
Statins Aggravate the Risk of Insulin Resistance in Human Muscle

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Long-Term Statin-Treatment Activates AMPKα in Human Skeletal Muscle
2.2. Short-Term Statin Treatment in Human Primary Myotubes Affects Glucose Metabolism via AMPKα Similar to the Situation in Human Skeletal Muscle
3. Discussion
4. Materials and Methods
4.1. Human Primary Myoblasts
4.2. Reagents, Buffers, and Antibodies
4.3. Glucose Uptake in Primary Muscle Cell Populations
4.4. Immunofluorescence
4.5. Protein Expression Using Immunoblot and Sandwich ELISA
4.6. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ward, N.C.; Watts, G.F.; Eckel, R.H. Statin Toxicity. Mechanistic Insights and Clinical Implications. Circ. Res. 2019, 124, 328–350. [Google Scholar] [CrossRef] [PubMed]
- Thakker, D.; Nair, S.; Pagada, A.; Jamdade, V.; Malik, A. Statin use and the risk of developing diabetes: A network meta-analysis. Pharmacoepidemiol. Drug Saf. 2016, 25, 1131–1149. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.U.; Rahman, H.; Okunrintemi, V.; Riaz, H.; Khan, M.S.; Sattur, S.; Kaluski, E.; Lincoff, A.M.; Martin, S.S.; Blaha, M.J. Association of Lowering Low-Density Lipoprotein Cholesterol With Contemporary Lipid-Lowering Therapies and Risk of Diabetes Mellitus: A Systematic Review and Meta-Analysis. J. Am. Heart Assoc. 2019, 8, e011581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinton, E.A. Statin-Related New-Onset Diabetes Appears Driven by Increased Insulin Resistance: Are There Clinical Implications? Arter. Thromb. Vasc. Biol. 2021, 41, 2798–2801. [Google Scholar] [CrossRef]
- Ko, M.J.; Jo, A.J.; Kim, Y.J.; Kang, S.H.; Cho, S.; Jo, S.; Park, C.; Yun, S.; Lee, W.J.; Park, D. Time- and Dose-Dependent Association of Statin Use With Risk of Clinically Relevant New-Onset Diabetes Mellitus in Primary Prevention: A Nationwide Observational Cohort Study. J. Am. Heart Assoc. 2019, 8, e011320. [Google Scholar] [CrossRef]
- Ridker, P.M.; Silvertown, J.D. Inflammation, C-Reactive Protein, and Atherothrombosis. J. Periodontol. 2008, 79, 1544–1551. [Google Scholar] [CrossRef] [Green Version]
- Cederberg, H.; Stančáková, A.; Yaluri, N.; Modi, S.R.; Kuusisto, J.; Laakso, M. Increased risk of diabetes with statin treatment is associated with impaired insulin sensitivity and insulin secretion: A 6 year follow-up study of the METSIM cohort. Diabetologia 2015, 58, 1109–1117. [Google Scholar] [CrossRef] [Green Version]
- Sattar, N.; Preiss, D.; Murray, H.M.; Welsh, P.; Buckley, B.M.; de Craen, A.J.M.; Seshasai, S.R.K.; Mcmurray, J.J.; Freeman, D.J.; Jukema, J.W.; et al. Statins and risk of incident diabetes: A collaborative meta-analysis of randomised statin trials. Lancet 2010, 375, 735–742. [Google Scholar] [CrossRef]
- Crandall, J.P.; Mather, K.; Rajpathak, S.N.; Goldberg, R.B.; Watson, K.; Foo, S.; Ratner, R.; Barrett-Connor, E.; Temprosa, M. Statin use and risk of developing diabetes: Results from the Diabetes Prevention Program. BMJ Open Diabetes Res. Care 2017, 5, e000438. [Google Scholar] [CrossRef] [Green Version]
- Preiss, D.; Seshasai, S.R.K.; Welsh, P.; Murphy, S.A.; Ho, J.; Waters, D.D.; Demicco, D.A.; Barter, P.; Cannon, C.P.; Sabatine, M.S.; et al. Risk of Incident Diabetes With Intensive-Dose Compared With Moderate-Dose Statin Therapy. JAMA 2011, 305, 2556–2564. [Google Scholar] [CrossRef] [Green Version]
- Betteridge, D.J.; Carmena, R. The diabetogenic action of statins—Mechanisms and clinical implications. Nat. Rev. Endocrinol. 2016, 12, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Ray, K.K.; Wiklund, O.; Corsini, A.; Catapano, A.L.; Bruckert, E.; De Backer, G.; A Hegele, R.; Hovingh, G.K.; A Jacobson, T.; et al. Adverse effects of statin therapy: Perception vs. the evidence—Focus on glucose homeostasis, cognitive, renal and hepatic function, haemorrhagic stroke and cataract. Eur. Hear. J. 2018, 39, 2526–2539. [Google Scholar] [CrossRef] [PubMed]
- Baron, A.D.; Brechtel, G.; Wallace, P.; Edelman, S.V. Rates and tissue sites of non-insulin- and insulin-mediated glucose uptake in humans. Am. J. Physiol. Metab. 1988, 255, E769–E774. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E.; Taddei, S.; Santoro, D.; Natali, A.; Boni, C.; Del Chiaro, D.; Buzzigoli, G. Independent stimulation of glucose metabolism and Na+-K+ exchange by insulin in the human forearm. Am. J. Physiol. Metab. 1988, 255, E953–E958. [Google Scholar] [CrossRef]
- Ryder, J.W.; Fahlman, R.; Wallberg-Henriksson, H.; Alessi, D.R.; Krook, A.; Zierath, J. Effect of Contraction on Mitogen-activated Protein Kinase Signal Transduction in Skeletal Muscle. J. Biol. Chem. 2000, 275, 1457–1462. [Google Scholar] [CrossRef] [Green Version]
- Abel, E.D.; Peroni, O.D.; Kim, J.K.; Kim, Y.-B.; Boss, O.; Hadro, E.; Minnemann, T.; Shulman, G.I.; Kahn, B.B. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature 2001, 409, 729–733. [Google Scholar] [CrossRef]
- Nakata, M.; Nagasaka, S.; Kusaka, I.; Matsuoka, H.; Ishibashi, S.; Yada, T. Effects of statins on the adipocyte maturation and expression of glucose transporter 4 (SLC2A4): Implications in glycaemic control. Diabetologia 2006, 49, 1881–1892. [Google Scholar] [CrossRef] [Green Version]
- Kotani, K.; Peroni, O.D.; Minokoshi, Y.; Boss, O.; Kahn, B.B. GLUT4 glucose transporter deficiency increases hepatic lipid production and peripheral lipid utilization. J. Clin. Investig. 2004, 114, 1666–1675. [Google Scholar] [CrossRef]
- Sanvee, G.M.; Panajatovic, M.V.; Bouitbir, J.; Krähenbühl, S. Mechanisms of insulin resistance by simvastatin in C2C12 myotubes and in mouse skeletal muscle. Biochem. Pharmacol. 2019, 164, 23–33. [Google Scholar] [CrossRef]
- Seshadri, S.; Rapaka, N.; Prajapati, B.; Mandaliya, D.; Patel, S.; Muggalla, C.S.; Kapadia, B.; Babu, P.P.; Misra, P.; Saxena, U. Statins exacerbate glucose intolerance and hyperglycemia in a high sucrose fed rodent model. Sci. Rep. 2019, 9, 8825. [Google Scholar] [CrossRef] [Green Version]
- Dugani, S.B.; Akinkuolie, A.O.; Paynter, N.; Glynn, R.J.; Ridker, P.M.; Mora, S. Association of Lipoproteins, Insulin Resistance, and Rosuvastatin With Incident Type 2 Diabetes Mellitus. JAMA Cardiol. 2016, 1, 136–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kain, V.; Kapadia, B.; Misra, P.; Saxena, U. Simvastatin may induce insulin resistance through a novel fatty acid mediated cholesterol independent mechanism. Sci. Rep. 2015, 5, srep13823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, K.K.; Quon, M.; Han, S.H.; Lee, Y.; Kim, S.J.; Shin, E.K. Atorvastatin Causes Insulin Resistance and Increases Ambient Glycemia in Hypercholesterolemic Patients. J. Am. Coll. Cardiol. 2010, 55, 1209–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbasi, F.; Lamendola, C.; Harris, C.S.; Harris, V.; Tsai, M.-S.; Tripathi, P.; Abbas, F.; Reaven, G.M.; Reaven, P.D.; Snyder, M.P.; et al. Statins Are Associated With Increased Insulin Resistance and Secretion. Arter. Thromb. Vasc. Biol. 2021, 41, 2786–2797. [Google Scholar] [CrossRef] [PubMed]
- Kohli, P.; Waters, D.D.; Nemr, R.; Arsenault, B.J.; Messig, M.; DeMicco, D.A.; Laskey, R.; Kastelein, J.J. Risk of New-Onset Diabetes and Cardiovascular Risk Reduction From High-Dose Statin Therapy in Pre-Diabetics and Non–Pre-Diabetics. J. Am. Coll. Cardiol. 2015, 65, 402–404. [Google Scholar] [CrossRef] [Green Version]
- Grunwald, S.A.; Popp, O.; Haafke, S.; Jedraszczak, N.; Grieben, U.; Saar, K.; Patone, G.; Kress, W.; Steinhagen-Thiessen, E.; Dittmar, G.; et al. Statin-induced myopathic changes in primary human muscle cells and reversal by a prostaglandin F2 alpha analogue. Sci. Rep. 2020, 10, 2158. [Google Scholar] [CrossRef] [Green Version]
- Lalli, C.A.; Pauli, J.R.; Prada, P.D.O.; Cintra, D.; Ropelle, E.; Velloso, L.A.; Saad, M.J.A. Statin modulates insulin signaling and insulin resistance in liver and muscle of rats fed a high-fat diet. Metabolism 2008, 57, 57–65. [Google Scholar] [CrossRef]
- Olofsson, S.-O.; Andersson, L.; Håversen, L.; Olsson, C.; Myhre, S.; Rutberg, M.; Mobini, R.; Li, L.; Lu, E.; Borén, J.; et al. The formation of lipid droplets: Possible role in the development of insulin resistance/type 2 diabetes. Prostaglandins Leukot. Essent. Fat. Acids 2011, 85, 215–218. [Google Scholar] [CrossRef]
- PPhillips, P.S.; Haas, R.H.; Bannykh, S.; Hathaway, S.; Gray, N.L.; Kimura, B.J.; Vladutiu, G.D.; England, J.D.; Scripps Mercy Clinical Research Center. Statin-Associated Myopathy with Normal Creatine Kinase Levels. Ann. Intern. Med. 2002, 137, 581. [Google Scholar] [CrossRef]
- Randle, P.; Garland, P.; Hales, C.; Newsholme, E. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 1, 785–789. [Google Scholar] [CrossRef]
- Clarke, P.R.; Hardie, D.G. Regulation of HMG-CoA reductase: Identification of the site phosphorylated by the AMP-activated protein kinase in vitro and in intact rat liver. EMBO J. 1990, 9, 2439–2446. Available online: http://www.ncbi.nlm.nih.gov/pubmed/2369897 (accessed on 3 September 2019). [CrossRef] [PubMed]
- Habegger, K.M.; Hoffman, N.; Ridenour, C.M.; Brozinick, J.T.; Elmendorf, J.S. AMPK Enhances Insulin-Stimulated GLUT4 Regulation via Lowering Membrane Cholesterol. Endocrinology 2012, 153, 2130–2141. [Google Scholar] [CrossRef] [PubMed]
- Takaguri, A.; Satoh, K.; Itagaki, M.; Tokumitsu, Y.; Ichihara, K. Effects of atorvastatin and pravastatin on signal transduction related to glucose uptake in 3T3L1 adipocytes. J. Pharmacol. Sci. 2008, 107, 80–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parpal, S.; Karlsson, M.; Thorn, H.; Strålfors, P. Cholesterol Depletion Disrupts Caveolae and Insulin Receptor Signaling for Metabolic Control via Insulin Receptor Substrate-1, but Not for Mitogen-activated Protein Kinase Control. J. Biol. Chem. 2001, 276, 9670–9678. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, M.; Thorn, H.; Parpal, S.; Strålfors, P.; Gustavsson, J. Insulin induces translocation of glucose transporter GLUT4 to plasma membrane caveolae in adipocytes. FASEB J. 2002, 16, 249–251. [Google Scholar] [CrossRef]
- Shang, L.; Chen, T.; Deng, Y.; Huang, Y.; Huang, Y.; Xian, J.; Lu, W.; Yang, L.; Huang, Q. Caveolin-3 promotes glycometabolism, growth and proliferation in muscle cells. PLoS ONE 2017, 12, e0189004. [Google Scholar] [CrossRef] [Green Version]
- Kotani, K.; Ogawa, W.; Hashiramoto, M.; Onishi, T.; Ohno, S.; Kasuga, M. Inhibition of Insulin-induced Glucose Uptake by Atypical Protein Kinase C Isotype-specific Interacting Protein in 3T3-L1 Adipocytes. J. Biol. Chem. 2000, 275, 26390–26395. [Google Scholar] [CrossRef] [Green Version]
- Crossland, H.; Constantin-Teodosiu, D.; Gardiner, S.M.; Constantin, D.; Greenhaff, P. A potential role for Akt/FOXO signalling in both protein loss and the impairment of muscle carbohydrate oxidation during sepsis in rodent skeletal muscle. J. Physiol. 2008, 586, 5589–5600. [Google Scholar] [CrossRef]
- Schiaffino, S.; Mammucari, C. Regulation of skeletal muscle growth by the IGF1-Akt/PKB pathway: Insights from genetic models. Skelet. Muscle 2011, 1, 4. [Google Scholar] [CrossRef]
- Bonifacio, A.; Sanvee, G.M.; Bouitbir, J.; Krähenbühl, S. The AKT/mTOR signaling pathway plays a key role in statin-induced myotoxicity. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 1841–1849. [Google Scholar] [CrossRef] [Green Version]
- Mullen, P.J.; Lüscher, B.; Scharnagl, H.; Krähenbühl, S.; Brecht, K. Effect of simvastatin on cholesterol metabolism in C2C12 myotubes and HepG2 cells, and consequences for statin-induced myopathy. Biochem. Pharmacol. 2010, 79, 1200–1209. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Wada, I.; Kimura, J. Inhibition of Rab1 GTPase and Endoplasmic Reticulum-to-Golgi Trafficking Underlies Statin’s Toxicity in Rat Skeletal Myofibers. J. Pharmacol. Exp. Ther. 2011, 338, 62–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gendaszewska-Darmach, E.; Garstka, M.A.; Błażewska, K.M. Targeting Small GTPases and Their Prenylation in Diabetes Mellitus. J. Med. Chem. 2021, 64, 9677–9710. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.-P.; Zhao, W. Different effects of statins on induction of diabetes mellitus: An experimental study. Drug Des. Dev. Ther. 2015, 9, 6211–6223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, M.; Van Der Donk, W.A.; Chen, J. Lanthionine synthetase C–like protein 2 (LanCL2) is a novel regulator of Akt. Mol. Biol. Cell 2014, 25, 3954–3961. [Google Scholar] [CrossRef]
- Carmena, R.; Betteridge, D.J. Diabetogenic Action of Statins: Mechanisms. Curr. Atheroscler. Rep. 2019, 21, 23. [Google Scholar] [CrossRef]
- Hyder, T.; Marti, J.L.G.; Nasrazadani, A.; Brufsky, A.M. Statins and endocrine resistance in breast cancer. Cancer Drug Resist 2021, 4, 356–364. [Google Scholar] [CrossRef]
- Sakamoto, K.; Holman, G.D. Emerging role for AS160/TBC1D4 and TBC1D1 in the regulation of GLUT4 traffic. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E29–E37. [Google Scholar]
- Alexandrov, K.; Wu, Y.; Blankenfeldt, W.; Waldmann, H.; Goody, R.S. 8—Organization and Function of the Rab Prenylation and Recycling Machinery. In The Enzymes; Academic Press: Cambridge, MA, USA, 2011; Volume 29, pp. 147–162. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Weber-Carstens, S.; Schneider, J.; Wollersheim, T.; Assmann, A.; Bierbrauer, J.; Marg, A.; Al Hasani, H.; Chadt, A.; Wenzel, K.; Koch, S.; et al. Critical Illness Myopathy and GLUT4. Am. J. Respir. Crit. Care Med. 2013, 187, 387–396. [Google Scholar] [CrossRef]
- Marg, A.; Schoewel, V.; Timmel, T.; Schulze, A.; Shah, C.; Daumke, O.; Spuler, S. Sarcolemmal Repair Is a Slow Process and Includes EHD2. Traffic 2012, 13, 1286–1294. [Google Scholar] [CrossRef] [PubMed]
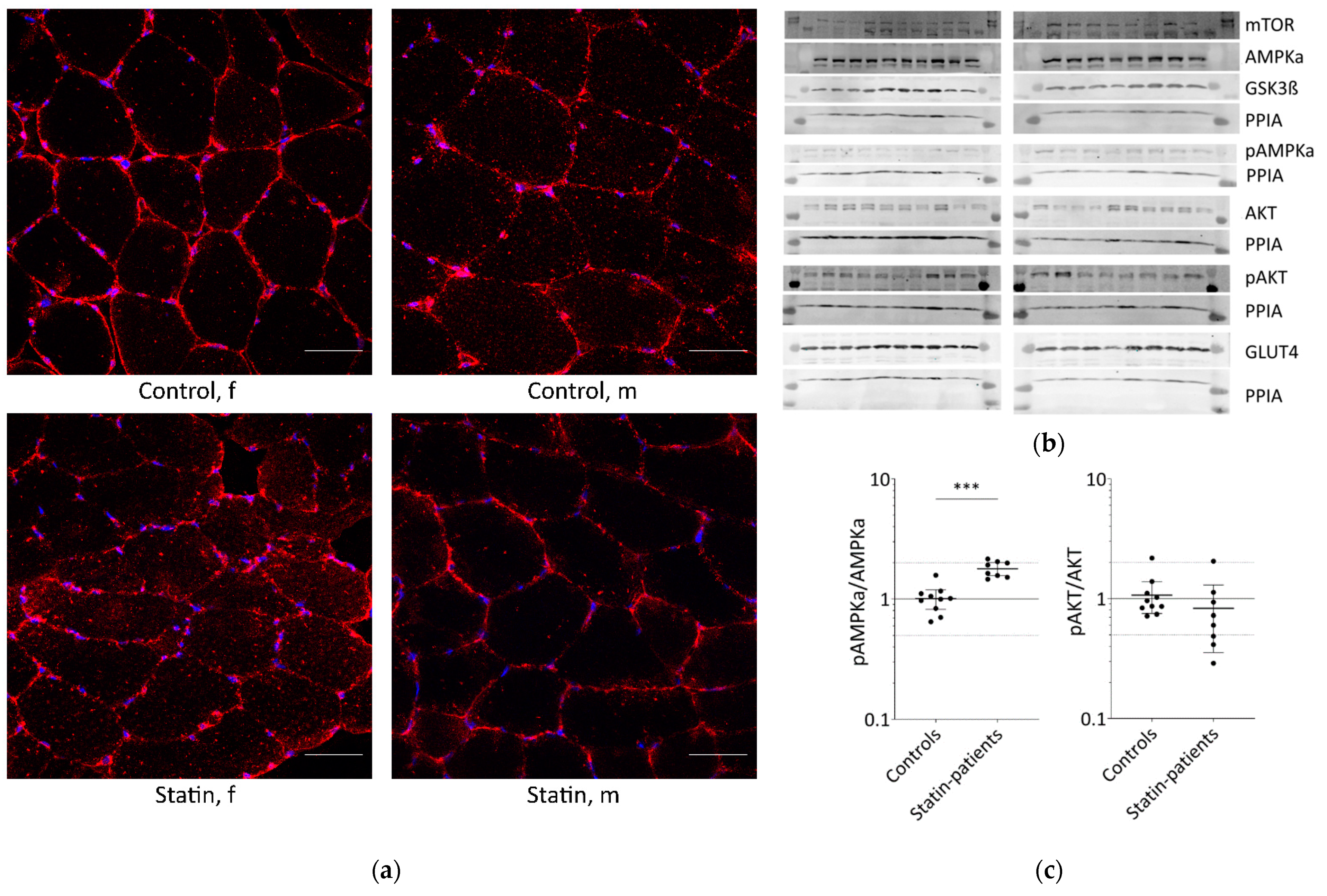


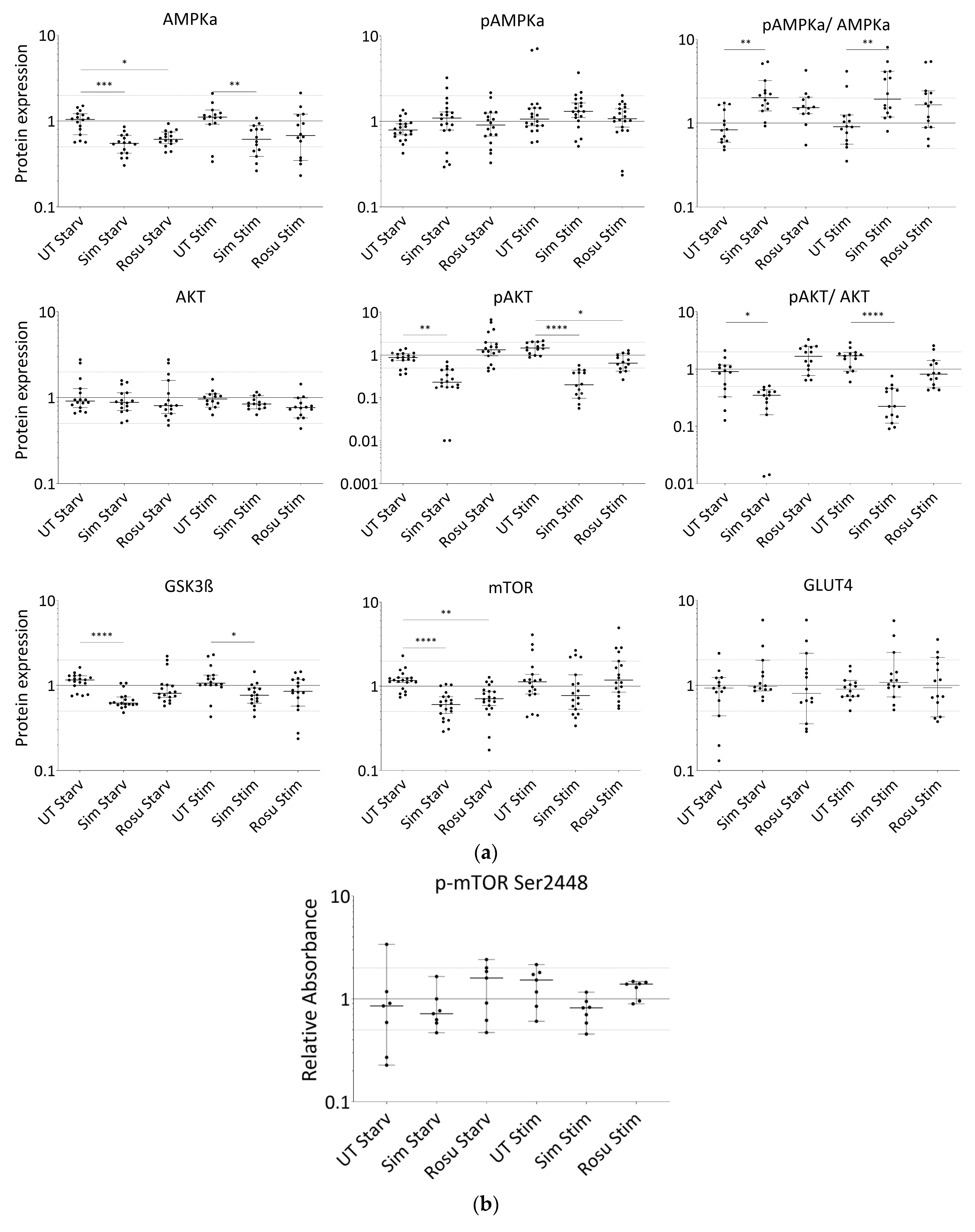


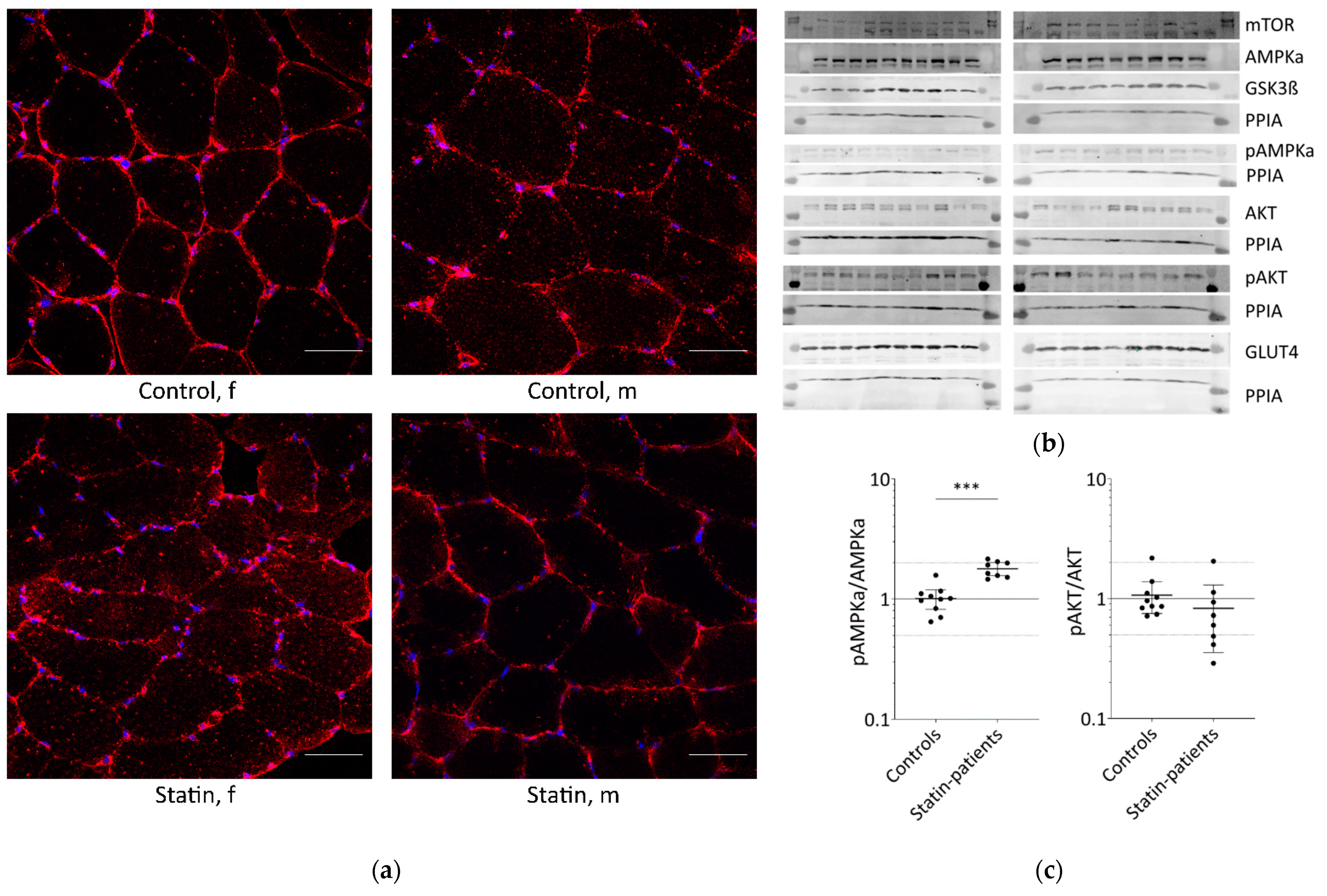{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Gender of Donor | Age of Donor | Medical History of Donor | Myopathological Findings in Biopsy Specimen |
|---|---|---|---|---|
| C1 | f | 47 | No statins | Normal # |
| C2 | f | 54 | No statins, myalgia of unknown origin, CK normal | Normal |
| C3 § | f | 51 | No statins, no myalgia, CK normal | Normal# |
| C4 § | f | 59 | No neuromuscular symptom, hip replacement surgery | Normal |
| C5 § | f | 51 | No statins, myalgia of unknown origin, CK normal | Normal; increased intracellular lipid droplets |
| C6 | f | 56 | No statins, myalgia of unknown origin, CK normal | Normal, rare atrophic fibers |
| C7 | m | 65 | No statins | Normal |
| C8 | m | 64 | No statins | Normal |
| C9 | m | 66 | No statins | Normal |
| C10 § | m | 67 | No neuromuscular symptom, hip replacement surgery | Normal |
| C11 § | m | 50 | No statins, myalgia of unknown origin, CK = 290 U/L | Normal; minor fiber type grouping # |
| C12 § | m | 56 | No statins, myalgia of unknown origin, CK = 513 U/L | Normal, rare central nuclei # |
| S1 | f | 58 | Statin myopathy, Pain rate = 4 Simvastatin 20 mg; CK = 254 U/L | Normal |
| S2 | f | 60 | Statin myopathy, Pain rate = 2 Simvastatin 20 mg; CK = 203 U/L | Normal |
| S3 | m | 71 | Statin myopathy, Pain rate = 2 Atorvastatin 10 mg; CK = 209 U/L | Normal |
| S4 | m | 64 | Statin myopathy, Pain rate = 3 Atorvastatin 10 mg; CK = 527 U/L | Normal |
| S5 | m | 62 | Statin myopathy, Pain rate = 4 Pravastatin 10 mg; CK = 688 U/L | Normal |
| S6 | m | 58 | Statin myopathy, Pain rate = 5 Atorvastatin 40 mg; CK= 430 U/L | Normal |
| S7 | m | 53 | Statin myopathy, Pain rate = 3 Pravastatin 5 mg; CK = 534 U/L | Normal |
| S8 § | m | 77 | Statin patient without side effects; Simvastatin 10 mg; CK = 195 U/L | Normal |
| Antibody | Supplier | Dilution |
|---|---|---|
| Akt (#9272) | Cell Signaling Technology (Beverly, MA, USA) | 1:2000 |
| phosphorylated Akt (Ser473; #9271) | 1:1000 | |
| AMPKα (#2603) | 1:1000 | |
| phosphorylated AMPKα (Thr172; #2535) | 1:1000 | |
| GSK3β (27C10) | 1:1000 | |
| mTOR (#2983) | 1:1000 | |
| Cyclophilin A/ PPIA (#ab41684) | Abcam (Cambridge, UK) | 1:5000 |
| GAPDH (#ab9484) | 1:10000 | |
| GLUT4 (#ab654); used in immunofluorescence staining and Western blotting with primary human myotubes | 1:3000 | |
| GLUT4 antiserum (1154 p); used in Western blot and immunohistochemical muscle tissue staining with human skeletal muscle tissue | kindly provided by Hoffmann-La Roche (Nutley, NJ, USA) to Hadi Al-Hasani and AG Schuermann (DIFE Potsdam, Nuthetal, Germany) | 1:1000 |
| CAV3 (sc-5310) | Santa Cruz (CA USA) | 1:150 |
| Alexa 488 and Alexa 568 | Invitrogen (Berlin, Germany) | 1:500 |
| Biotin goat anti-rabbit ab and Cy3-conjugated streptavidin | Jackson ImmunoResearch Laboratories (West Grove, PA, USA) | 1:200 |
| ECL™ IgG, HRP linked | GE-Healthcare (Waukesha, WI , USA) | 1:2000 |
| IRDye 800 | Rockland (Washington, DC, USA) | 1:5000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grunwald, S.A.; Haafke, S.; Grieben, U.; Kassner, U.; Steinhagen-Thiessen, E.; Spuler, S. Statins Aggravate the Risk of Insulin Resistance in Human Muscle. Int. J. Mol. Sci. 2022, 23, 2398. https://doi.org/10.3390/ijms23042398
Grunwald SA, Haafke S, Grieben U, Kassner U, Steinhagen-Thiessen E, Spuler S. Statins Aggravate the Risk of Insulin Resistance in Human Muscle. International Journal of Molecular Sciences. 2022; 23(4):2398. https://doi.org/10.3390/ijms23042398
Chicago/Turabian StyleGrunwald, Stefanie A., Stefanie Haafke, Ulrike Grieben, Ursula Kassner, Elisabeth Steinhagen-Thiessen, and Simone Spuler. 2022. "Statins Aggravate the Risk of Insulin Resistance in Human Muscle" International Journal of Molecular Sciences 23, no. 4: 2398. https://doi.org/10.3390/ijms23042398
APA StyleGrunwald, S. A., Haafke, S., Grieben, U., Kassner, U., Steinhagen-Thiessen, E., & Spuler, S. (2022). Statins Aggravate the Risk of Insulin Resistance in Human Muscle. International Journal of Molecular Sciences, 23(4), 2398. https://doi.org/10.3390/ijms23042398