
Identification of Novel and Potent Indole-Based Benzenesulfonamides as Selective Human Carbonic Anhydrase II Inhibitors: Design, Synthesis, In Vitro, and In Silico Studies
 ,
,  , , ,
, , ,  and
and
Abstract
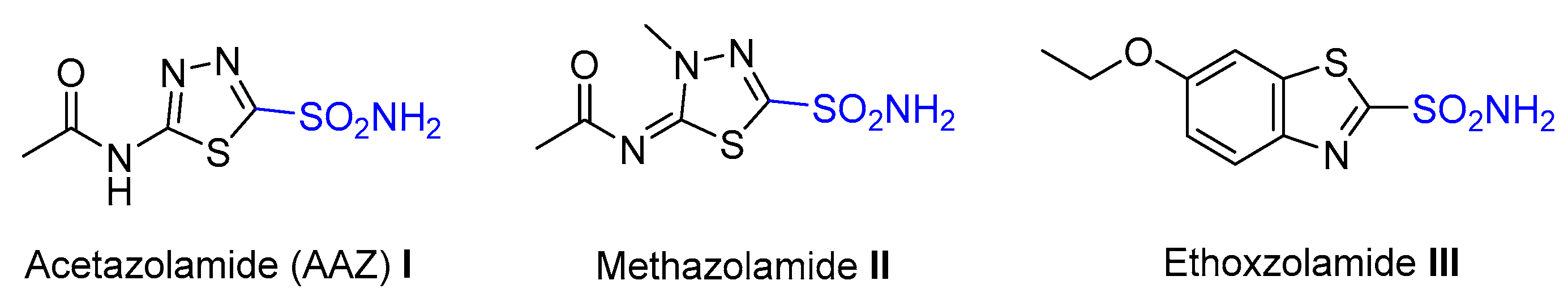
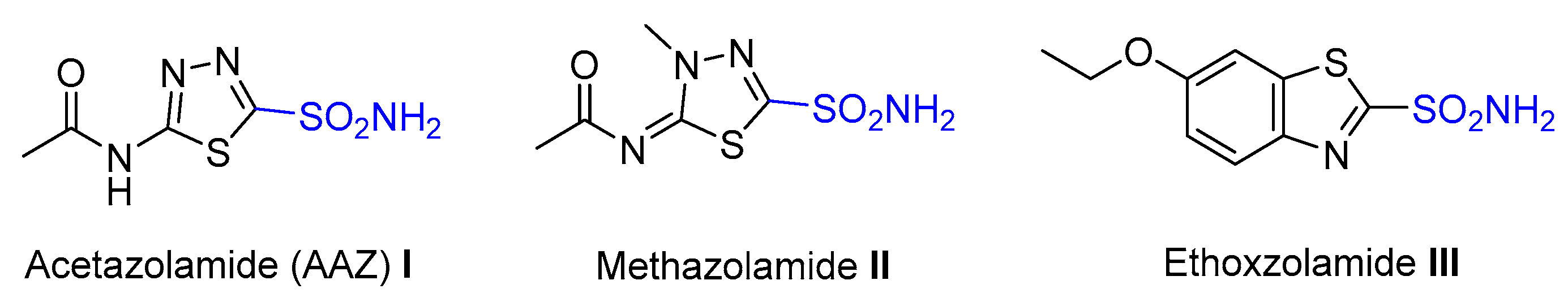
:1. Introduction
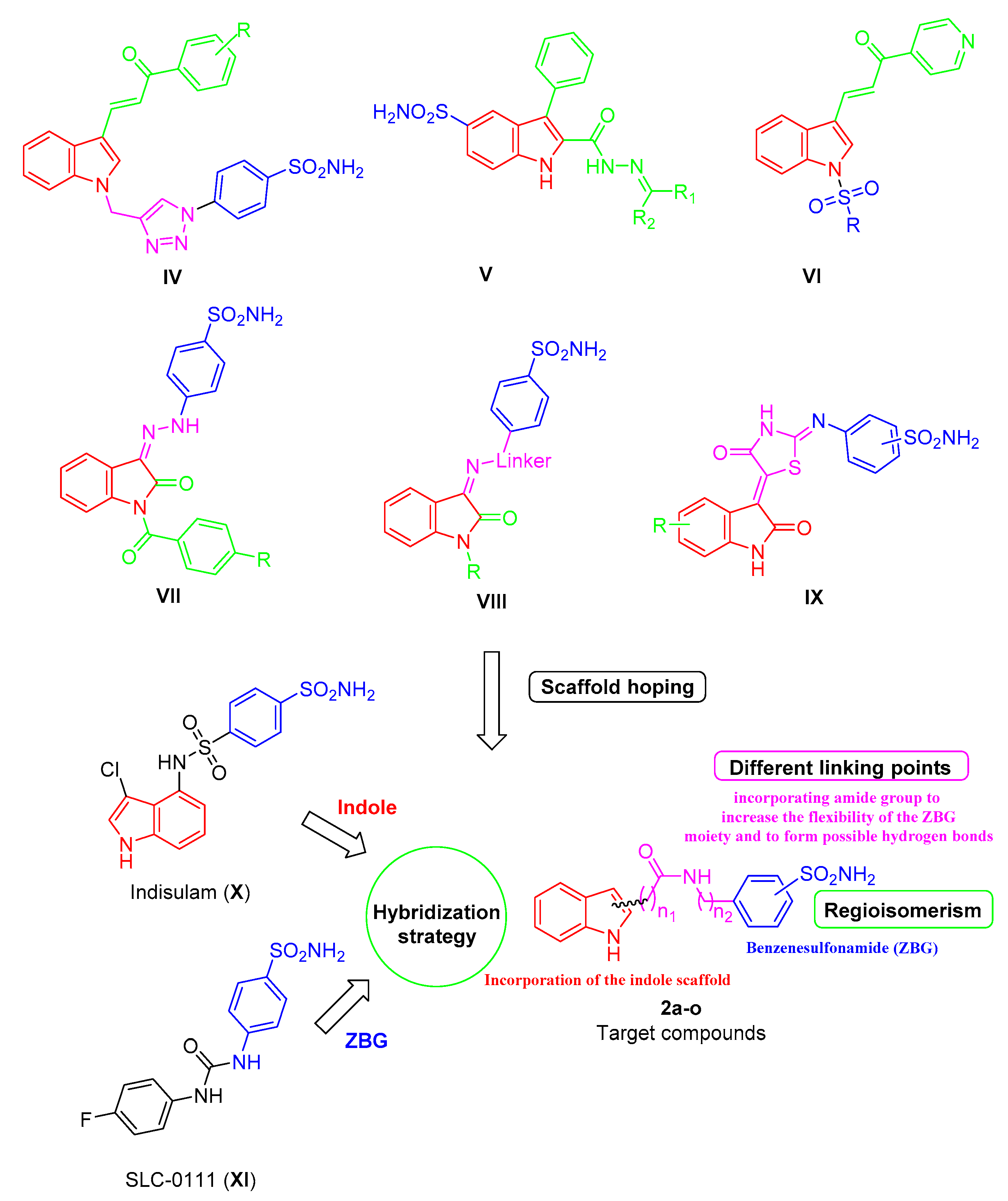
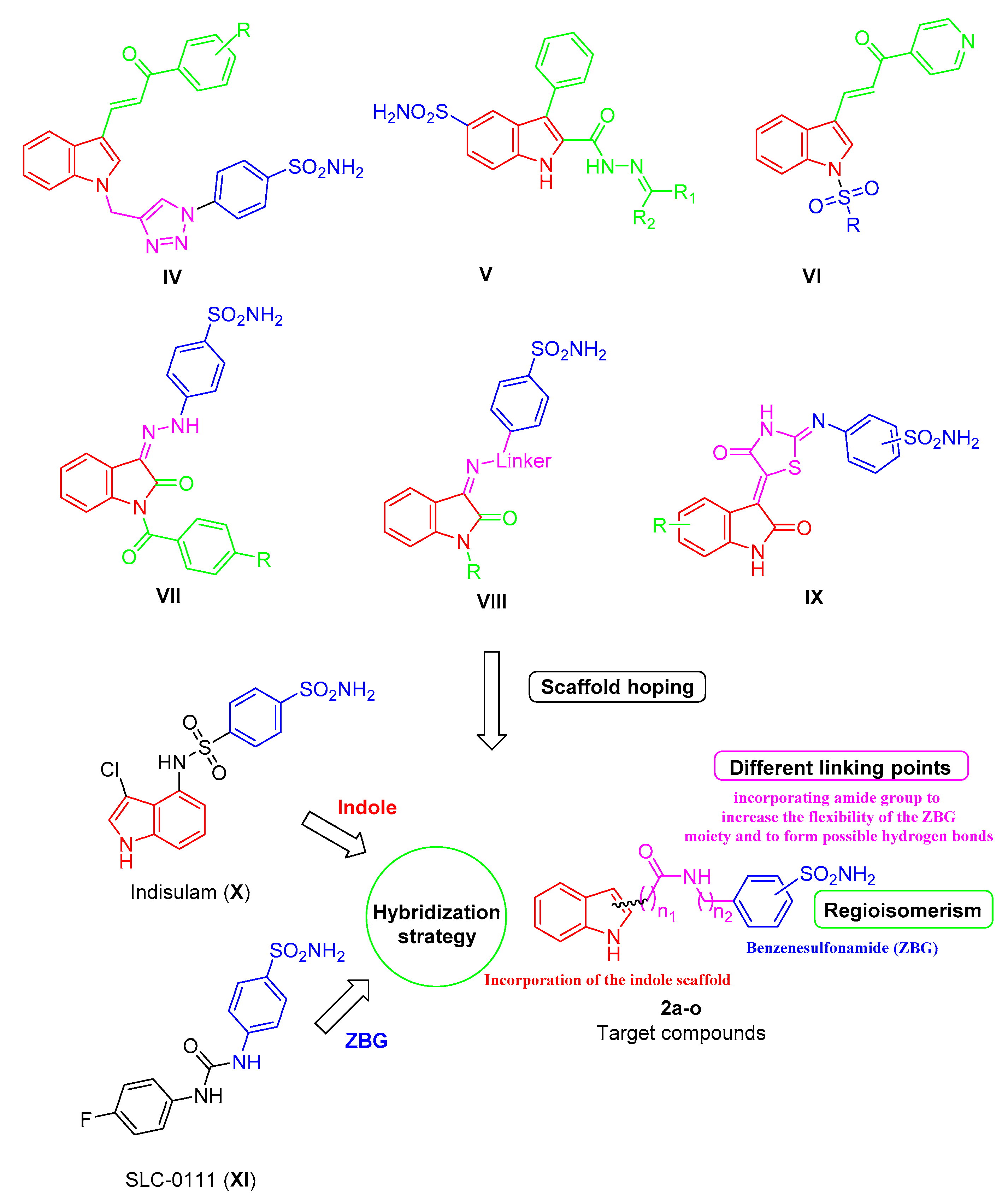
2. Results and Discussion
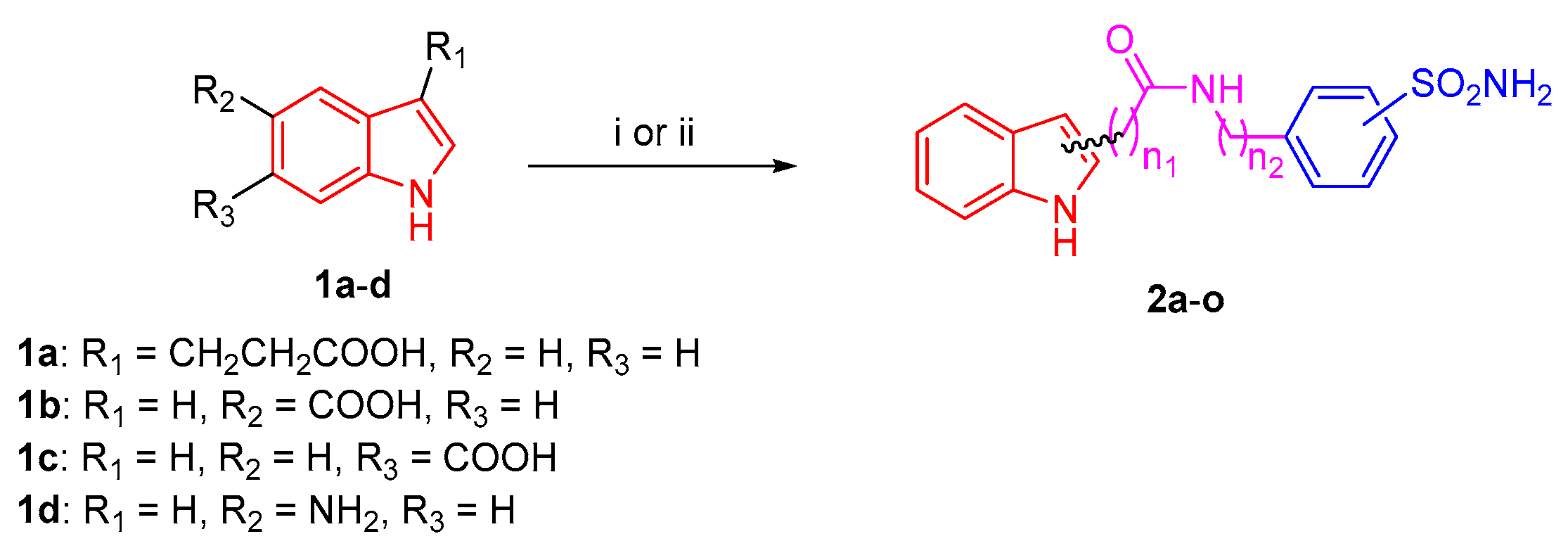
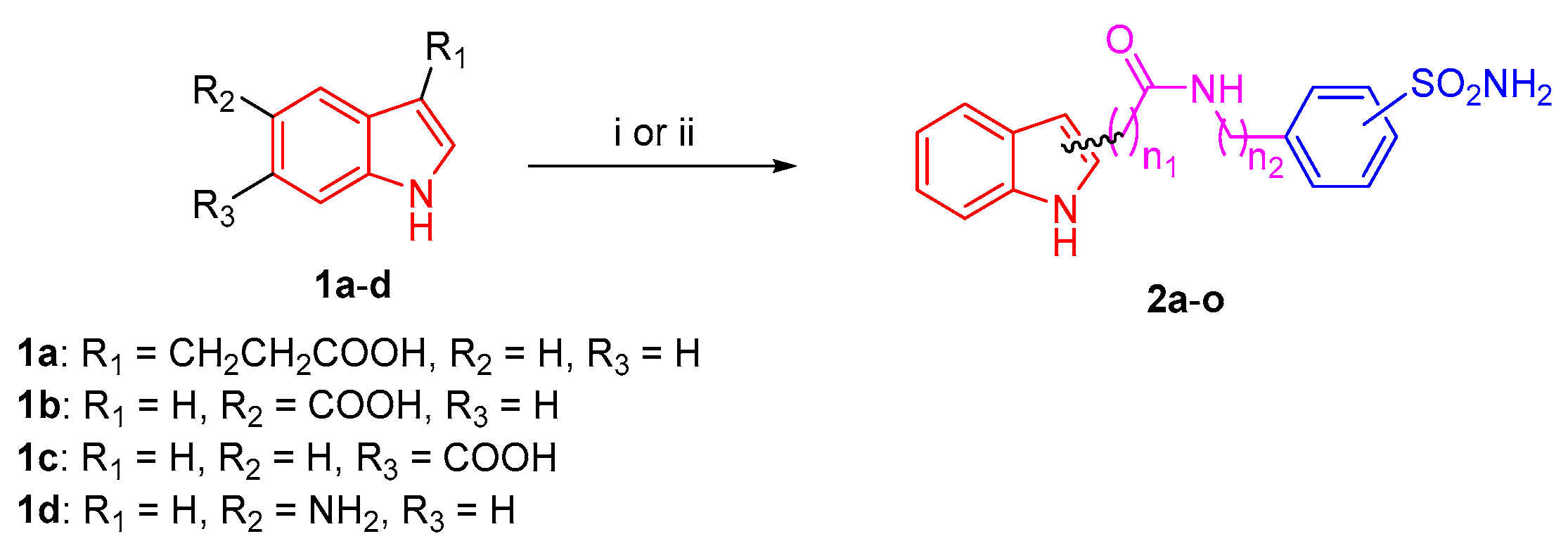
2.1. Chemical Synthesis
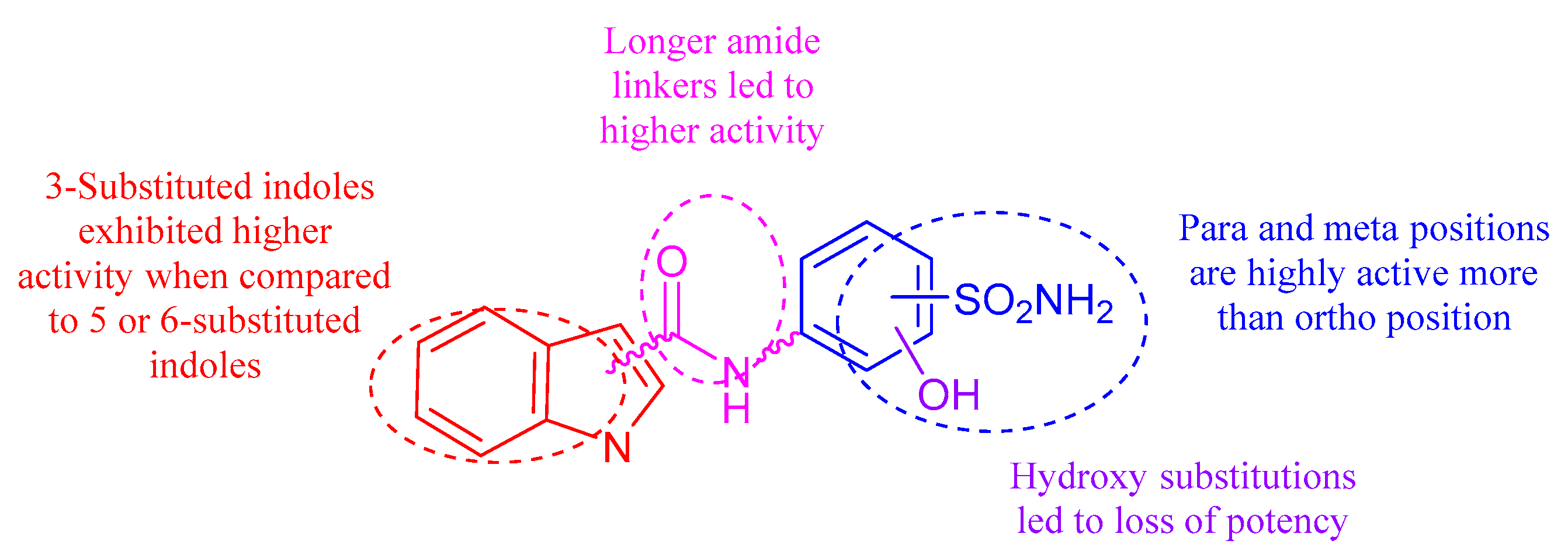
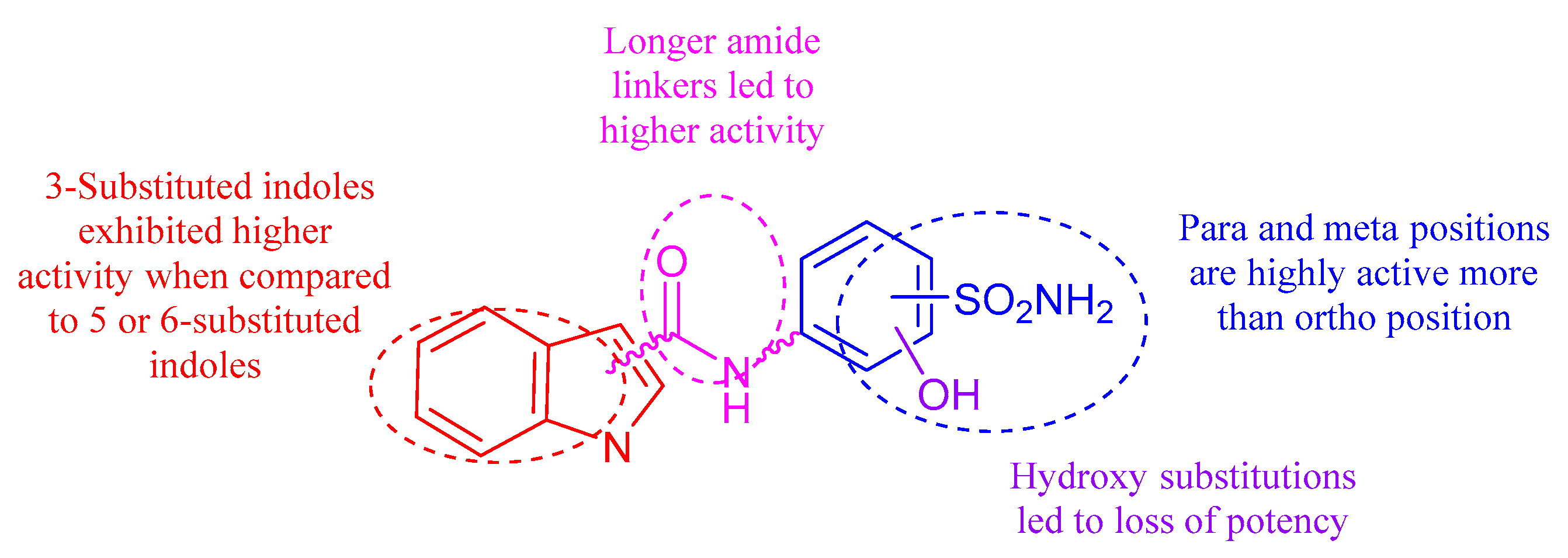
2.2. Carbonic Anhydrase Inhibition
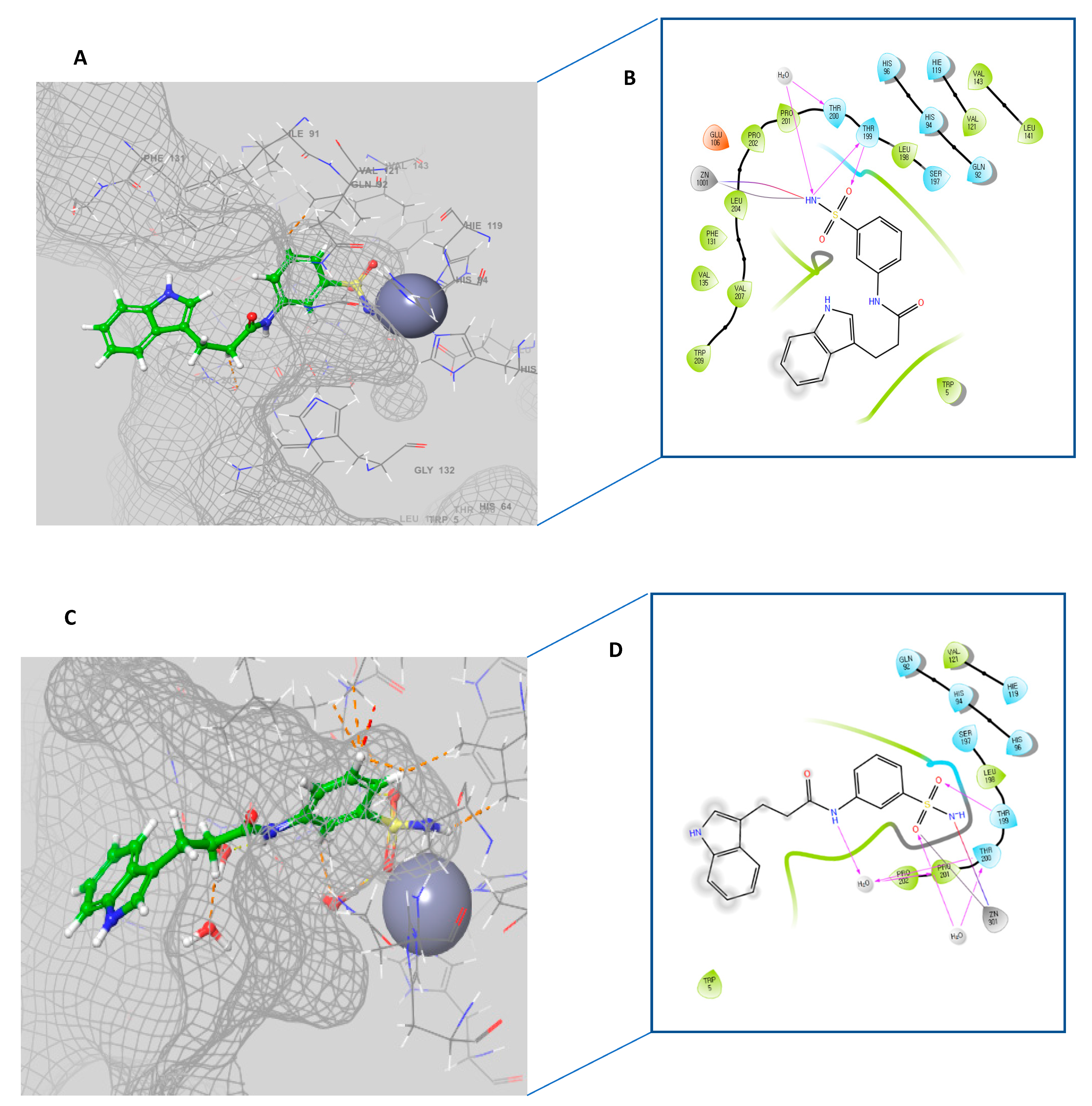
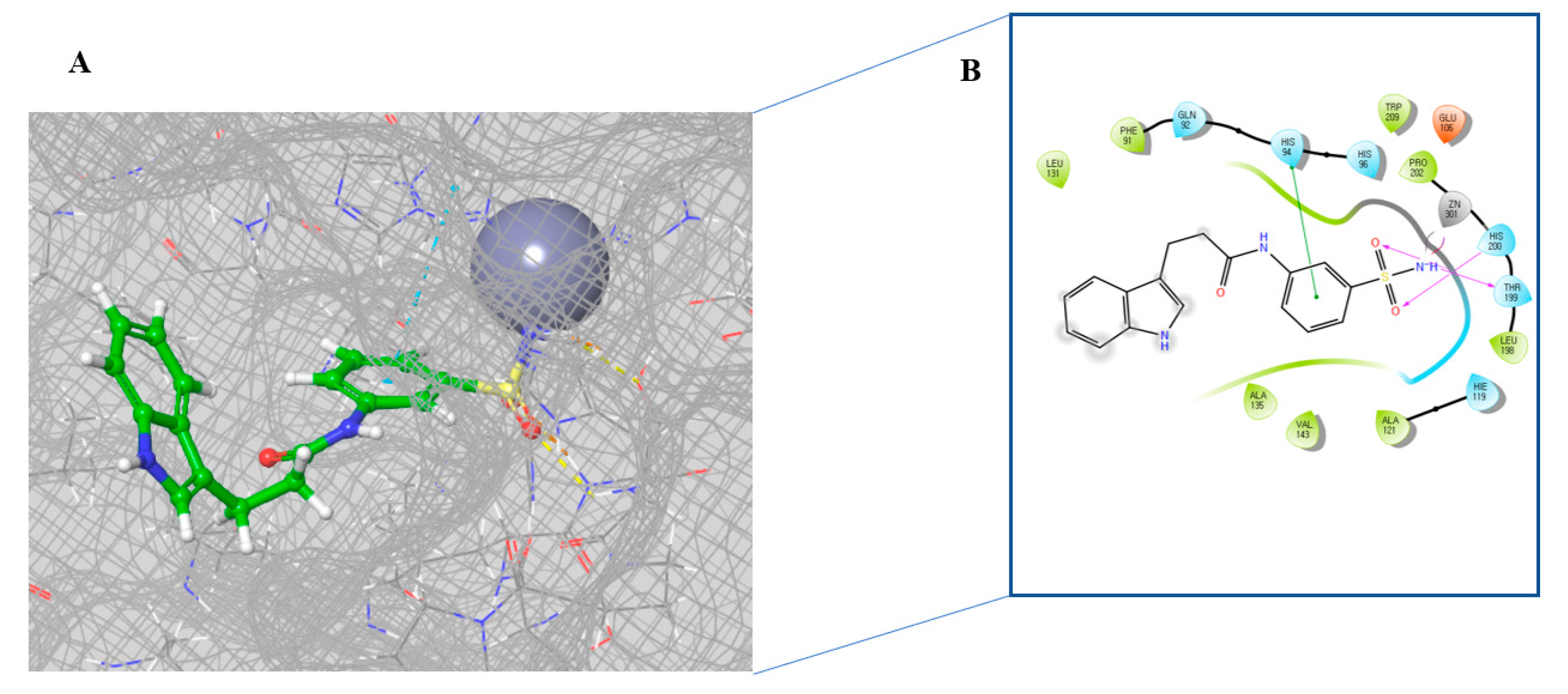
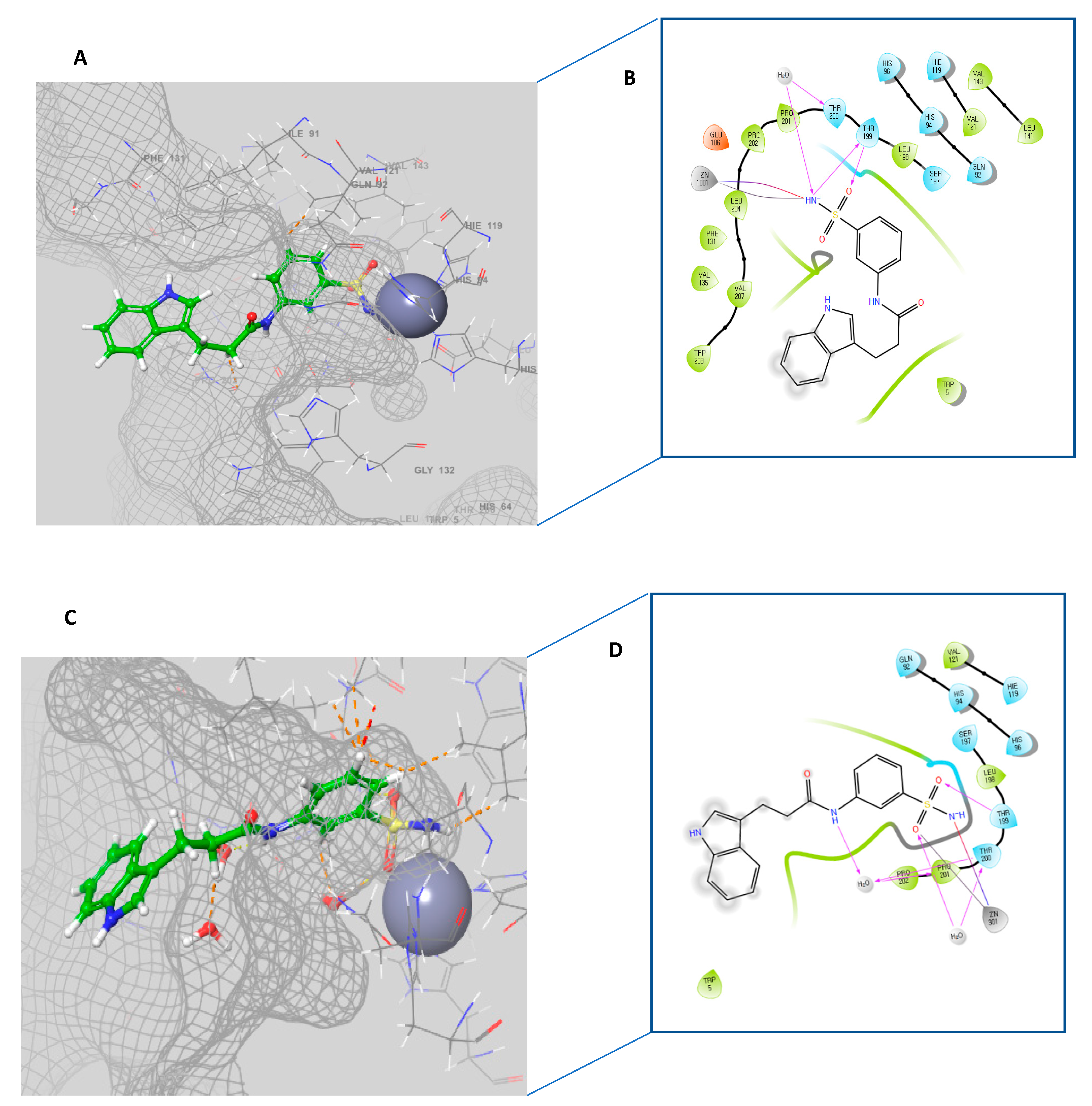
2.3. Molecular Docking Study
3. Materials and Methods
3.1. Chemistry
3.2. Carbonic Anhydrase Inhibition
3.3. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nocentini, A.; Supuran, C.T.; Capasso, C. An overview on the recently discovered iota-carbonic anhydrases. J. Enzym. Inhib. Med. Chem. 2021, 36, 1988–1995. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Topal, F.; Aksu, K.; Gulcin, I.; Tümer, F.; Goksu, S. Inhibition Profiles of Some Symmetric Sulfamides Derived from Phenethylamines on Human Carbonic Anhydrase, I.; and II Isoenzymes. Chem. Biodivers. 2021, 18, e2100422. [Google Scholar] [CrossRef] [PubMed]
- Durdagi, S.; Şentürk, M.; Ekinci, D.; Balaydın, H.T.; Göksu, S.; Küfrevioğlu, Ö.İ.; Innocenti, A.; Scozzafava, A.; Supuran, C.T. Kinetic and docking studies of phenol-based inhibitors of carbonic anhydrase isoforms I, II, IX and XII evidence a new binding mode within the enzyme active site. Bioorg. Med. Chem. 2011, 19, 1381–1389. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T.; Scozzafava, A. Carbonic anhydrase inhibitors and their therapeutic potential. Expert Opin. Ther. Pat. 2000, 10, 575–600. [Google Scholar] [CrossRef]
- Mohamed, M.M.; Sloane, B.F. multifunctional enzymes in cancer. Nat. Rev. Cancer 2006, 6, 764–775. [Google Scholar] [CrossRef]
- Haapasalo, J.; Nordfors, K.; Järvelä, S.; Bragge, H.; Rantala, I.; Parkkila, A.-K.; Haapasalo, H.; Parkkila, S. Carbonic anhydrase II in the endothelium of glial tumors: A potential target for therapy. Neuro-Oncol. 2007, 9, 308–313. [Google Scholar] [CrossRef] [Green Version]
- Cuffaro, D.; Nuti, E.; Rossello, A. An overview of carbohydrate-based carbonic anhydrase inhibitors. J. Enzym. Inhib. Med. Chem. 2020, 35, 1906–1922. [Google Scholar] [CrossRef]
- Awadallah, F.M.; Bua, S.; Mahmoud, W.R.; Nada, H.H.; Nocentini, A.; Supuran, C.T. Inhibition studies on a panel of human carbonic anhydrases with N1-substituted secondary sulfonamides incorporating thiazolinone or imidazolone-indole tails. J. Enzym. Inhib. Med. Chem. 2018, 33, 629–638. [Google Scholar] [CrossRef] [Green Version]
- Supuran, C.T. Carbon- versus sulphur-based zinc binding groups for carbonic anhydrase inhibitors? J. Enzym. Inhib. Med. Chem. 2018, 33, 485–495. [Google Scholar] [CrossRef] [Green Version]
- Sanku, R.K.K. Development and Biological Evaluation of Carbonic Anhydrase Modulators as Potential Nootropics and Anticancer Agents. Ph.D. Thesis, Temple University, Ann Arbor, MI, USA, 2018. [Google Scholar]
- Supuran, C.T. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin. Drug Discov. 2017, 12, 61–88. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Exploring the multiple binding modes of inhibitors to carbonic anhydrases for novel drug discovery. Expert Opin. Drug Discov. 2020, 15, 671–686. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, M.; Kondeti, B.; McKenna, R. Insights towards sulfonamide drug specificity in α-carbonic anhydrases. Bioorg. Med. Chem. 2013, 21, 1526–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carta, F.; Supuran, C.T.; Scozzafava, A. Sulfonamides and their isosters as carbonic anhydrase inhibitors. Future Med. Chem. 2014, 6, 1149–1165. [Google Scholar] [CrossRef] [PubMed]
- Chiaramonte, N.; Bua, S.; Ferraroni, M.; Nocentini, A.; Bonardi, A.; Bartolucci, G.; Durante, M.; Lucarini, L.; Chiapponi, D.; Dei, S.; et al. 2-Benzylpiperazine: A new scaffold for potent human carbonic anhydrase inhibitors. Synthesis, enzyme inhibition, enantioselectivity, computational and crystallographic studies and in vivo activity for a new class of intraocular pressure lowering agents. Eur. J. Med. Chem. 2018, 151, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Swain, B.; Thacker, P.S.; Sigalapalli, D.K.; Yadav, P.P.; Angeli, A.; Supuran, C.T.; Arifuddin, M. Synthesis and carbonic anhydrase inhibition studies of sulfonamide based indole-1,2,3-triazole chalcone hybrids. Bioorg. Chem. 2020, 99, 103839. [Google Scholar] [CrossRef] [PubMed]
- Demir-Yazıcı, K.; Bua, S.; Akgüneş, N.M.; Akdemir, A.; Supuran, C.T.; Güzel-Akdemir, Ö. Indole-Based Hydrazones Containing A Sulfonamide Moiety as Selective Inhibitors of Tumor-Associated Human Carbonic Anhydrase Isoforms IX and XII. Int. J. Mol. Sci. 2019, 20, 2354. [Google Scholar] [CrossRef] [Green Version]
- Peerzada, M.N.; Khan, P.; Ahmad, K.; Hassan, M.I.; Azam, A. Synthesis, characterization and biological evaluation of tertiary sulfonamide derivatives of pyridyl-indole based heteroaryl chalcone as potential carbonic anhydrase IX inhibitors and anticancer agents. Eur. J. Med. Chem. 2018, 155, 13–23. [Google Scholar] [CrossRef]
- George, R.F.; Bua, S.; Supuran, C.T.; Awadallah, F.M. Synthesis of some N-aroyl-2-oxindole benzenesulfonamide conjugates with carbonic anhydrase inhibitory activity. Bioorg. Chem. 2020, 96, 103635. [Google Scholar] [CrossRef]
- Assi, R.; Kantarjian, H.M.; Kadia, T.M.; Pemmaraju, N.; Jabbour, E.; Jain, N.; Daver, N.; Estrov, Z.; Uehara, T.; Owa, T.; et al. Final results of a phase 2, open-label study of indisulam, idarubicin, and cytarabine in patients with relapsed or refractory acute myeloid leukemia and high-risk myelodysplastic syndrome. Cancer 2018, 124, 2758–2765. [Google Scholar] [CrossRef] [Green Version]
- Bozdag, M.; Carta, F.; Ceruso, M.; Ferraroni, M.; McDonald, P.C.; Dedhar, S.; Supuran, C.T. Discovery of 4-Hydroxy-3-(3-(phenylureido)benzenesulfonamides as SLC-0111 Analogues for the Treatment of Hypoxic Tumors Overexpressing Carbonic Anhydrase IX. J. Med. Chem. 2018, 61, 6328–6338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macalino, S.J.Y.; Gosu, V.; Hong, S.; Choi, S. Role of computer-aided drug design in modern drug discovery. Arch. Pharmacal Res. 2015, 38, 1686–1701. [Google Scholar] [CrossRef] [PubMed]
- Guedes, I.A.; de Magalhães, C.S.; Dardenne, L.E.J.B.R. Receptor–ligand molecular docking. Biophys. Rev. 2014, 6, 75–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Li, Y.; Yang, Y.; Du, J.; Zhang, S.; Yang, L. In silico research to assist the investigation of carboxamide derivatives as potent TRPV1 antagonists. Mol. Biosyst. 2015, 11, 2885–2899. [Google Scholar] [CrossRef] [PubMed]
- Elkamhawy, A.; Kim, N.Y.; Hassan, A.H.E.; Park, J.E.; Yang, J.E.; Oh, K.S.; Lee, B.H.; Lee, M.Y.; Shin, K.J.; Lee, K.T.; et al. Design, synthesis and biological evaluation of novel thiazolidinedione derivatives as irreversible allosteric IKK-β modulators. Eur. J. Med. Chem. 2018, 157, 691–704. [Google Scholar] [CrossRef]
- Park, J.E.; Elkamhawy, A.; Hassan, A.H.E.; Pae, A.N.; Lee, J.; Paik, S.; Park, B.G.; Roh, E.J. Synthesis and evaluation of new pyridyl/pyrazinyl thiourea derivatives: Neuroprotection against amyloid-β-induced toxicity. Eur. J. Med. Chem. 2017, 141, 322–334. [Google Scholar] [CrossRef]
- Elkamhawy, A.; Hassan, A.H.E.; Paik, S.; Sup Lee, Y.; Lee, H.H.; Shin, J.S.; Lee, K.T.; Roh, E.J. EGFR inhibitors from cancer to inflammation: Discovery of 4-fluoro-N-(4-(3-(trifluoromethyl)phenoxy)pyrimidin-5-yl)benzamide as a novel anti-inflammatory EGFR inhibitor. Bioorg. Chem. 2019, 86, 112–118. [Google Scholar] [CrossRef]
- Al-Sanea, M.M.; Elkamhawy, A.; Zakaria, A.; Park, B.S.; Kwon, Y.; Lee, S.H.; Lee, S.W.; Kim, I.T. Synthesis and in vitro screening of phenylbipyridinylpyrazole derivatives as potential antiproliferative agents. Molecules 2015, 20, 1031–1045. [Google Scholar] [CrossRef] [Green Version]
- Maresca, A.; Temperini, C.; Vu, H.; Pham, N.B.; Poulsen, S.-A.; Scozzafava, A.; Quinn, R.J.; Supuran, C.T. Non-Zinc Mediated Inhibition of Carbonic Anhydrases: Coumarins Are a New Class of Suicide Inhibitors. J. Am. Chem. Soc. 2009, 131, 3057–3062. [Google Scholar] [CrossRef] [Green Version]
- Alafeefy, A.M.; Ceruso, M.; Al-Jaber, N.A.; Parkkila, S.; Vermelho, A.B.; Supuran, C.T. A new class of quinazoline-sulfonamides acting as efficient inhibitors against the α-carbonic anhydrase from Trypanosoma cruzi. J. Enzym. Inhib. Med. Chem. 2015, 30, 581–585. [Google Scholar] [CrossRef]
- Menchise, V.; De Simone, G.; Alterio, V.; Di Fiore, A.; Pedone, C.; Scozzafava, A.; Supuran, C.T. Carbonic Anhydrase Inhibitors: Stacking with Phe131 Determines Active Site Binding Region of Inhibitors As Exemplified by the X-ray Crystal Structure of a Membrane-Impermeant Antitumor Sulfonamide Complexed with Isozyme II. J. Med. Chem. 2005, 48, 5721–5727. [Google Scholar] [CrossRef]
- Margheri, F.; Ceruso, M.; Carta, F.; Laurenzana, A.; Maggi, L.; Lazzeri, S.; Simonini, G.; Annunziato, F.; Del Rosso, M.; Supuran, C.T.; et al. Overexpression of the transmembrane carbonic anhydrase isoforms IX and XII in the inflamed synovium. J. Enzym. Inhib. Med. Chem. 2016, 31, 60–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozdag, M.; Ferraroni, M.; Nuti, E.; Vullo, D.; Rossello, A.; Carta, F.; Scozzafava, A.; Supuran, C.T. Combining the tail and the ring approaches for obtaining potent and isoform-selective carbonic anhydrase inhibitors: Solution and X-ray crystallographic studies. Bioorg, Med. Chem. 2014, 22, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Bozdag, M.; Farooq, U.; Angeli, A.; Carta, F.; Berto, P.; Zanotti, G.; Supuran, C.T. Benzylaminoethyureido-Tailed Benzenesulfonamides: Design, Synthesis, Kinetic and X-ray Investigations on Human Carbonic Anhydrases. Int. J. Mol. Sci. 2020, 21, 2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahon, B.P.; Lomelino, C.L.; Ladwig, J.; Rankin, G.M.; Driscoll, J.M.; Salguero, A.L.; Pinard, M.A.; Vullo, D.; Supuran, C.T.; Poulsen, S.-A.; et al. Mapping Selective Inhibition of the Cancer-Related Carbonic Anhydrase IX Using Structure–Activity Relationships of Glucosyl-Based Sulfamates. J. Med. Chem. 2015, 58, 6630–6638. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd | Starting | Benzenesulfonamide Reagent | Chemical Structure |
|---|---|---|---|
| 2a | 1a |  | 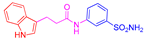 |
| 2b | 1a | 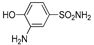 | 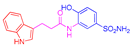 |
| 2c | 1a |  | 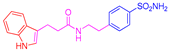 |
| 2d | 1a | 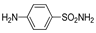 | 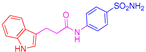 |
| 2e | 1b | 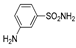 | 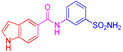 |
| 2f | 1b | 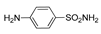 | 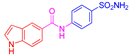 |
| 2g | 1b | 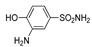 | 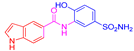 |
| 2h | 1b | 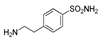 | 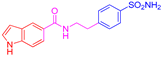 |
| 2i | 1b | 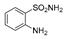 |  |
| 2j | 1c |  | 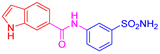 |
| 2k | 1c | 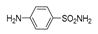 | 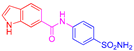 |
| 2l | 1c | 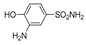 | 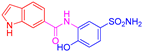 |
| 2m | 1c | 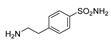 | 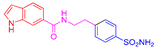 |
| 2n | 1c | 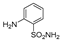 | 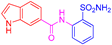 |
| 2o | 1d | 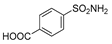 | 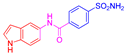 |
| Cpd | Ki (nM) | |||
|---|---|---|---|---|
| hCA I | hCA II | hCA IX | hCA XII | |
| 2a | 79.8 | 5.9 | 205.8 | 54.2 |
| 2b | 2796 | 7.3 | 286.5 | 85.1 |
| 2c | 217.3 | 9.0 | 221.6 | 36.9 |
| 2d | 97.6 | 7.1 | 165.5 | 42.2 |
| 2e | 5575 | 45.5 | 766.3 | 90.9 |
| 2f | 3105 | 16.0 | 165.1 | 257.0 |
| 2g | 4772 | 679.9 | 279.6 | 85.3 |
| 2h | 553.7 | 8.6 | 239.6 | 235.6 |
| 2i | 7453 | 71.9 | 635.0 | 89.4 |
| 2j | 5397 | 94.8 | 654.0 | 40.5 |
| 2k | 4655 | 833.4 | 427.6 | 83.8 |
| 2l | 4275 | 681.6 | 423.0 | 73.7 |
| 2m | 252.9 | 38.8 | 277.9 | 85.1 |
| 2n | 7511 | 670.1 | 487.3 | 36.9 |
| 2o | 56.6 | 7.5 | 169.6 | 66.9 |
| AAZ | 250.0 | 12.1 | 25.7 | 5.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elkamhawy, A.; Woo, J.; Nada, H.; Angeli, A.; Bedair, T.M.; Supuran, C.T.; Lee, K. Identification of Novel and Potent Indole-Based Benzenesulfonamides as Selective Human Carbonic Anhydrase II Inhibitors: Design, Synthesis, In Vitro, and In Silico Studies. Int. J. Mol. Sci. 2022, 23, 2540. https://doi.org/10.3390/ijms23052540
Elkamhawy A, Woo J, Nada H, Angeli A, Bedair TM, Supuran CT, Lee K. Identification of Novel and Potent Indole-Based Benzenesulfonamides as Selective Human Carbonic Anhydrase II Inhibitors: Design, Synthesis, In Vitro, and In Silico Studies. International Journal of Molecular Sciences. 2022; 23(5):2540. https://doi.org/10.3390/ijms23052540
Chicago/Turabian StyleElkamhawy, Ahmed, Jiyu Woo, Hossam Nada, Andrea Angeli, Tarek M. Bedair, Claudiu T. Supuran, and Kyeong Lee. 2022. "Identification of Novel and Potent Indole-Based Benzenesulfonamides as Selective Human Carbonic Anhydrase II Inhibitors: Design, Synthesis, In Vitro, and In Silico Studies" International Journal of Molecular Sciences 23, no. 5: 2540. https://doi.org/10.3390/ijms23052540
APA StyleElkamhawy, A., Woo, J., Nada, H., Angeli, A., Bedair, T. M., Supuran, C. T., & Lee, K. (2022). Identification of Novel and Potent Indole-Based Benzenesulfonamides as Selective Human Carbonic Anhydrase II Inhibitors: Design, Synthesis, In Vitro, and In Silico Studies. International Journal of Molecular Sciences, 23(5), 2540. https://doi.org/10.3390/ijms23052540