
The Association between Hypoxia-Induced Low Activity and Apoptosis Strongly Resembles That between TTX-Induced Silencing and Apoptosis

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
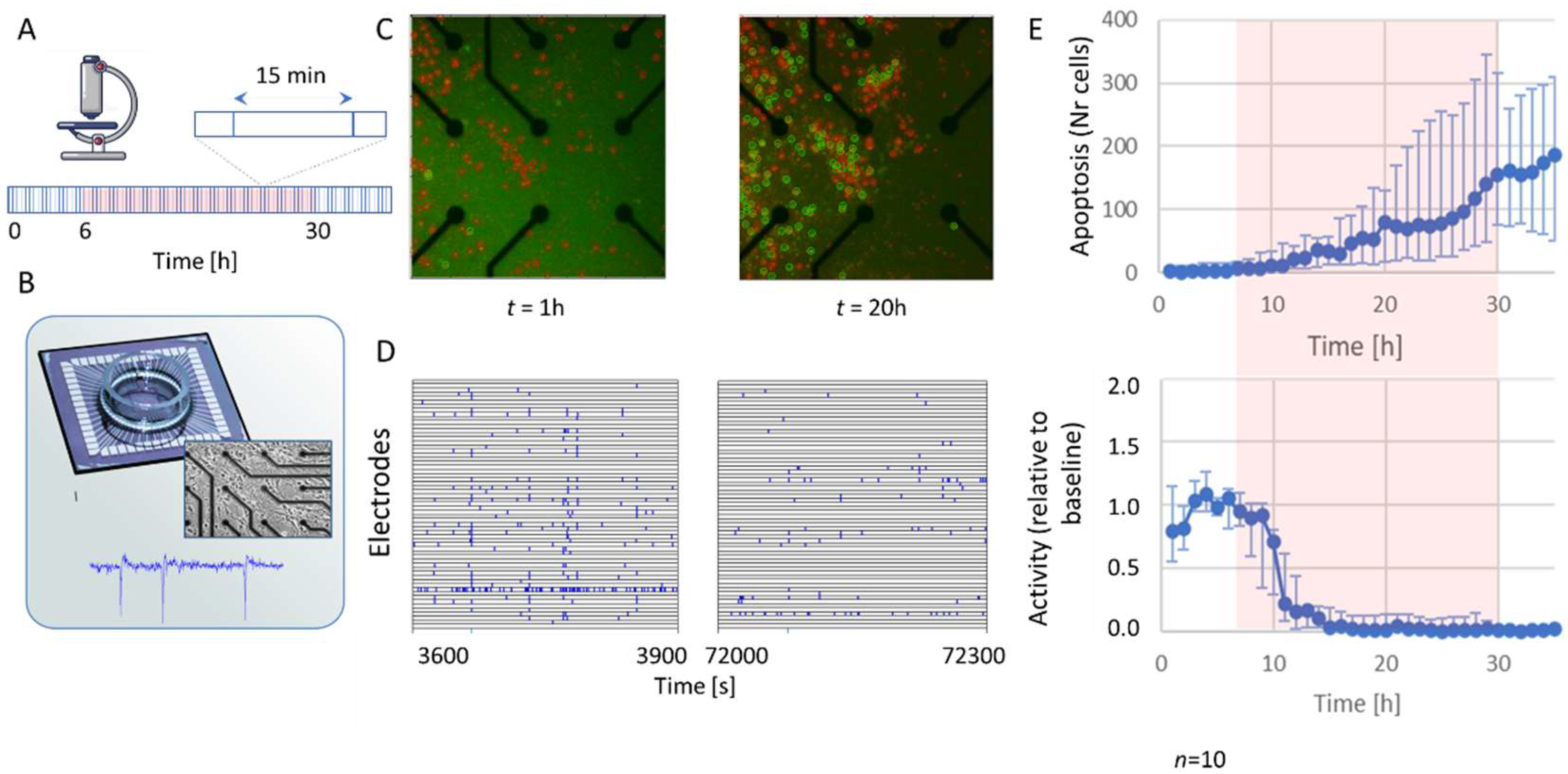
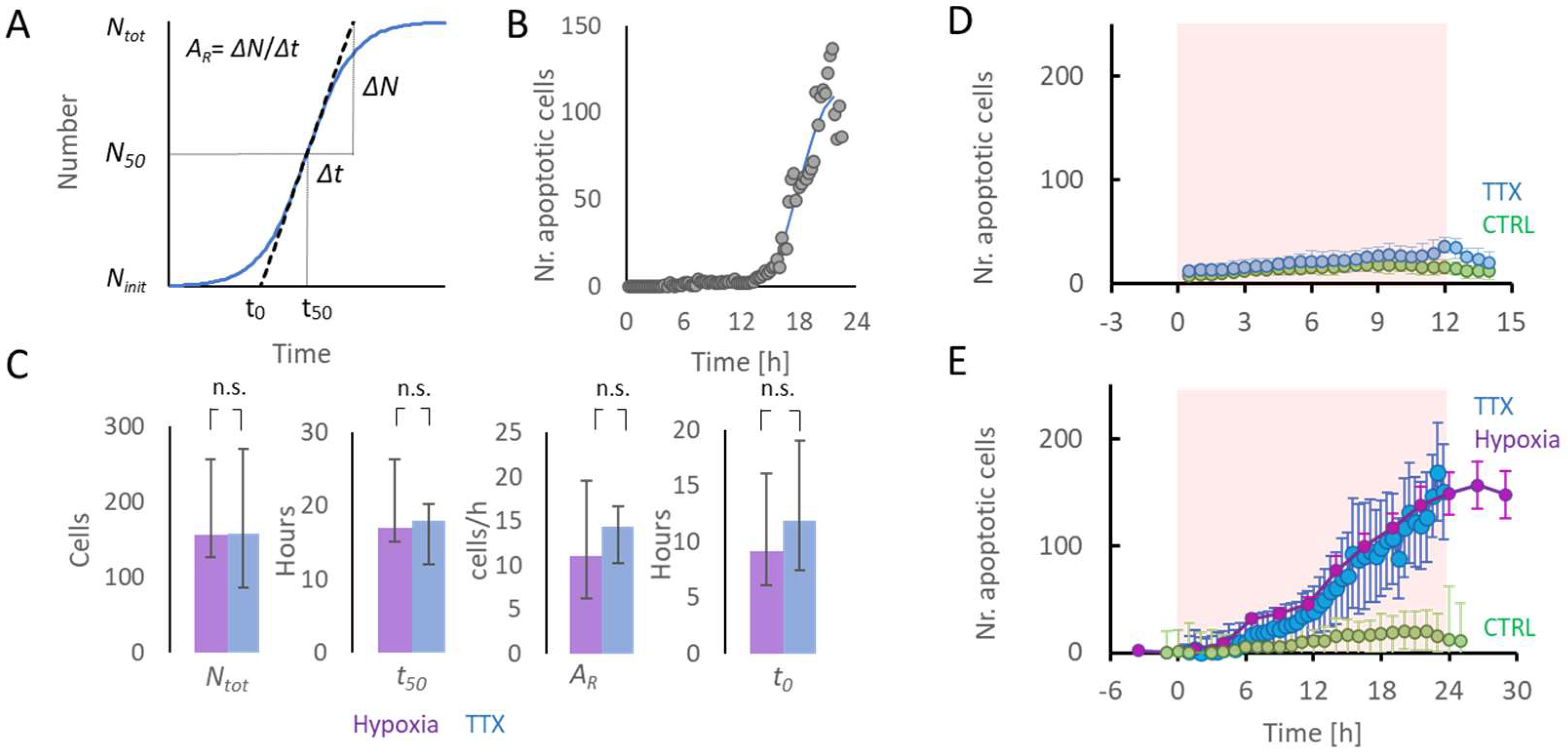
2.1. Activity and Apoptosis during Hypoxia
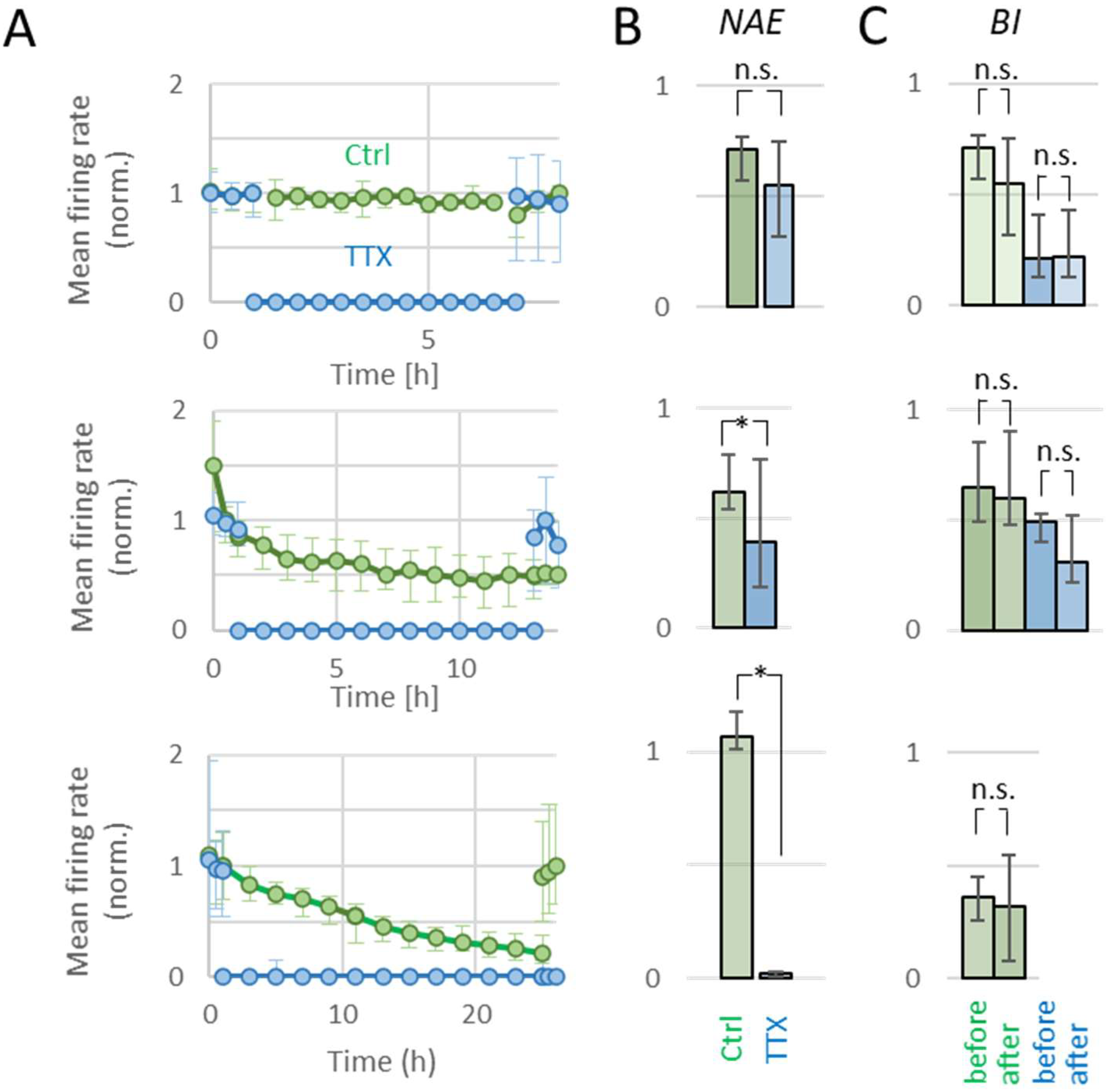
2.2. Activity and Apoptosis during TTX
2.2.1. Three and Six Hours of TTX
2.2.2. Twelve Hours of TTX
2.2.3. Twenty-Four Hours of TTX
2.3. Initiation of Apoptosis Associated with TTX- or Hypoxia-Induced Low Activity
2.4. Relation between Remaining Activity during Hypoxia and Apoptosis
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Maintenance of Normoxia and Induction of Hypoxia
4.3. Data Acquisition
4.3.1. Artifact Detection
4.3.2. Exclusion Criteria
4.4. Readouts
4.4.1. Array-Wide Fire Rate (AWFR)
4.4.2. Burstiness Index (BI)
4.4.3. Number of Active Electrodes (NAE)
4.4.4. Apoptosis Imaging
4.5. Pharmacologically Induced Inactivity
4.6. Onset of Apoptosis
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Li, L.; Scott, C.A.; Rothwell, P.M. Trends in Stroke Incidence in High-Income Countries in the 21st Century: Population-Based Study and Systematic Review. Stroke 2020, 51, 1372–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bose, A.J.N.R.; Henkes, H.; Alfke, K.; Reith, W.; Mayer, T.E.; Berlis, A.; Branca, V.; Sit, S.P. The Penumbra System: A mechanical device for the treatment of acute stroke due to thromboembolism. AJNR Am. J. Neuroradiol. 2008, 29, 1409–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillemin, K.; Krasnow, M.A. The hypoxic response: Huffing and HIFing. Cell 1997, 89, 9–12. [Google Scholar] [CrossRef] [Green Version]
- Krijnen, P.A.J.; Nijmeijer, R.; Meijer, C.J.L.M.; Visser, C.A.; Hack, C.E.; Niessen, H.W.M. Apoptosis in myocardial ischaemia and infarction. J. Clin. Pathol. 2002, 55, 801–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozeman, A.D.; Wermer, M.J.; Vos, J.A.; à Nijeholt, G.J.L.; Beumer, D.; Berkhemer, O.A.; Dippel, D.W.; Algra, A.; Boiten, J.; Schonewille, W.J.; et al. Evolution of Intra-arterial Therapy for Acute Ischemic Stroke in The Netherlands: MR CLEAN Pretrial Experience. J. Stroke Cerebrovasc. Dis. 2016, 25, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, S.; Stanwell, P.; Cordato, D.; Attia, J.; Levi, C. Reperfusion therapy in acute ischemic stroke: Dawn of a new era? BMC Neurol. 2018, 18, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima, F.O.; Furie, K.L.; Silva, G.S.; Lev, M.H.; Camargo, É.C.; Singhal, A.B.; Harris, G.J.; Halpern, E.F.; Koroshetz, W.J.; Smith, W.S.; et al. Prognosis of untreated strokes due to anterior circulation proximal intracranial arterial occlusions detected by use of computed tomography angiography. JAMA Neurol. 2014, 71, 151–157. [Google Scholar] [CrossRef]
- Radak, D.; Katsiki, N.; Resanovic, I.; Jovanovic, A.; Sudar-Milovanovic, E.; Zafirovic, S.; AMousad, S.; RIsenovic, E. Apoptosis and Acute Brain Ischemia in Ischemic Stroke. Curr. Vasc. Pharmacol. 2017, 15, 115–122. [Google Scholar] [CrossRef]
- Onténiente, B.; Rasika, S.; Benchoua, A.; Guégan, C. Molecular pathways in cerebral ischemia: Cues to novel therapeutic strategies. Mol. Neurobiol. 2003, 27, 33–72. [Google Scholar] [CrossRef]
- Linnik, M.D.; Zobrist, R.H.; Hatfield, M.D. Evidence supporting a role for programmed cell death in focal cerebral ischemia in rats. Stroke 1993, 24, 2002–2008. [Google Scholar] [CrossRef] [Green Version]
- Ferrer, I.; Planas, A.M. Signaling of cell death and cell survival following focal cerebral ischemia: Life and death struggle in the penumbra. J. Neuropathol. Exp. Neurol. 2003, 62, 329–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, B.A.; Bresnahan, J.C.; Beattie, M.S. Apoptosis in Nervous System Injury. In Encyclopedia of Neuroscience; Squire, L.R., Ed.; Academic Press: Oxford, UK, 2009; pp. 523–529. [Google Scholar]
- Hofmeijer, J.; Mulder, A.T.; Farinha, A.C.; van Putten, M.J.; le Feber, J. Mild hypoxia affects synaptic connectivity incultured neuronal networks. Brain Res. 2014, 1557, 180–189. [Google Scholar] [CrossRef] [PubMed]
- le Feber, J.; Tzafi Pavlidou, S.; Erkamp, N.; van Putten, M.J.; Hofmeijer, J. Progression of Neuronal Damage in an In Vitro Model of the Ischemic Penumbra. PLoS ONE 2016, 11, e0147231. [Google Scholar] [CrossRef] [PubMed]
- Gottron, M.A.; Lo, D.C. The Na+/K+-ATPase as a Drug Target for Ischemic Stroke. In New Strategies in Stroke Intervention: Ionic Transporters, Pumps, and New Channels; Annunziato, L., Ed.; Humana Press: Totowa, NJ, USA, 2010; pp. 129–151. [Google Scholar]
- le Feber, J.; Erkamp, N.; Van Putten, M.J.; Hofmeijer, J. Loss and recovery of functional connectivity in cultured cortical networks exposed to hypoxia. J. Neurophysiol. 2017, 118, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I. Apoptosis: Future targets for neuroprotective strategies. Cereb. Dis. 2006, 21, 9–20. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Heck, N.; Golbs, A.; Riedemann, T.; Sun, J.J.; Lessmann, V.; Luhmann, H.J. Activity-dependent regulation of neuronal apoptosis in neonatal mouse cerebral cortex. Cereb. Cortex 2008, 18, 1335–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dribben, W.H.; Eisenman, L.N.; Mennerick, S. Magnesium induces neuronal apoptosis by suppressing excitability. Cell Death Dis. 2010, 1, e63. [Google Scholar] [CrossRef] [Green Version]
- Hobbiss, A.F.; Ramiro-Cortés, Y.; Israely, I. Homeostatic Plasticity Scales Dendritic Spine Volumes and Changes the Threshold and Specificity of Hebbian Plasticity. iScience 2018, 8, 161–174. [Google Scholar] [CrossRef] [Green Version]
- Wrosch, J.K.; Einem, V.V.; Breininger, K.; Dahlmanns, M.; Maier, A.; Kornhuber, J.; Groemer, T.W. Rewiring of neuronal networks during synaptic silencing. Sci. Rep. 2017, 7, 11724. [Google Scholar] [CrossRef]
- Golbs, A.; Nimmervoll, B.; Sun, J.J.; Sava, I.E.; Luhmann, H.J. Control of programmed cell death by distinct electrical activity patterns. Cereb. Cortex 2011, 21, 1192–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoyanova, I.I.; Hofmeijer, J.; van Putten, M.J.; le Feber, J. Acyl Ghrelin Improves Synapse Recovery in an In Vitro Model of Postanoxic Encephalopathy. Mol. Neurobiol. 2016, 53, 6136–6143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muzzi, L.; Hassink, G.; Levers, M.; Jansman, M.; Frega, M.; Hofmeijer, J.; van Putten, M.; le Feber, J. Mild stimulation improves neuronal survival in an in-vitro model of the ischemic penumbra. J. Neural. Eng. 2019, 7, 016001. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Carnahan, J.; Greenberg, M. Requirement for BDNF in activity-dependent survival of cortical neurons. Science 1994, 263, 1618–1623. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.H.; Lee, C.M.; Xie, W.; Cui, B.; Poo, M.M. Activity-dependent BDNF release via endocytic pathways is regulated by synaptotagmin-6 and complexin. Proc. Natl. Acad. Sci. USA 2015, 112, E4475–E4484. [Google Scholar] [CrossRef] [Green Version]
- Lein, P.J.; Supasai, S.; Guignet, M. Chapter 9—Apoptosis as a Mechanism of Developmental Neurotoxicity. In Handbook of Developmental Neurotoxicology, 2nd ed.; Slikker, W., Paule, M.G., Wang, C., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 91–112. [Google Scholar]
- Bredesen, D.E.; Mehlen, P.; Rabizadeh, S. Receptors that mediate cellular dependence. Cell Death Differ. 2005, 12, 1031–1043. [Google Scholar] [CrossRef] [Green Version]
- Blanquie, O.; Yang, J.W.; Kilb, W.; Sharopov, S.; Sinning, A.; Luhmann, H.J. Electrical activity controls area-specific expression of neuronal apoptosis in the mouse developing cerebral cortex. eLife 2017, 6, e27696. [Google Scholar] [CrossRef]
- Wang, Q.; Shen, F.Y.; Zou, R.; Zheng, J.J.; Yu, X.; Wang, Y.W. Ketamine-induced apoptosis in the mouse cerebral cortex follows similar characteristic of physiological apoptosis and can be regulated by neuronal activity. Mol. Brain 2017, 10, 24. [Google Scholar] [CrossRef] [Green Version]
- Ikonomidou, C.; Bosch, F.; Miksa, M.; Bittigau, P.; Vockler, J.; Dikranian, K.; Tenkova, T.I.; Stefovska, V.; Turski, L.; Olney, J.W. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 1999, 283, 70–74. [Google Scholar] [CrossRef]
- Ikonomidou, C.; Bittigau, P.; Ishimaru, M.J.; Wozniak, D.F.; Koch, C.; Genz, K.; Price, M.T.; Stefovska, V.; Horster, F.; Tenkova, T.; et al. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science 2000, 287, 1056–1060. [Google Scholar] [CrossRef]
- Lebedeva, J.; Zakharov, A.; Ogievetsky, E.; Minlebaeva, A.; Kurbanov, R.; Gerasimova, E.; Sitdikova, G.; Khazipov, R. Inhibition of Cortical Activity and Apoptosis Caused by Ethanol in Neonatal Rats In Vivo. Cereb. Cortex 2017, 27, 1068–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luhmann, H.J.; Sinning, A.; Yang, J.W.; Reyes-Puerta, V.; Stüttgen, M.C.; Kirischuk, S.; Kilb, W. Spontaneous Neuronal Activity in Developing Neocortical Networks: From Single Cells to Large-Scale Interactions. Front. Neural Circuits 2016, 10, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fishbein, I.; Segal, M. Miniature synaptic currents become neurotoxic to chronically silenced neurons. Cereb. Cortex 2007, 17, 1292–1306. [Google Scholar] [CrossRef] [Green Version]
- Schonfeld-Dado, E.; Fishbein, I.; Segal, M. Degeneration of cultured cortical neurons following prolonged inactivation: Molecular mechanisms. J. Neurochem. 2009, 110, 1203–1213. [Google Scholar] [CrossRef] [PubMed]
- Hazan, L.; Ziv, N.E. Activity Dependent and Independent Determinants of Synaptic Size Diversity. J. Neurosci. 2020, 40, 2828. [Google Scholar] [CrossRef]
- Banasiak, K.J.; Burenkova, O.; Haddad, G.G. Activation of voltage-sensitive sodium channels during oxygen deprivation leads to apoptotic neuronal death. Neuroscience 2004, 126, 31–44. [Google Scholar] [CrossRef]
- Brahma, M.K.; Dohare, P.; Varma, S.; Rath, S.K.; Garg, P.; Biswal, P.K.; Chowdhury, P.D.; Ray, M. The neuronal apoptotic death in global cerebral ischemia in gerbil: Important role for sodium channel modulator. J. Neurosci. Res. 2009, 87, 1400–1411. [Google Scholar] [CrossRef]
- Schock, S.C.; Jolin-Dahel, K.S.; Schock, P.C.; Theiss, S.; Arbuthnott, G.W.; Garcia-Munoz, M.; Staines, W.A. Development of dissociated cryopreserved rat cortical neurons in vitro. J. Neurosci. Methods 2012, 205, 324–333. [Google Scholar] [CrossRef]
- Charlesworth, P.; Cotterill, E.; Morton, A.; Grant, S.G.; Eglen, S.J. Quantitative differences in developmental profiles of spontaneous activity in cortical and hippocampal cultures. Neural Dev. 2015, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Belle, A.M.; Enright, H.A.; Sales, A.P.; Kulp, K.; Osburn, J.; Kuhn, E.A.; Fischer, N.O.; Wheeler, E.K. Evaluation of in vitro neuronal platforms as surrogates for in vivo whole brain systems. Sci. Rep. 2018, 8, 10820. [Google Scholar] [CrossRef] [Green Version]
- Potter, S.M.; DeMarse, T.B. A new approach to neural cell culture for long-term studies. J. Neurosci. Methods 2001, 110, 17–24. [Google Scholar] [CrossRef]
- Eguchi, Y.; Srinivasan, A.; Tomaselli, K.J.; Shimizu, S.; Tsujimoto, Y. ATP-dependent steps in apoptotic signal transduction. Cancer Res. 1999, 59, 2174–2181. [Google Scholar]
- Wang, X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001, 15, 2922–2933. [Google Scholar] [PubMed]
- Pires Monteiro, S.; Voogd, E.; Muzzi, L.; De Vecchis, G.; Mossink, B.; Levers, M.; Hassink, G.; Van Putten, M.; Le Feber, J.; Hofmeijer, J.; et al. Neuroprotective effect of hypoxic preconditioning and neuronal activation in a in vitro human model of the ischemic penumbra. J. Neural Eng. 2021, 18, 036016. [Google Scholar] [CrossRef] [PubMed]
- Nimmervoll, B.; White, R.; Yang, J.W.; An, S.; Henn, C.; Sun, J.J.; Luhmann, H.J. LPS-induced microglial secretion of TNFα increases activity-dependent neuronal apoptosis in the neonatal cerebral cortex. Cereb. Cortex 2013, 23, 1742–1755. [Google Scholar] [CrossRef] [PubMed]
- Gualtieri, F.; Marinelli, C.; Longo, D.; Pugnaghi, M.; Nichelli, P.F.; Meletti, S.; Biagini, G. Hypoxia Markers are Expressed in Interneurons Exposed to Recurrent Seizures. Neuromol. Med. 2013, 15, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Lucchi, C.; Marinelli, C.; Longo, D.; Pugnaghi, M.; Nichelli, P.F.; Meletti, S.; Biagini, G. Ischemic-hypoxic mechanisms leading to hippocampal dysfunction as a consequence of status epilepticus. Epilepsy Behav. 2015, 49, 47–54. [Google Scholar] [CrossRef]
- Hwang, S.; Ham, S.; Lee, S.E.; Lee, Y.; Lee, G.H. Hypoxia regulates the level of glutamic acid decarboxylase enzymes and interrupts inhibitory synapse stability in primary cultured neurons. NeuroToxicology 2017, 65, 221–230. [Google Scholar] [CrossRef]
- le Feber, J.; Dummer, A.; Hassink, G.C.; van Putten, M.J.A.M.; Hofmeijer, J. Evolution of excitation-inhibition ratio in cortical cultures exposed to hypoxia. Front. Cell Neurosci. 2018, 12, 183. [Google Scholar] [CrossRef]
- Nair, P.K.; Buerk, D.G.; Halsey, J.H.J. Comparisons of Oxygen Metabolism and Tissue pO2 in Cortex and Hippocampus of Gerbil Brain. Stroke 1987, 18, 616–622. [Google Scholar] [CrossRef] [Green Version]
- Grote, J.; Laue, O.; Eiring, P.; Wehler, M. Evaluation of brain tissue O2 supply based on results of pO2 measurements with needle and surface microelectrodes. J. Auton. Nerv. Syst. 1996, 57, 168–172. [Google Scholar] [CrossRef]
- Zhu, P.J.; Krnjević, K. Anoxia selectively depresses excitatory synaptic transmission in hippocampal slices. Neurosci. Lett. 1994, 166, 27–30. [Google Scholar] [CrossRef]
- Romijn, H.J.; van Huizen, F.; Wolters, P.S. Towards an improved serum-free, chemically defined medium for long-term culturing of cerebral cortex tissue. Neurosc. Biobehav. Rev. 1984, 8, 301–334. [Google Scholar] [CrossRef]
- Wagenaar, D.A.; Nadasdy, Z.; Potter, S.M. Persistent dynamic attractors in activity patterns of cultured neuronal networks. Phys. Rev. E 2006, 73, 051907. [Google Scholar] [CrossRef] [Green Version]
- Kamioka, H.; Maeda, E.; Jimbo, Y.; Robinson, H.P.; Kawana, A. Spontaneous periodic synchronized bursting during formation of mature patterns of connections in cortical cultures. Neurosci. Lett. 1996, 206, 109–112. [Google Scholar] [CrossRef]
- Wagenaar, D.A.; DeMarse, T.B.; Potter, S.M. MeaBench: A toolset for multi-electrode data acquisition and on-line analysis. In Proceedings of the 2nd International IEEE EMBS Conference on Neural Engineering, Arlington, VA, USA, 16–19 March 2005. [Google Scholar]
- Lagarias, J.C.; Reeds, J.A.; Wright, M.H.; Wright, P.H. Convergence Properties of the Nelder--Mead Simplex Method in Low Dimensions. SIAM J. Optim. 1998, 9, 112–147. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taxis di Bordonia e Valnigra, D.; Hassink, G.C.; Levers, M.R.; Frega, M.; Hofmeijer, J.; van Putten, M.J.A.M.; le Feber, J. The Association between Hypoxia-Induced Low Activity and Apoptosis Strongly Resembles That between TTX-Induced Silencing and Apoptosis. Int. J. Mol. Sci. 2022, 23, 2754. https://doi.org/10.3390/ijms23052754
Taxis di Bordonia e Valnigra D, Hassink GC, Levers MR, Frega M, Hofmeijer J, van Putten MJAM, le Feber J. The Association between Hypoxia-Induced Low Activity and Apoptosis Strongly Resembles That between TTX-Induced Silencing and Apoptosis. International Journal of Molecular Sciences. 2022; 23(5):2754. https://doi.org/10.3390/ijms23052754
Chicago/Turabian StyleTaxis di Bordonia e Valnigra, Domitilla, Gerco C. Hassink, Marloes R. Levers, Monica Frega, Jeannette Hofmeijer, Michel J. A. M. van Putten, and Joost le Feber. 2022. "The Association between Hypoxia-Induced Low Activity and Apoptosis Strongly Resembles That between TTX-Induced Silencing and Apoptosis" International Journal of Molecular Sciences 23, no. 5: 2754. https://doi.org/10.3390/ijms23052754
APA StyleTaxis di Bordonia e Valnigra, D., Hassink, G. C., Levers, M. R., Frega, M., Hofmeijer, J., van Putten, M. J. A. M., & le Feber, J. (2022). The Association between Hypoxia-Induced Low Activity and Apoptosis Strongly Resembles That between TTX-Induced Silencing and Apoptosis. International Journal of Molecular Sciences, 23(5), 2754. https://doi.org/10.3390/ijms23052754