Egli, A.; Hirsch, H.H.; Weisser, M.; Garzoni, C.; van Delden, C.; Bochud, P.Y.; Manuel, O.; et al. Human MicroRNA Responses Predict Cytomegalovirus Replication Following Solid Organ Transplantation. J. Infect. Dis. 2017, 215, 537–546. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hancock, M.H.; Hook, L.M.; Mitchell, J.; Nelson, J.A. Human Cytomegalovirus MicroRNAs miR-US5-1 and miR-UL112-3p Block Proinflammatory Cytokine Production in Response to NF-kappaB-Activating Factors through Direct Downregulation of IKKalpha and IKKbeta. MBio 2017, 8, e00109-17. [Google Scholar] [CrossRef]
- Hook, L.; Hancock, M.; Landais, I.; Grabski, R.; Britt, W.; Nelson, J.A. Cytomegalovirus microRNAs. Curr. Opin. Virol. 2014, 7, 40–46. [Google Scholar] [CrossRef]
- Nachmani, D.; Lankry, D.; Wolf, D.G.; Mandelboim, O. The human cytomegalovirus microRNA miR-UL112 acts synergistically with a cellular microRNA to escape immune elimination. Nat. Immunol. 2010, 11, 806–813. [Google Scholar] [CrossRef]
- Wang, F.Z.; Weber, F.; Croce, C.; Liu, C.G.; Liao, X.; Pellett, P.E. Human cytomegalovirus infection alters the expression of cellular microRNA species that affect its replication. J. Virol. 2008, 82, 9065–9074. [Google Scholar] [CrossRef]
- Lee, K.H.; Lim, B.J.; Ferreira, V.H.; Min, S.Y.; Hong, Y.M.; Jo, J.H.; Han, S.H. Expression of human miR-200b-3p and -200c-3p in cytomegalovirus-infected tissues. Biosci. Rep. 2018, 38, 1–9. [Google Scholar] [CrossRef]
- Buzdin, A.A.; Artcibasova, A.V.; Fedorova, N.F.; Suntsova, M.V.; Garazha, A.V.; Sorokin, M.I.; Allina, D.; Shalatonin, M.; Borisov, N.M.; Zhavoronkov, A.A.; et al. Early stage of cytomegalovirus infection suppresses host microRNA expression regulation in human fibroblasts. Cell Cycle 2016, 15, 3378–3389. [Google Scholar] [CrossRef] [PubMed]
- Poole, E.; McGregor Dallas, S.R.; Colston, J.; Joseph, R.S.V.; Sinclair, J. Virally induced changes in cellular microRNAs maintain latency of human cytomegalovirus in CD34+ progenitors. J. Gen. Virol. 2011, 92, 1539–1549. [Google Scholar] [CrossRef]
- Stinski, M.F.; Isomura, H. Role of the cytomegalovirus major immediate early enhancer in acute infection and reactivation from latency. Med. Microbiol. Immunol. 2008, 197, 223–231. [Google Scholar] [CrossRef]
- Yurochko, A.D.; Kowalik, T.F.; Huong, S.M.; Huang, E.S. Human cytomegalovirus upregulates NF-kappa B activity by transactivating the NF-kappa B p105/p50 and p65 promoters. J. Virol. 1995, 69, 5391–5400. [Google Scholar] [CrossRef] [PubMed]
- Kowalik, T.F.; Wing, B.; Haskill, J.S.; Azizkhan, J.C.; Baldwin, A.S., Jr.; Huang, E.S. Multiple mechanisms are implicated in the regulation of NF-kappa B activity during human cytomegalovirus infection. Proc. Natl. Acad. Sci. USA 1993, 90, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- Sambucetti, L.C.; Cherrington, J.M.; Wilkinson, G.W.; Mocarski, E.S. NF-kappa B activation of the cytomegalovirus enhancer is mediated by a viral transactivator and by T cell stimulation. EMBO J. 1989, 8, 4251–4258. [Google Scholar] [CrossRef]
- Poole, E.; King, C.A.; Sinclair, J.H.; Alcami, A. The UL144 gene product of human cytomegalovirus activates NFkappaB via a TRAF6-dependent mechanism. EMBO J. 2006, 25, 4390–4399. [Google Scholar] [CrossRef]
- DeMeritt, I.B.; Podduturi, J.P.; Tilley, A.M.; Nogalski, M.T.; Yurochko, A.D. Prolonged activation of NF-kappaB by human cytomegalovirus promotes efficient viral replication and late gene expression. Virology 2006, 346, 15–31. [Google Scholar] [CrossRef]
- Goodwin, C.M.; Ciesla, J.H.; Munger, J. Who’s Driving? Human Cytomegalovirus, Interferon, and NFκB Signaling. Viruses 2018, 10, 447. [Google Scholar] [CrossRef]
- DeMeritt, I.B.; Milford, L.E.; Yurochko, A.D. Activation of the NF-kappaB pathway in human cytomegalovirus-infected cells is necessary for efficient transactivation of the major immediate-early promoter. J. Virol. 2004, 78, 4498–4507. [Google Scholar] [CrossRef]
- Prösch, S.; Wuttke, R.; Krüger, D.H.; Volk, H.D. NF-kappaB—A potential therapeutic target for inhibition of human cytomegalovirus (re)activation? Biol. Chem. 2002, 383, 1601–1609. [Google Scholar] [CrossRef] [PubMed]
- Haller, D.; Russo, M.P.; Sartor, R.B.; Jobin, C. IKK beta and phosphatidylinositol 3-kinase/Akt participate in non-pathogenic Gram-negative enteric bacteria-induced RelA phosphorylation and NF-kappa B activation in both primary and intestinal epithelial cell lines. J. Biol. Chem. 2002, 277, 38168–38178. [Google Scholar] [CrossRef] [PubMed]
- Yoboua, F.; Martel, A.; Duval, A.; Mukawera, E.; Grandvaux, N. Respiratory syncytial virus-mediated NF-kappa B p65 phosphorylation at serine 536 is dependent on RIG-I, TRAF6, and IKK beta. J. Virol. 2010, 84, 7267–7277. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Tang, E.; Guan, K.; Wang, C.Y. IKK beta plays an essential role in the phosphorylation of RelA/p65 on serine 536 induced by lipopolysaccharide. J. Immunol. 2003, 170, 5630–5635. [Google Scholar] [CrossRef]
- Giridharan, S.; Srinivasan, M. Mechanisms of NF-κB p65 and strategies for therapeutic manipulation. J. Inflamm. Res. 2018, 11, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Milanovic, M.; Kracht, M.; Schmitz, M.L. The cytokine-induced conformational switch of nuclear factor κB p65 is mediated by p65 phosphorylation. Biochem. J. 2014, 457, 401–413. [Google Scholar] [CrossRef]
- Song, Y.J.; Jen, K.Y.; Soni, V.; Kieff, E.; Cahir-McFarland, E. IL-1 receptor-associated kinase 1 is critical for latent membrane protein 1-induced p65/RelA serine 536 phosphorylation and NF-kappaB activation. Proc. Natl. Acad. Sci. USA 2006, 103, 2689–2694. [Google Scholar] [CrossRef]
- Sanders, R.L.; Del Rosario, C.J.; White, E.A.; Spector, D.H. Internal deletions of IE2 86 and loss of the late IE2 60 and IE2 40 proteins encoded by human cytomegalovirus affect the levels of UL84 protein but not the amount of UL84 mRNA or the loading and distribution of the mRNA on polysomes. J. Virol. 2008, 82, 11383–11397. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hou, W.; Torres, L.; Cruz-Cosme, R.; Arroyo, F.; Irizarry, L.; Luciano, D.; Marquez, A.; Rivera, L.L.; Sala, A.L.; Luo, M.H.; et al. Two Polypyrimidine Tracts in Intron 4 of the Major Immediate Early Gene Are Critical for Gene Expression Switching from IE1 to IE2 and for Replication of Human Cytomegalovirus. J. Virol. 2016, 90, 7339–7349. [Google Scholar] [CrossRef]
- Lin, Y.T.; Prendergast, J.; Grey, F. The host ubiquitin-dependent segregase VCP/p97 is required for the onset of human cytomegalovirus replication. PLoS Pathog. 2017, 13, e1006329. [Google Scholar] [CrossRef]
- Stenberg, R.M.; Thomsen, D.R.; Stinski, M.F. Structural analysis of the major immediate early gene of human cytomegalovirus. J. Virol. 1984, 49, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Stenberg, R.M.; Witte, P.R.; Stinski, M.F. Multiple spliced and unspliced transcripts from human cytomegalovirus immediate-early region 2 and evidence for a common initiation site within immediate-early region 1. J. Virol. 1985, 56, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Gawn, J.M.; Greaves, R.F. Absence of IE1 p72 protein function during low-multiplicity infection by human cytomegalovirus results in a broad block to viral delayed-early gene expression. J. Virol. 2002, 76, 4441–4455. [Google Scholar] [CrossRef] [PubMed]
- Mocarski, E.S.; Kemble, G.W.; Lyle, J.M.; Greaves, R.F. A deletion mutant in the human cytomegalovirus gene encoding IE1(491aa) is replication defective due to a failure in autoregulation. Proc. Natl. Acad. Sci. USA 1996, 93, 11321–11326. [Google Scholar] [CrossRef] [PubMed]
- Arend, K.C.; Ziehr, B.; Vincent, H.A.; Moorman, N.J. Multiple Transcripts Encode Full-Length Human Cytomegalovirus IE1 and IE2 Proteins during Lytic Infection. J. Virol. 2016, 90, 8855–8865. [Google Scholar] [CrossRef] [PubMed]
- Arend, K.C.; Lenarcic, E.M.; Moorman, N.J. The 5′ Untranslated Region of the Major Immediate Early mRNA Is Necessary for Efficient Human Cytomegalovirus Replication. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Eickhoff, J.E.; Cotten, M. NF-kappaB activation can mediate inhibition of human cytomegalovirus replication. J. Gen. Virol. 2005, 86, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Christian, F.; Smith, E.L.; Carmody, R.J. The Regulation of NF-κB Subunits by Phosphorylation. Cells 2016, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Poole, E.; Atkins, E.; Nakayama, T.; Yoshie, O.; Groves, I.; Alcami, A.; Sinclair, J. NF-kappaB-mediated activation of the chemokine CCL22 by the product of the human cytomegalovirus gene UL144 escapes regulation by viral IE86. J. Virol. 2008, 82, 4250–4256. [Google Scholar] [CrossRef]
- Luganini, A.; Caposio, P.; Mondini, M.; Landolfo, S.; Gribaudo, G. New cell-based indicator assays for the detection of human cytomegalovirus infection and screening of inhibitors of viral immediate-early 2 protein activity. J. Appl. Microbiol. 2008, 105, 1791–1801. [Google Scholar] [CrossRef]
- Mishima, T.; Sadovsky, E.; Gegick, M.E.; Sadovsky, Y. Determinants of effective lentivirus-driven microRNA expression in vivo. Sci. Rep. 2016, 6, 33345. [Google Scholar] [CrossRef] [PubMed]
- Tatenhorst, L.; Rescher, U.; Gerke, V.; Paulus, W. Knockdown of annexin 2 decreases migration of human glioma cells in vitro. Neuropathol. Appl. Neurobiol. 2006, 32, 271–277. [Google Scholar] [CrossRef]
- Wan, J.; Guo, A.A.; Chowdhury, I.; Guo, S.; Hibbert, J.; Wang, G.; Liu, M. TRPM7 Induces Mechanistic Target of Rap1b Through the Downregulation of miR-28-5p in Glioma Proliferation and Invasion. Front. Oncol. 2019, 9, 1413. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Qin, T.; Li, J.; Wang, L.; Zhang, Q.; Jiang, Z.; Mao, J. MicroRNA-140-5p inhibits invasion and angiogenesis through targeting VEGF-A in breast cancer. Cancer Gene Ther. 2017, 24, 386–392. [Google Scholar] [CrossRef]
- Hart, M.; Kern, F.; Backes, C.; Rheinheimer, S.; Fehlmann, T.; Keller, A.; Meese, E. The deterministic role of 5-mers in microRNA-gene targeting. RNA Biol. 2018, 15, 1–7. [Google Scholar] [CrossRef] [PubMed]
- McGeary, S.E.; Lin, K.S.; Shi, C.Y.; Pham, T.M.; Bisaria, N.; Kelley, G.M.; Bartel, D.P. The biochemical basis of microRNA targeting efficacy. Science 2019, 366. [Google Scholar] [CrossRef]
- Moore, M.J.; Scheel, T.K.; Luna, J.M.; Park, C.Y.; Fak, J.J.; Nishiuchi, E.; Rice, C.M.; Darnell, R.B. miRNA-target chimeras reveal miRNA 3’-end pairing as a major determinant of Argonaute target specificity. Nat. Commun. 2015, 6, 8864. [Google Scholar] [CrossRef]
- Bjerke, G.A.; Yi, R. Integrated analysis of directly captured microRNA targets reveals the impact of microRNAs on mammalian transcriptome. RNA 2020, 26, 306–323. [Google Scholar] [CrossRef]
- Ryan, B.C.; Werner, T.S.; Howard, P.L.; Chow, R.L. ImiRP: A computational approach to microRNA target site mutation. BMC Bioinform. 2016, 17, 190. [Google Scholar] [CrossRef] [PubMed]
- Brennecke, J.; Stark, A.; Russell, R.B.; Cohen, S.M. Principles of microRNA-target recognition. PLoS Biol. 2005, 3, e85. [Google Scholar] [CrossRef] [PubMed]
- Compton, T. An immortalized human fibroblast cell line is permissive for human cytomegalovirus infection. J. Virol. 1993, 67, 3644–3648. [Google Scholar] [CrossRef] [PubMed]
- Zavala, A.G.; O’Dowd, J.M.; Fortunato, E.A. Infection of a Single Cell Line with Distinct Strains of Human Cytomegalovirus Can Result in Large Variations in Virion Production and Facilitate Efficient Screening of Virus Protein Function. J. Virol. 2015, 90, 2523–2535. [Google Scholar] [CrossRef]
- Bradley, A.J.; Lurain, N.S.; Ghazal, P.; Trivedi, U.; Cunningham, C.; Baluchova, K.; Gatherer, D.; Wilkinson, G.W.G.; Dargan, D.J.; Davison, A.J. High-throughput sequence analysis of variants of human cytomegalovirus strains Towne and AD169. J. Gen. Virol. 2009, 90, 2375–2380. [Google Scholar] [CrossRef] [PubMed]
- Michelson, S.; Alcami, J.; Kim, S.J.; Danielpour, D.; Bachelerie, F.; Picard, L.; Bessia, C.; Paya, C.; Virelizier, J.L. Human cytomegalovirus infection induces transcription and secretion of transforming growth factor beta 1. J. Virol. 1994, 68, 5730–5737. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Ball, C.B.; Collins, G.; Hu, Q.; Luse, D.S.; Price, D.H.; Meier, J.L. Human cytomegalovirus IE2 drives transcription initiation from a select subset of late infection viral promoters by host RNA polymerase II. PLoS Pathog. 2020, 16, e1008402. [Google Scholar] [CrossRef]
- Du, G.; Stinski, M.F. Interaction network of proteins associated with human cytomegalovirus IE2-p86 protein during infection: A proteomic analysis. PLoS ONE 2013, 8, e81583. [Google Scholar] [CrossRef]
- Hwang, E.S.; Zhang, Z.; Cai, H.; Huang, D.Y.; Huong, S.M.; Cha, C.Y.; Huang, E.S. Human cytomegalovirus IE1-72 protein interacts with p53 and inhibits p53-dependent transactivation by a mechanism different from that of IE2-86 protein. J. Virol. 2009, 83, 12388–12398. [Google Scholar] [CrossRef] [PubMed]
- Bodaghi, B.; Slobbe-van Drunen, M.E.; Topilko, A.; Perret, E.; Vossen, R.C.; van Dam-Mieras, M.C.; Zipeto, D.; Virelizier, J.L.; LeHoang, P.; Bruggeman, C.A.; et al. Entry of human cytomegalovirus into retinal pigment epithelial and endothelial cells by endocytosis. Investig. Ophthalmol. Vis. Sci. 1999, 40, 2598–2607. [Google Scholar]
- Zhang, Q.; Lai, M.M.; Lou, Y.Y.; Guo, B.H.; Wang, H.Y.; Zheng, X.Q. Transcriptome altered by latent human cytomegalovirus infection on THP-1 cells using RNA-seq. Gene 2016, 594, 144–150. [Google Scholar] [CrossRef]
- Zheng, X.; Gao, Y.; Zhang, Q.; Liu, Y.; Peng, Y.; Fu, M.; Ji, Y. Identification of transcription factor AML-1 binding site upstream of human cytomegalovirus UL111A gene. PLoS ONE 2015, 10, e0117773. [Google Scholar] [CrossRef]
- Reeves, M.B.; MacAry, P.A.; Lehner, P.J.; Sissons, J.G.; Sinclair, J.H. Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc. Natl. Acad. Sci. USA 2005, 102, 4140–4145. [Google Scholar] [CrossRef] [PubMed]
- Mercorelli, B.; Lembo, D.; Palù, G.; Loregian, A. Early inhibitors of human cytomegalovirus: State-of-art and therapeutic perspectives. Pharmacol. Ther. 2011, 131, 309–329. [Google Scholar] [CrossRef] [PubMed]
- Wiebusch, L.; Truss, M.; Hagemeier, C. Inhibition of human cytomegalovirus replication by small interfering RNAs. J. Gen. Virol. 2004, 85, 179–184. [Google Scholar] [CrossRef]
- Nazarov, P.V.; Kreis, S. Integrative approaches for analysis of mRNA and microRNA high-throughput data. Comput. Struct. Biotechnol. J. 2021, 19, 1154–1162. [Google Scholar] [CrossRef]
- Torres, L.; Tang, Q. Immediate-Early (IE) gene regulation of cytomegalovirus: IE1- and pp71-mediated viral strategies against cellular defenses. Virol. Sin. 2014, 29, 343–352. [Google Scholar] [CrossRef]
- Jamaluddin, M.; Tian, B.; Boldogh, I.; Garofalo, R.P.; Brasier, A.R. Respiratory syncytial virus infection induces a reactive oxygen species-MSK1-phospho-Ser-276 RelA pathway required for cytokine expression. J. Virol. 2009, 83, 10605–10615. [Google Scholar] [CrossRef] [PubMed]
- Caposio, P.; Musso, T.; Luganini, A.; Inoue, H.; Gariglio, M.; Landolfo, S.; Gribaudo, G. Targeting the NF-kappaB pathway through pharmacological inhibition of IKK2 prevents human cytomegalovirus replication and virus-induced inflammatory response in infected endothelial cells. Antivir. Res. 2007, 73, 175–184. [Google Scholar] [CrossRef]
- Hancock, M.H.; Nelson, J.A. Modulation of the NFκb Signalling Pathway by Human Cytomegalovirus. Virol. (Hyderabad) 2017, 1, 104. [Google Scholar]
- Siggers, T.; Chang, A.B.; Teixeira, A.; Wong, D.; Williams, K.J.; Ahmed, B.; Ragoussis, J.; Udalova, I.A.; Smale, S.T.; Bulyk, M.L. Principles of dimer-specific gene regulation revealed by a comprehensive characterization of NF-κB family DNA binding. Nat. Immunol. 2011, 13, 95–102. [Google Scholar] [CrossRef]
- Hoffmann, A.; Natoli, G.; Ghosh, G. Transcriptional regulation via the NF-kappaB signaling module. Oncogene 2006, 25, 6706–6716. [Google Scholar] [CrossRef]
- Mattioli, I.; Geng, H.; Sebald, A.; Hodel, M.; Bucher, C.; Kracht, M.; Schmitz, M.L. Inducible phosphorylation of NF-kappa B p65 at serine 468 by T cell costimulation is mediated by IKK epsilon. J. Biol. Chem. 2006, 281, 6175–6183. [Google Scholar] [CrossRef] [PubMed]
- Buss, H.; Dörrie, A.; Schmitz, M.L.; Hoffmann, E.; Resch, K.; Kracht, M. Constitutive and interleukin-1-inducible phosphorylation of p65 NF-{kappa}B at serine 536 is mediated by multiple protein kinases including I{kappa}B kinase (IKK)-{alpha}, IKK{beta}, IKK{epsilon}, TRAF family member-associated (TANK)-binding kinase 1 (TBK1), and an unknown kinase and couples p65 to TATA-binding protein-associated factor II31-mediated interleukin-8 transcription. J. Biol. Chem. 2004, 279, 55633–55643. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T.; Bebien, M.; Liu, G.Y.; Nizet, V.; Karin, M. IKKalpha limits macrophage NF-kappaB activation and contributes to the resolution of inflammation. Nature 2005, 434, 1138–1143. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, H.; Chiba, H.; Miyoshi, H.; Sugita, T.; Toriumi, W. IkappaB kinases phosphorylate NF-kappaB p65 subunit on serine 536 in the transactivation domain. J. Biol. Chem. 1999, 274, 30353–30356. [Google Scholar] [CrossRef]
- Pradere, J.P.; Hernandez, C.; Koppe, C.; Friedman, R.A.; Luedde, T.; Schwabe, R.F. Negative regulation of NF-kappaB p65 activity by serine 536 phosphorylation. Sci. Signal. 2016, 9, ra85. [Google Scholar] [CrossRef]
- Mattioli, I.; Sebald, A.; Bucher, C.; Charles, R.P.; Nakano, H.; Doi, T.; Kracht, M.; Schmitz, M.L. Transient and selective NF-kappa B p65 serine 536 phosphorylation induced by T cell costimulation is mediated by I kappa B kinase beta and controls the kinetics of p65 nuclear import. J. Immunol. 2004, 172, 6336–6344. [Google Scholar] [CrossRef]
- Arcangeletti, M.C.; Vasile Simone, R.; Rodighiero, I.; De Conto, F.; Medici, M.C.; Maccari, C.; Chezzi, C.; Calderaro, A. Human cytomegalovirus reactivation from latency: Validation of a “switch” model in vitro. Virol. J. 2016, 13, 1–15. [Google Scholar] [CrossRef]
- Gan, X.; Wang, H.; Yu, Y.; Yi, W.; Zhu, S.; Li, E.; Liang, Y. Epigenetically repressing human cytomegalovirus lytic infection and reactivation from latency in THP-1 model by targeting H3K9 and H3K27 histone demethylases. PLoS ONE 2017, 12, e0175390. [Google Scholar] [CrossRef]
- Poma, E.E.; Kowalik, T.F.; Zhu, L.; Sinclair, J.H.; Huang, E.S. The human cytomegalovirus IE1-72 protein interacts with the cellular p107 protein and relieves p107-mediated transcriptional repression of an E2F-responsive promoter. J. Virol. 1996, 70, 7867–7877. [Google Scholar] [CrossRef] [PubMed]
- Furnari, B.A.; Poma, E.; Kowalik, T.F.; Huong, S.M.; Huang, E.S. Human cytomegalovirus immediate-early gene 2 protein interacts with itself and with several novel cellular proteins. J. Virol. 1993, 67, 4981–4991. [Google Scholar] [CrossRef]
- He, Y.S.; Xu, L.; Huang, E.S. Characterization of human cytomegalovirus UL84 early gene and identification of its putative protein product. J. Virol. 1992, 66, 1098–1108. [Google Scholar] [CrossRef] [PubMed]
- Hayden, R.T.; Preiksaitis, J.; Tong, Y.; Pang, X.; Sun, Y.; Tang, L.; Cook, L.; Pounds, S.; Fryer, J.; Caliendo, A.M. Commutability of the First World Health Organization International Standard for Human Cytomegalovirus. J. Clin. Microbiol. 2015, 53, 3325–3333. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PCR Primers | Sequence (5′→3′) | ||
|---|---|---|---|
| HCMV Genes | Forward | Reverse | |
| UL122 | CAACATGATCATCCACGCTG | GAGACTTGTTCCTCAGGTCC | |
| UL123 | AAGCGGCCTCTGATAACCAAGCC | AGCACCATCCTCCTCTTCCTCTGG | |
| UL83 | TGCCCTGGATGCGATACTG | AGGACCTGACGATGACCG | |
| MIEP | Upstream | GGTCAAAACAGCGTGGATGG | CGTGTACGGTGGGAGGTCTA |
| Downstream | CCGTAAGTTATGTAACGCGGA | ACCGCCATGTTGACATTGATT | |
| Synthetic oligonucleotides a | Sequence (5′→3′) | ||
| Recombinant plasmids | |||
| Wild-type UL123-3′UTR-pEGFP | GAATTCACTATTGTATATATATATCAGTTACTGTTATGGATCCCACGTCACTATTGTATACTCTATATTATACTCTATGTTATACTCTGTAATCCTACTCAATAAACTCGAG | ||
| Mutant UL123-3′UTR-pEGFP b | GAATTCACTATTGTATATATATATCAGTTACTGTTATGGATCCCACGTCACTATTGTATACTCTACGCGCTACTCTATGTTATACTCTGTAATCCTACTCAATAAACTCGAG | ||
| UL122-123- 3′UTR-pEGFP | CTCGAGTTTATTGAGTAGGATTACAGAGTATAACATAGAGTATAATATAGAGTATACAATAGTGACGTGGGATCCATAACAGTAACTGATATATATATACAATAGTGAATTCAGCCAGCAAGACAGCGATCTCGAGGTGAAAAACTGGAAAGAGAGACATGGACTCTTGTACATAGTGATTCCCCGTGACAGTATTAACGTGTGGTGAGAATGCTGTTTAATAAAAGAATTC | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, Y.-M.; Min, S.Y.; Kim, D.; Kim, S.; Seo, D.; Lee, K.H.; Han, S.H. Human MicroRNAs Attenuate the Expression of Immediate Early Proteins and HCMV Replication during Lytic and Latent Infection in Connection with Enhancement of Phosphorylated RelA/p65 (Serine 536) That Binds to MIEP. Int. J. Mol. Sci. 2022, 23, 2769. https://doi.org/10.3390/ijms23052769
Hong Y-M, Min SY, Kim D, Kim S, Seo D, Lee KH, Han SH. Human MicroRNAs Attenuate the Expression of Immediate Early Proteins and HCMV Replication during Lytic and Latent Infection in Connection with Enhancement of Phosphorylated RelA/p65 (Serine 536) That Binds to MIEP. International Journal of Molecular Sciences. 2022; 23(5):2769. https://doi.org/10.3390/ijms23052769
Chicago/Turabian StyleHong, Yeon-Mi, Seo Yeon Min, Dayeong Kim, Subin Kim, Daekwan Seo, Kyoung Hwa Lee, and Sang Hoon Han. 2022. "Human MicroRNAs Attenuate the Expression of Immediate Early Proteins and HCMV Replication during Lytic and Latent Infection in Connection with Enhancement of Phosphorylated RelA/p65 (Serine 536) That Binds to MIEP" International Journal of Molecular Sciences 23, no. 5: 2769. https://doi.org/10.3390/ijms23052769
APA StyleHong, Y.-M., Min, S. Y., Kim, D., Kim, S., Seo, D., Lee, K. H., & Han, S. H. (2022). Human MicroRNAs Attenuate the Expression of Immediate Early Proteins and HCMV Replication during Lytic and Latent Infection in Connection with Enhancement of Phosphorylated RelA/p65 (Serine 536) That Binds to MIEP. International Journal of Molecular Sciences, 23(5), 2769. https://doi.org/10.3390/ijms23052769