
JAK2-Mediated Phosphorylation of Stress-Induced Phosphoprotein-1 (STIP1) in Human Cells

 , , and
, , and
Abstract
1. Introduction
2. Result
2.1. JAK2 Phosphorylates STIP1 at Tyrosine 134 and 152
2.2. STIP1 Phosphorylation Is Essential in Maintaining the JAK2/STAT3 Signaling Pathway
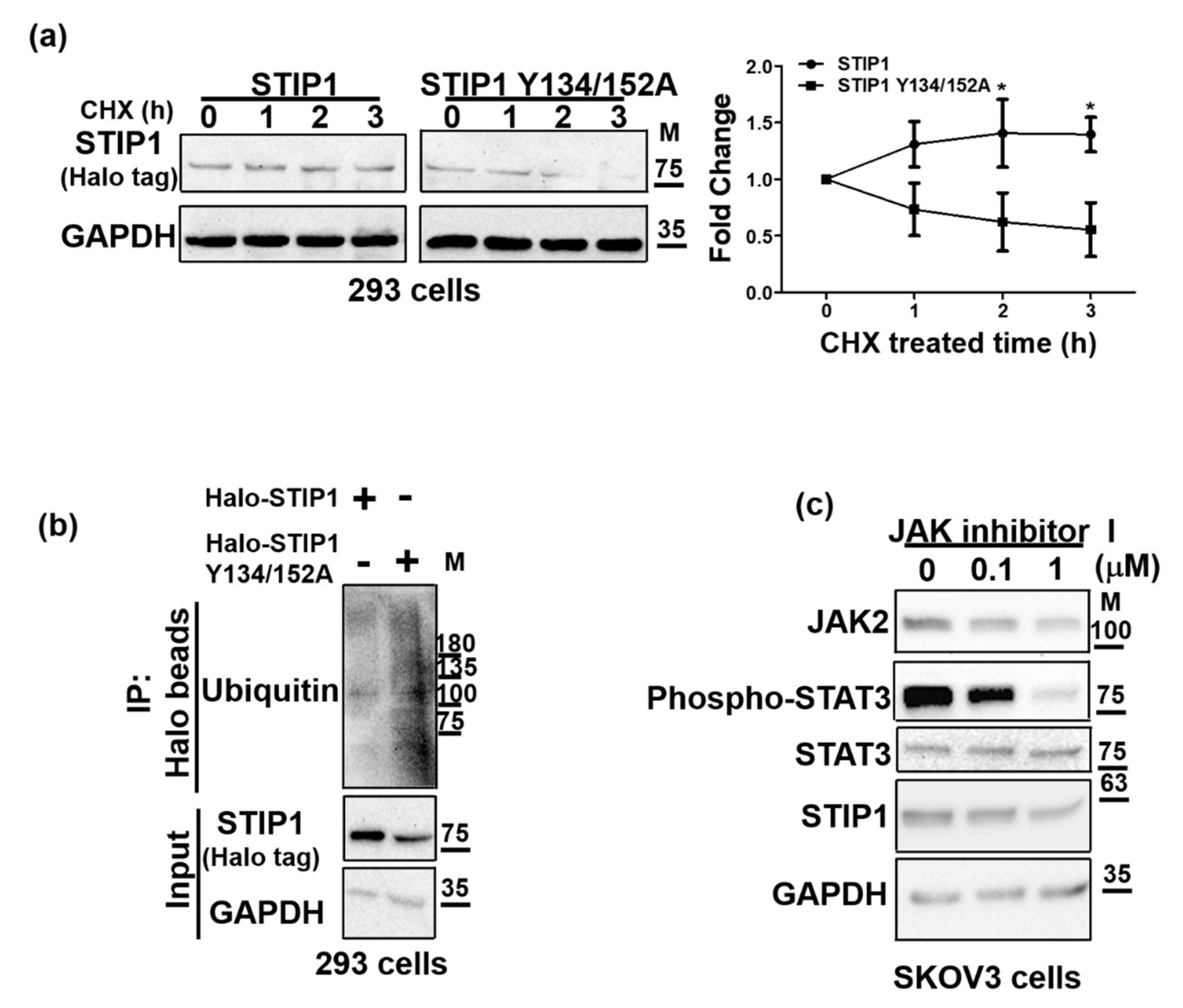
2.3. JAK2-Mediated STIP1 Phosphorylation Maintains Its Protein Stability
2.4. JAK2-Mediated STIP1 Phosphorylation Inhibits Cisplatin-Induced Cell Death
2.5. JAK2-Mediated STIP1 Phosphorylation Regulates Nuclear-Cytoplasmic Shuttling and Its Secretion in the Extracellular Space
2.6. STIP1 and JAK2 Expression Levels Are Positively Correlated to Each Other in Human Serous Ovarian Cancer
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Materials
4.2. Collection of Human Ovarian Cancer Specimens
4.3. DNA Constructs
4.4. DNA Transfection
4.5. Cell Viability Assay
4.6. Western Blot
4.7. Immunoprecipitation
4.8. Confocal Microscopy
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Longshaw, V.M.; Chapple, J.P.; Balda, M.S.; Cheetham, M.E.; Blatch, G.L. Nuclear translocation of the Hsp70/Hsp90 organizing protein mSTI1 is regulated by cell cycle kinases. J. Cell Sci. 2004, 117, 701–710. [Google Scholar] [CrossRef]
- Carrigan, P.E.; Riggs, D.L.; Chinkers, M.; Smith, D.F. Functional comparison of human and Drosophila Hop reveals novel role in steroid receptor maturation. J. Biol. Chem. 2005, 280, 8906–8911. [Google Scholar] [CrossRef] [PubMed]
- Odunuga, O.O.; Longshaw, V.M.; Blatch, G.L. Hop: More than an Hsp70/Hsp90 adaptor protein. BioEssays 2004, 26, 1058–1068. [Google Scholar] [CrossRef] [PubMed]
- Beraldo, F.H.; Soares, I.N.; Goncalves, D.F.; Fan, J.; Thomas, A.A.; Santos, T.G.; Mohammad, A.H.; Roffe, M.; Calder, M.D.; Nikolova, S.; et al. Stress-inducible phosphoprotein 1 has unique cochaperone activity during development and regulates cellular response to ischemia via the prion protein. FASEB J 2013, 27, 3594–3607. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Xing, B.; Sun, Y.; Du, X.; Lu, M.; Hao, C.; Lu, Z.; Mi, W.; Wu, S.; Wei, H.; et al. Proteome analysis of hepatocellular carcinoma by two-dimensional difference gel electrophoresis: Novel protein markers in hepatocellular carcinoma tissues. Mol. Cell Proteom. 2007, 6, 1798–1808. [Google Scholar] [CrossRef] [PubMed]
- Walsh, N.; O’Donovan, N.; Kennedy, S.; Henry, M.; Meleady, P.; Clynes, M.; Dowling, P. Identification of pancreatic cancer invasion-related proteins by proteomic analysis. Proteome Sci. 2009, 7, 3. [Google Scholar] [CrossRef]
- Chao, A.; Lai, C.H.; Tsai, C.L.; Hsueh, S.; Hsueh, C.; Lin, C.Y.; Chou, H.H.; Lin, Y.J.; Chen, H.W.; Chang, T.C.; et al. Tumor stress-induced phosphoprotein1 (STIP1) as a prognostic biomarker in ovarian cancer. PLoS ONE 2013, 8, e57084. [Google Scholar] [CrossRef]
- Wang, T.H.; Chao, A.; Tsai, C.L.; Chang, C.L.; Chen, S.H.; Lee, Y.S.; Chen, J.K.; Lin, Y.J.; Chang, P.Y.; Wang, C.J.; et al. Stress-induced phosphoprotein 1 as a secreted biomarker for human ovarian cancer promotes cancer cell proliferation. Mol. Cell Proteom. 2010, 9, 1873–1884. [Google Scholar] [CrossRef]
- Kubota, H.; Yamamoto, S.; Itoh, E.; Abe, Y.; Nakamura, A.; Izumi, Y.; Okada, H.; Iida, M.; Nanjo, H.; Itoh, H.; et al. Increased expression of co-chaperone HOP with HSP90 and HSC70 and complex formation in human colonic carcinoma. Cell Stress Chaperones 2010, 15, 1003–1011. [Google Scholar] [CrossRef]
- Wu, R.; Liu, F.; Peng, P.; Qiu, H.; Xiong, H.; Yu, S.; Huang, X.; Zhang, H.; Zhuang, L. Tumor stress-induced phosphoprotein 1 as a prognostic biomarker for breast cancer. Ann. Transl. Med. 2018, 6, 302. [Google Scholar] [CrossRef]
- Padden, J.; Megger, D.A.; Bracht, T.; Reis, H.; Ahrens, M.; Kohl, M.; Eisenacher, M.; Schlaak, J.F.; Canbay, A.E.; Weber, F.; et al. Identification of novel biomarker candidates for the immunohistochemical diagnosis of cholangiocellular carcinoma. Mol. Cell Proteom. 2014, 13, 2661–2672. [Google Scholar] [CrossRef]
- Walsh, N.; Larkin, A.; Swan, N.; Conlon, K.; Dowling, P.; McDermott, R.; Clynes, M. RNAi knockdown of Hop (Hsp70/Hsp90 organising protein) decreases invasion via MMP-2 down regulation. Cancer Lett. 2011, 306, 180–189. [Google Scholar] [CrossRef]
- Wang, J.H.; Gong, C.; Guo, F.J.; Zhou, X.; Zhang, M.S.; Qiu, H.; Chao, T.F.; Liu, Y.; Qin, L.; Xiong, H.H. Knockdown of STIP1 inhibits the invasion of CD133positive cancer stemlike cells of the osteosarcoma MG63 cell line via the PI3K/Akt and ERK1/2 pathways. Int. J. Mol. Med. 2020, 46, 2251–2259. [Google Scholar] [CrossRef]
- Jing, Y.; Liang, W.; Liu, J.; Zhang, L.; Wei, J.; Zhu, Y.; Yang, J.; Ji, K.; Zhang, Y.; Huang, Z. Stress-induced phosphoprotein 1 promotes pancreatic cancer progression through activation of the FAK/AKT/MMP signaling axis. Pathol. Res. Pract. 2019, 215, 152564. [Google Scholar] [CrossRef]
- Tsai, C.L.; Chao, A.S.; Jung, S.M.; Lin, C.Y.; Chao, A.; Wang, T.H. Stress-induced phosphoprotein 1 acts as a scaffold protein for glycogen synthase kinase-3 beta-mediated phosphorylation of lysine-specific demethylase 1. Oncogenesis 2018, 7, 31. [Google Scholar] [CrossRef]
- Zhang, S.; Shao, J.; Su, F. Prognostic significance of STIP1 expression in human cancer: A meta-analysis. Clin. Chim. Acta 2018, 486, 168–176. [Google Scholar] [CrossRef]
- Tsai, C.L.; Tsai, C.N.; Lin, C.Y.; Chen, H.W.; Lee, Y.S.; Chao, A.; Wang, T.H.; Wang, H.S.; Lai, C.H. Secreted stress-induced phosphoprotein 1 activates the ALK2-SMAD signaling pathways and promotes cell proliferation of ovarian cancer cells. Cell Rep. 2012, 2, 283–293. [Google Scholar] [CrossRef]
- Chen, Z.; Xu, L.; Su, T.; Ke, Z.; Peng, Z.; Zhang, N.; Peng, S.; Zhang, Q.; Liu, G.; Wei, G.; et al. Autocrine STIP1 signaling promotes tumor growth and is associated with disease outcome in hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2017, 493, 365–372. [Google Scholar] [CrossRef]
- Leonard, W.J.; O’Shea, J.J. Jaks and STATs: Biological implications. Annu. Rev. Immunol. 1998, 16, 293–322. [Google Scholar] [CrossRef]
- Kumar, N.; Kuang, L.; Villa, R.; Kumar, P.; Mishra, J. Mucosal Epithelial Jak Kinases in Health and Diseases. Mediat. Inflamm. 2021, 2021, 6618924. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Jaskiewicz, A.; Domoradzki, T.; Pajak, B. Targeting the JAK2/STAT3 Pathway-Can We Compare It to the Two Faces of the God Janus? Int. J. Mol. Sci. 2020, 21, 8261. [Google Scholar] [CrossRef]
- Tsai, C.L.; Chao, A.; Jung, S.M.; Tsai, C.N.; Lin, C.Y.; Chen, S.H.; Sue, S.C.; Wang, T.H.; Wang, H.S.; Lai, C.H. Stress-induced phosphoprotein-1 maintains the stability of JAK2 in cancer cells. Oncotarget 2016, 7, 50548–50563. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Yan, Z.; Zhang, G.; Wang, X.; Pan, Y.; Huang, M. STIP1 Regulates Proliferation and Migration of Lung Adenocarcinoma Through JAK2/STAT3 Signaling Pathway. Cancer Manag. Res. 2019, 11, 10061–10072. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Wen, J.; Lin, B.; Xia, E.; Zheng, C.; Ye, L.; Wang, Y.; Wang, O.; Chen, Y. Stress-induced phosphoprotein 1 facilitates breast cancer cell progression and indicates poor prognosis for breast cancer patients. Hum. Cell 2021, 34, 901–917. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Cao, N.; Mu, L.; Cao, W. Stress induced phosphoprotein 1 promotes tumor growth and metastasis of melanoma via modulating JAK2/STAT3 pathway. Biomed Pharmacother. 2019, 116, 108962. [Google Scholar] [CrossRef] [PubMed]
- Vainchenker, W.; Constantinescu, S.N. JAK/STAT signaling in hematological malignancies. Oncogene 2013, 32, 2601–2613. [Google Scholar] [CrossRef]
- Morelli, A.P.; Tortelli, T.C., Jr.; Mancini, M.C.S.; Pavan, I.C.B.; Silva, L.G.S.; Severino, M.B.; Granato, D.C.; Pestana, N.F.; Ponte, L.G.S.; Peruca, G.F.; et al. STAT3 contributes to cisplatin resistance, modulating EMT markers, and the mTOR signaling in lung adenocarcinoma. Neoplasia 2021, 23, 1048–1058. [Google Scholar] [CrossRef]
- Liang, R.; Chen, X.; Chen, L.; Wan, F.; Chen, K.; Sun, Y.; Zhu, X. STAT3 signaling in ovarian cancer: A potential therapeutic target. J. Cancer 2020, 11, 837–848. [Google Scholar] [CrossRef]
- Chaitanya, G.V.; Steven, A.J.; Babu, P.P. PARP-1 cleavage fragments: Signatures of cell-death proteases in neurodegeneration. Cell Commun. Signal. 2010, 8, 31. [Google Scholar] [CrossRef]
- Soares, I.N.; Caetano, F.A.; Pinder, J.; Rodrigues, B.R.; Beraldo, F.H.; Ostapchenko, V.G.; Durette, C.; Pereira, G.S.; Lopes, M.H.; Queiroz-Hazarbassanov, N.; et al. Regulation of stress-inducible phosphoprotein 1 nuclear retention by protein inhibitor of activated STAT PIAS1. Mol. Cell Proteom. 2013, 12, 3253–3270. [Google Scholar] [CrossRef]
- Ma, X.L.; Tang, W.G.; Yang, M.J.; Xie, S.H.; Wu, M.L.; Lin, G.; Lu, R.Q. Serum STIP1, a Novel Indicator for Microvascular Invasion, Predicts Outcomes and Treatment Response in Hepatocellular Carcinoma. Front Oncol. 2020, 10, 511. [Google Scholar] [CrossRef]
- Lackie, R.E.; Maciejewski, A.; Ostapchenko, V.G.; Marques-Lopes, J.; Choy, W.Y.; Duennwald, M.L.; Prado, V.F.; Prado, M.A.M. The Hsp70/Hsp90 Chaperone Machinery in Neurodegenerative Diseases. Front Neurosci. 2017, 11, 254. [Google Scholar] [CrossRef]
- Wang, J.; You, H.; Qi, J.; Yang, C.; Ren, Y.; Cheng, H. Autocrine and paracrine STIP1 signaling promote osteolytic bone metastasis in renal cell carcinoma. Oncotarget 2017, 8, 17012–17026. [Google Scholar] [CrossRef][Green Version]
- Zhang, X.; Hu, F.; Li, G.; Li, G.; Yang, X.; Liu, L.; Zhang, R.; Zhang, B.; Feng, Y. Human colorectal cancer-derived mesenchymal stem cells promote colorectal cancer progression through IL-6/JAK2/STAT3 signaling. Cell Death Dis. 2018, 9, 25. [Google Scholar] [CrossRef]
- Park, S.Y.; Lee, C.J.; Choi, J.H.; Kim, J.H.; Kim, J.W.; Kim, J.Y.; Nam, J.S. The JAK2/STAT3/CCND2 Axis promotes colorectal Cancer stem cell persistence and radioresistance. J. Exp. Clin. Cancer Res. 2019, 38, 399. [Google Scholar] [CrossRef]
- Abubaker, K.; Luwor, R.B.; Zhu, H.; McNally, O.; Quinn, M.A.; Burns, C.J.; Thompson, E.W.; Findlay, J.K.; Ahmed, N. Inhibition of the JAK2/STAT3 pathway in ovarian cancer results in the loss of cancer stem cell-like characteristics and a reduced tumor burden. BMC Cancer 2014, 14, 317. [Google Scholar] [CrossRef]
- Qureshy, Z.; Johnson, D.E.; Grandis, J.R. Targeting the JAK/STAT pathway in solid tumors. J. Cancer Metastasis Treat. 2020, 6, 27. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Morgan, E.L.; Macdonald, A. JAK2 Inhibition Impairs Proliferation and Sensitises Cervical Cancer Cells to Cisplatin-Induced Cell Death. Cancers 2019, 11, 1934. [Google Scholar] [CrossRef]
- Wang, M.; Lin, T.; Wang, Y.; Gao, S.; Yang, Z.; Hong, X.; Chen, G. CXCL12 suppresses cisplatin-induced apoptosis through activation of JAK2/STAT3 signaling in human non-small-cell lung cancer cells. Onco Targets Ther. 2017, 10, 3215–3224. [Google Scholar] [CrossRef]
- Danni, X.; Jiangzheng, Z.; Huamao, S.; Yinglian, P.; Changcheng, Y.; Yanda, L. Chaperonin containing TCP1 subunit 3 (CCT3) promotes cisplatin resistance of lung adenocarcinoma cells through targeting the Janus kinase 2/signal transducers and activators of transcription 3 (JAK2/STAT3) pathway. Bioengineered 2021, 12, 7335–7347. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.S.; Li, J.; Chen, E.; Kent, D.G.; Park, H.J.; Green, A.R. JAK2V617F mediates resistance to DNA damage-induced apoptosis by modulating FOXO3A localization and Bcl-xL deamidation. Oncogene 2016, 35, 2235–2246. [Google Scholar] [CrossRef] [PubMed]
- Hoter, A.; El-Sabban, M.E.; Naim, H.Y. The HSP90 Family: Structure, Regulation, Function, and Implications in Health and Disease. Int. J. Mol. Sci. 2018, 19, 2560. [Google Scholar] [CrossRef] [PubMed]
- Sreedhar, A.S.; Kalmar, E.; Csermely, P.; Shen, Y.F. Hsp90 isoforms: Functions, expression and clinical importance. FEBS Lett. 2004, 562, 11–15. [Google Scholar] [CrossRef]
- Bhattacharya, K.; Picard, D. The Hsp70-Hsp90 go-between Hop/Stip1/Sti1 is a proteostatic switch and may be a drug target in cancer and neurodegeneration. Cell Mol. Life Sci. 2021, 78, 7257–7273. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.J.; Yao, J.; Si, J.M. Nuclear JAK2: Form and function in cancer. Anat. Rec. 2011, 294, 1446–1459. [Google Scholar] [CrossRef]
- Ding, N.; Miller, S.A.; Savant, S.S.; O’Hagan, H.M. JAK2 regulates mismatch repair protein-mediated epigenetic alterations in response to oxidative damage. Environ. Mol. Mutagen. 2019, 60, 308–319. [Google Scholar] [CrossRef]
- Hajj, G.N.; Arantes, C.P.; Dias, M.V.; Roffe, M.; Costa-Silva, B.; Lopes, M.H.; Porto-Carreiro, I.; Rabachini, T.; Lima, F.R.; Beraldo, F.H.; et al. The unconventional secretion of stress-inducible protein 1 by a heterogeneous population of extracellular vesicles. Cell Mol. Life Sci. 2013, 70, 3211–3227. [Google Scholar] [CrossRef]
- Caetano, F.A.; Lopes, M.H.; Hajj, G.N.; Machado, C.F.; Pinto Arantes, C.; Magalhaes, A.C.; Vieira Mde, P.; Americo, T.A.; Massensini, A.R.; Priola, S.A.; et al. Endocytosis of prion protein is required for ERK1/2 signaling induced by stress-inducible protein 1. J. Neurosci. 2008, 28, 6691–6702. [Google Scholar] [CrossRef]
- Weidenauer, L.; Quadroni, M. Phosphorylation in the Charged Linker Modulates Interactions and Secretion of Hsp90beta. Cells 2021, 10, 1701. [Google Scholar] [CrossRef]
- Marubayashi, S.; Koppikar, P.; Taldone, T.; Abdel-Wahab, O.; West, N.; Bhagwat, N.; Caldas-Lopes, E.; Ross, K.N.; Gonen, M.; Gozman, A.; et al. HSP90 is a therapeutic target in JAK2-dependent myeloproliferative neoplasms in mice and humans. J. Clin. Investig. 2010, 120, 3578–3593. [Google Scholar] [CrossRef]
- Fridman, J.S.; Sarlis, N.J. The interplay between inhibition of JAK2 and HSP90. JAK-STAT 2012, 1, 77–79. [Google Scholar] [CrossRef][Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequence (5′→3′) |
|---|---|
| JAK2 V617 F | TGGAGTATGTTTCTGTGGAGAC |
| JAK2 V617 R | TAATTTAAAACCAAATGCTTGTG |
| STIP1 Y134A F | GCCTAATCTGGCTCAGAAGTTGGAG |
| STIP1 Y134A R | ATGTTGAAAGGGTTCATGAATTTTC |
| STIP1 Y152A F | TGATCCTACCGCCCGGGAGCTG |
| STIP1 Y152A R | CTGAGTAGTGTCCTTGTC |
| STIP1 Y296A F | CCGAGAAGACGCTCGACAGATTGC |
| STIP1 Y296A R | TTTTCTCTCCCCACTTCAATG |
| STIP1 Y303A F | TGCCAAAGCAGCTGCTCGAATTG |
| STIP1 Y303A R | ATCTGTCGATAGTCTTCTC |
| STIP1 Y404A F | AGCTGCCTGCGCCACCAAACTC |
| STIP1 Y404A R | CGATTGCTGTATAATTTGGC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chao, A.; Liao, M.-J.; Chen, S.-H.; Lee, Y.-S.; Tsai, C.-N.; Lin, C.-Y.; Tsai, C.-L. JAK2-Mediated Phosphorylation of Stress-Induced Phosphoprotein-1 (STIP1) in Human Cells. Int. J. Mol. Sci. 2022, 23, 2420. https://doi.org/10.3390/ijms23052420
Chao A, Liao M-J, Chen S-H, Lee Y-S, Tsai C-N, Lin C-Y, Tsai C-L. JAK2-Mediated Phosphorylation of Stress-Induced Phosphoprotein-1 (STIP1) in Human Cells. International Journal of Molecular Sciences. 2022; 23(5):2420. https://doi.org/10.3390/ijms23052420
Chicago/Turabian StyleChao, Angel, Min-Jie Liao, Shun-Hua Chen, Yun-Shien Lee, Chi-Neu Tsai, Chiao-Yun Lin, and Chia-Lung Tsai. 2022. "JAK2-Mediated Phosphorylation of Stress-Induced Phosphoprotein-1 (STIP1) in Human Cells" International Journal of Molecular Sciences 23, no. 5: 2420. https://doi.org/10.3390/ijms23052420
APA StyleChao, A., Liao, M.-J., Chen, S.-H., Lee, Y.-S., Tsai, C.-N., Lin, C.-Y., & Tsai, C.-L. (2022). JAK2-Mediated Phosphorylation of Stress-Induced Phosphoprotein-1 (STIP1) in Human Cells. International Journal of Molecular Sciences, 23(5), 2420. https://doi.org/10.3390/ijms23052420