
Diabetes and Ischemic Stroke: An Old and New Relationship an Overview of the Close Interaction between These Diseases

 , , , ,
, , , , 
Abstract
1. Introduction
2. Diabetes Mellitus and Atherosclerosis: Vascular Complications
2.1. Definition of Diabetes Mellitus
- Type 1 diabetes (T1D), characterized by pancreatic β-cell destruction with absolute insulin deficiency, usually caused by immunological disorders (this form includes LADA, latent autoimmune diabetes of the adults, with late-onset);
- Type 2 diabetes (T2D), with a broad spectrum of variations ranging from the prevailing insulin resistance with relative insulin deficit to predominant insulin secretion defect with insulin resistance;
- Gestational diabetes, connoted by the occurrence of diabetes during pregnancy and its resolution at the end of the gestational period, though some complications developed in this phase may be irreversible;
- Other specific diabetes types (including disorders of the exocrine pancreas like pancreatitis or cystic fibrosis, endocrinopathies like Cushing syndrome or pheochromocytoma, drug-induced by glucocorticoids or neuroleptics, genetic forms of β-cell dysfunction-MODY, genetic syndromes with altered glycemic metabolism, and infectious diseases) [2].
2.2. Definition of Atherosclerosis
2.3. Vascular Complications of Diabetes Mellitus
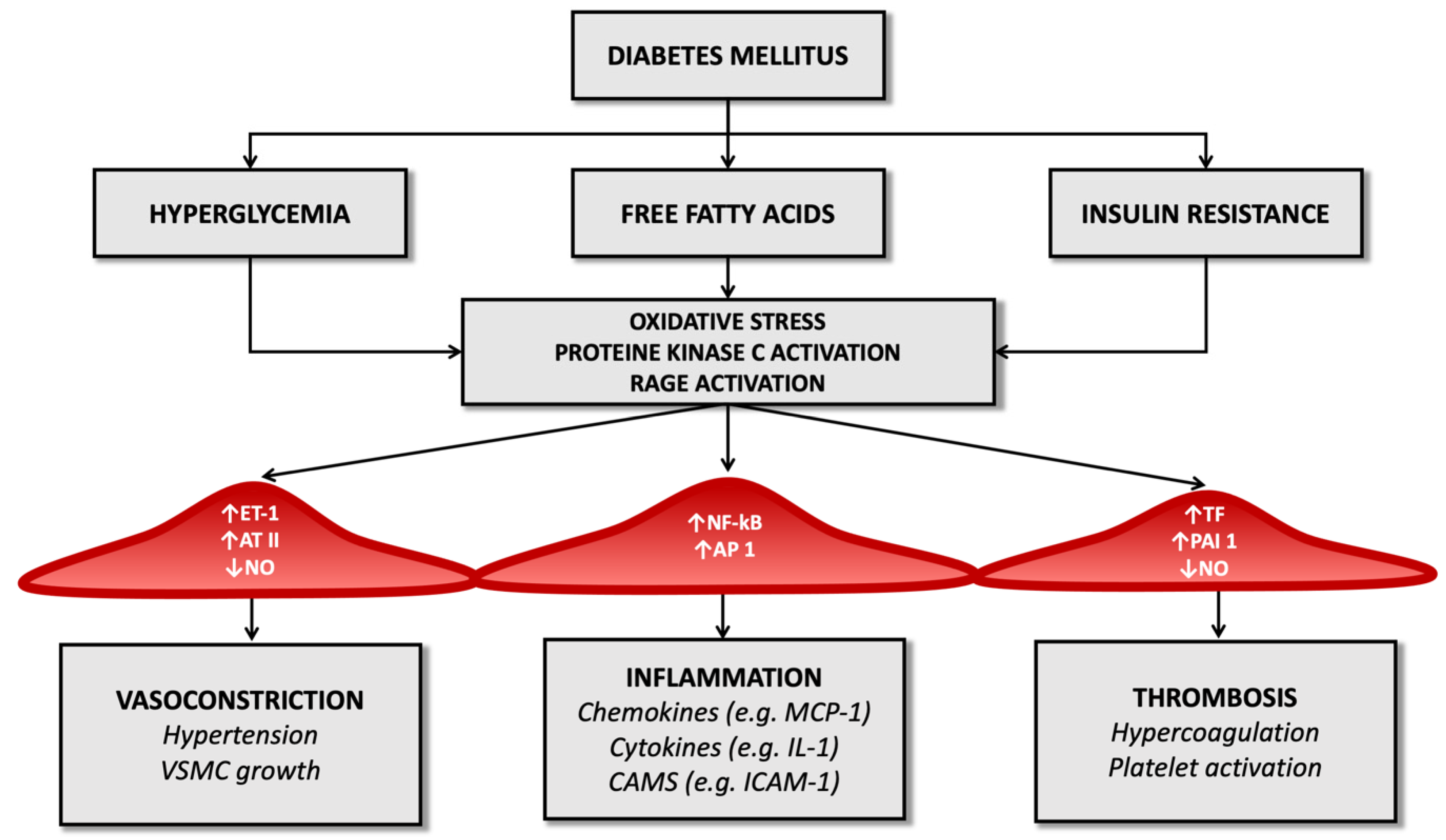
3. Molecular Pathology of Vascular Damage in Diabetes Mellitus and Atherosclerosis
4. Epidemiology of Ischemic Stroke in T2DM Patients
5. Most Common Stroke Subtypes in Diabetes
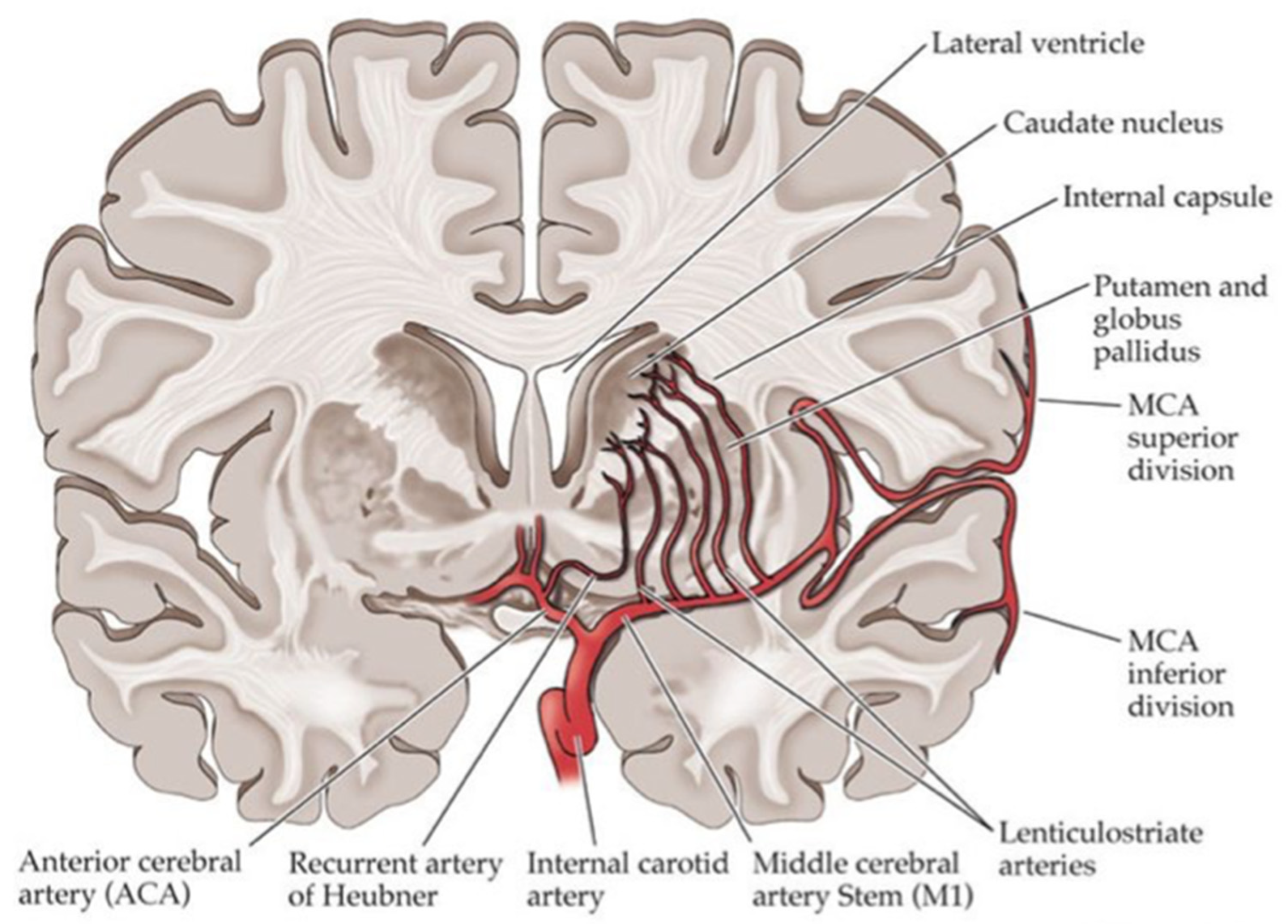
Small Vessel Disease in Diabetes Patients
- lacunae (formerly defined as silent lacunar infarcts);
- recent small subcortical infarcts (RSSI; formerly categorized as acute lacunar stroke);
- white matter hyperintensity (WMH);
- microbleeds;
- enlarged perivascular space.
- deep WMLs, which are separate from the cerebral ventricle and located in subcortical white matter;
- periventricular WMLs that are connected to the ventricular system.
6. Effects of Hyperglycemia during Acute Ischemic Stroke
7. Diabetic Foot Syndrome: A Burden of Cardiovascular and Cerebrovascular Risk in Diabetic Patients
7.1. Cardiovascular Morbidity and DFS
7.2. Immune-Inflammatory Features of DFS
8. Conclusions
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Petersmann, A.; Müller-Wieland, D.; Müller, U.A.; Landgraf, R.; Nauck, M.; Freckmann, G.; Heinemann, L.; Schleicher, E. Definition, Classification and Diagnosis of Diabetes Mellitus. Exp. Clin. Endocrinol. Diabetes 2019, 127, S1–S7. [Google Scholar] [CrossRef] [PubMed]
- Kerner, W.; Brückel, J.; German Diabetes Association. Definition, Classification and Diagnosis of Diabetes Mellitus. Exp. Clin. Endocrinol. Diabetes 2014, 122, 384–386. [Google Scholar] [CrossRef] [PubMed]
- Cerf, M.E. Beta Cell Dysfunction and Insulin Resistance. Front. Endocrinol. 2013, 4, 37. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global Aetiology and Epidemiology of Type 2 Diabetes Mellitus and Its Complications. Nat. Rev. Endocrinol. 2018, 14, 88–98. [Google Scholar] [CrossRef]
- Christensen, A.A.; Gannon, M. The Beta Cell in Type 2 Diabetes. Curr. Diabetes Rep. 2019, 19, 81. [Google Scholar] [CrossRef]
- Yamamoto, W.R.; Bone, R.N.; Sohn, P.; Syed, F.; Reissaus, C.A.; Mosley, A.L.; Wijeratne, A.B.; True, J.D.; Tong, X.; Kono, T.; et al. Endoplasmic Reticulum Stress Alters Ryanodine Receptor Function in the Murine Pancreatic β Cell. J. Biol. Chem. 2019, 294, 168–181. [Google Scholar] [CrossRef]
- Halban, P.A.; Polonsky, K.S.; Bowden, D.W.; Hawkins, M.A.; Ling, C.; Mather, K.J.; Powers, A.C.; Rhodes, C.J.; Sussel, L.; Weir, G.C. β-Cell Failure in Type 2 Diabetes: Postulated Mechanisms and Prospects for Prevention and Treatment. Diabetes Care 2014, 37, 1751–1758. [Google Scholar] [CrossRef]
- Fuchsberger, C.; Flannick, J.; Teslovich, T.M.; Mahajan, A.; Agarwala, V.; Gaulton, K.J.; Ma, C.; Fontanillas, P.; Moutsianas, L.; McCarthy, D.J.; et al. The Genetic Architecture of Type 2 Diabetes. Nature 2016, 536, 41–47. [Google Scholar] [CrossRef]
- McCarthy, M.I. Genomics, Type 2 Diabetes, and Obesity. N. Engl. J. Med. 2010, 363, 2339–2350. [Google Scholar] [CrossRef]
- Diabetes Atlas 2017 Is Now Online. Available online: https://ncdalliance.org/resources/diabetes-atlas-2017-is-now-online (accessed on 16 January 2022).
- NCD Risk Factor Collaboration (NCD-RisC). Worldwide Trends in Diabetes since 1980: A Pooled Analysis of 751 Population-Based Studies with 4.4 Million Participants. Lancet 2016, 387, 1513–1530. [Google Scholar] [CrossRef]
- American Diabetes Association. Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 2013, 37, S81–S90. [Google Scholar] [CrossRef]
- Gillett, M.J. International Expert Committee Report on the Role of the A1C Assay in the Diagnosis of Diabetes. Clin. Biochem. Rev. 2009, 30, 197–200. [Google Scholar] [PubMed]
- Hsueh, W.A.; Orloski, L.; Wyne, K. Prediabetes: The Importance of Early Identification and Intervention. Postgrad. Med. 2010, 122, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group; Lachin, J.M.; Genuth, S.; Cleary, P.; Davis, M.D.; Nathan, D.M. Retinopathy and Nephropathy in Patients with Type 1 Diabetes Four Years after a Trial of Intensive Therapy. N. Engl. J. Med. 2000, 342, 381–389. [Google Scholar] [CrossRef]
- Guthrie, R.A.; Guthrie, D.W. Pathophysiology of Diabetes Mellitus. Crit. Care Nurs. Q. 2004, 27, 113–125. [Google Scholar] [CrossRef]
- Schmidt, A.M. Highlighting Diabetes Mellitus: The Epidemic Continues. Arter. Thromb. Vasc. Biol. 2018, 38, e1–e8. [Google Scholar] [CrossRef]
- Lontchi-Yimagou, E.; Sobngwi, E.; Matsha, T.E.; Kengne, A.P. Diabetes Mellitus and Inflammation. Curr. Diabetes Rep. 2013, 13, 435–444. [Google Scholar] [CrossRef]
- Falk, E. Pathogenesis of Atherosclerosis. J. Am. Coll. Cardiol. 2006, 47, C7–C12. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis—An Inflammatory Disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Tavafi, M. Complexity of Diabetic Nephropathy Pathogenesis and Design of Investigations. J. Ren. Inj. Prev. 2013, 2, 59–62. [Google Scholar] [CrossRef]
- Glass, C.K.; Witztum, J.L. Atherosclerosis: The Road Ahead. Cell 2001, 104, 503–516. [Google Scholar] [CrossRef]
- Libby, P. Inflammation in Atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Kragel, A.H.; Reddy, S.G.; Wittes, J.T.; Roberts, W.C. Morphometric Analysis of the Composition of Atherosclerotic Plaques in the Four Major Epicardial Coronary Arteries in Acute Myocardial Infarction and in Sudden Coronary Death. Circulation 1989, 80, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.M.; Virmani, R.; Rosenfeld, M.E. The Good Smooth Muscle Cells in Atherosclerosis. Curr. Atheroscler. Rep. 2000, 2, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Kolodgie, F.D.; Petrov, A.; Virmani, R.; Narula, N.; Verjans, J.W.; Weber, D.K.; Hartung, D.; Steinmetz, N.; Vanderheyden, J.L.; Vannan, M.A.; et al. Targeting of Apoptotic Macrophages and Experimental Atheroma with Radiolabeled Annexin V. Circulation 2003, 108, 3134–3139. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.-J.; Libby, P. Progression of Atheroma: A Struggle between Death and Procreation. Arter. Thromb. Vasc. Biol. 2002, 22, 1370–1380. [Google Scholar] [CrossRef]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in Atherosclerosis: A Dynamic Balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef]
- Schaar, J.A.; Muller, J.E.; Falk, E.; Virmani, R.; Fuster, V.; Serruys, P.W.; Colombo, A.; Stefanadis, C.; Casscells, S.W.; Moreno, P.R.; et al. Terminology for High-Risk and Vulnerable Coronary Artery Plaques. Eur. Heart J. 2004, 25, 1077–1082. [Google Scholar] [CrossRef]
- Mann, J.; Davies, M. Mechanisms of Progression in Native Coronary Artery Disease: Role of Healed Plaque Disruption. Heart 1999, 82, 265–268. [Google Scholar] [CrossRef]
- Fuster, V.; Moreno, P.R.; Fayad, Z.A.; Corti, R.; Badimon, J.J. Atherothrombosis and High-Risk Plaque: Part I: Evolving Concepts. J. Am. Coll. Cardiol. 2005, 46, 937–954. [Google Scholar] [CrossRef]
- Calabro, P.; Chang, D.W.; Willerson, J.T.; Yeh, E.T.H. Release of C-Reactive Protein in Response to Inflammatory Cytokines by Human Adipocytes: Linking Obesity to Vascular Inflammation. J. Am. Coll. Cardiol. 2005, 46, 1112–1113. [Google Scholar] [CrossRef] [PubMed]
- Emerging Risk Factors Collaboration. C-Reactive Protein, Fibrinogen, and Cardiovascular Disease Prediction. N. Engl. J. Med. 2012, 367, 1310–1320. [Google Scholar] [CrossRef] [PubMed]
- Heyden, S.; Gerber, C.J. Atherosclerotic Cerebrovascular Disease —Its Nature and Management. Am. J. Med. 1969, 46, 763–773. [Google Scholar] [CrossRef]
- Królewski, A.S.; Czyzyk, A.; Janeczko, D.; Kopczyński, J. Mortality from Cardiovascular Diseases among Diabetics. Diabetologia 1977, 13, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Abbott, R.D.; Donahue, R.P.; MacMahon, S.W.; Reed, D.M.; Yano, K. Diabetes and the Risk of Stroke. JAMA 1987, 257, 949–952. [Google Scholar] [CrossRef] [PubMed]
- Stamler, J.; Vaccaro, O.; Neaton, J.D.; Wentworth, D. Diabetes, Other Risk Factors, and 12-Yr Cardiovascular Mortality for Men Screened in the Multiple Risk Factor Intervention Trial. Diabetes Care 1993, 16, 434–444. [Google Scholar] [CrossRef]
- You, R.X.; McNeil, J.J.; O’Malley, H.M.; Davis, S.M.; Thrift, A.G.; Donnan, G.A. Risk Factors for Stroke Due to Cerebral Infarction in Young Adults. Stroke 1997, 28, 1913–1918. [Google Scholar] [CrossRef]
- Rohr, J.; Kittner, S.; Feeser, B.; Hebel, J.R.; Whyte, M.G.; Weinstein, A.; Kanarak, N.; Buchholz, D.; Earley, C.; Johnson, C.; et al. Traditional Risk Factors and Ischemic Stroke in Young Adults: The Baltimore-Washington Cooperative Young Stroke Study. Arch. Neurol. 1996, 53, 603–607. [Google Scholar] [CrossRef]
- Hill, M.D. Stroke and Diabetes Mellitus. Handb. Clin. Neurol. 2014, 126, 167–174. [Google Scholar] [CrossRef]
- Folsom, A.R.; Rasmussen, M.L.; Chambless, L.E.; Howard, G.; Cooper, L.S.; Schmidt, M.I.; Heiss, G. Prospective Associations of Fasting Insulin, Body Fat Distribution, and Diabetes with Risk of Ischemic Stroke. The Atherosclerosis Risk in Communities (ARIC) Study Investigators. Diabetes Care 1999, 22, 1077–1083. [Google Scholar] [CrossRef]
- Goldstein, L.B.; Adams, R.; Becker, K.; Furberg, C.D.; Gorelick, P.B.; Hademenos, G.; Hill, M.; Howard, G.; Howard, V.J.; Jacobs, B.; et al. Primary Prevention of Ischemic Stroke: A Statement for Healthcare Professionals from the Stroke Council of the American Heart Association. Circulation 2001, 103, 163–182. [Google Scholar] [CrossRef] [PubMed]
- Himmelmann, A.; Hansson, L.; Svensson, A.; Harmsen, P.; Holmgren, C.; Svanborg, A. Predictors of Stroke in the Elderly. Acta Med. Scand. 1988, 224, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Kuusisto, J.; Mykkänen, L.; Pyörälä, K.; Laakso, M. Non-Insulin-Dependent Diabetes and Its Metabolic Control Are Important Predictors of Stroke in Elderly Subjects. Stroke 1994, 25, 1157–1164. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Anderson, T.J. The Ten Most Commonly Asked Questions about Endothelial Function in Cardiology. Cardiol. Rev. 2001, 9, 250–252. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, R.; Meinberg, E.G.; Stanley, J.C.; Gordon, D.; Webb, R.C. Nitric Oxide Reversibly Inhibits the Migration of Cultured Vascular Smooth Muscle Cells. Circ. Res. 1996, 78, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Kubes, P.; Suzuki, M.; Granger, D.N. Nitric Oxide: An Endogenous Modulator of Leukocyte Adhesion. Proc. Natl. Acad. Sci. USA 1991, 88, 4651–4655. [Google Scholar] [CrossRef] [PubMed]
- De Vriese, A.S.; Verbeuren, T.J.; Van de Voorde, J.; Lameire, N.H.; Vanhoutte, P.M. Endothelial Dysfunction in Diabetes. Endothel. Dysfunct. Diabetes. Br. J. Pharmacol. 2000, 130, 963–974. [Google Scholar] [CrossRef]
- Milstien, S.; Katusic, Z. Oxidation of Tetrahydrobiopterin by Peroxynitrite: Implications for Vascular Endothelial Function. Biochem. Biophys. Res. Commun. 1999, 263, 681–684. [Google Scholar] [CrossRef]
- Hennes, M.M.; O’Shaughnessy, I.M.; Kelly, T.M.; LaBelle, P.; Egan, B.M.; Kissebah, A.H. Insulin-Resistant Lipolysis in Abdominally Obese Hypertensive Individuals. Hypertension 1996, 28, 120–126. [Google Scholar] [CrossRef]
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High Glucose Level and Free Fatty Acid Stimulate Reactive Oxygen Species Production through Protein Kinase C--Dependent Activation of NAD(P)H Oxidase in Cultured Vascular Cells. Diabetes 2000, 49, 1939–1945. [Google Scholar] [CrossRef]
- Hopfner, R.L.; Gopalakrishnan, V. Endothelin: Emerging Role in Diabetic Vascular Complications. Diabetologia 1999, 42, 1383–1394. [Google Scholar] [CrossRef] [PubMed]
- Quehenberger, P.; Bierhaus, A.; Fasching, P.; Muellner, C.; Klevesath, M.; Hong, M.; Stier, G.; Sattler, M.; Schleicher, E.; Speiser, W.; et al. Endothelin 1 Transcription Is Controlled by Nuclear Factor-KappaB in AGE-Stimulated Cultured Endothelial Cells. Diabetes 2000, 49, 1561–1570. [Google Scholar] [CrossRef] [PubMed]
- Katakami, N. Mechanism of Development of Atherosclerosis and Cardiovascular Disease in Diabetes Mellitus. J. Atheroscler. Thromb. 2018, 25, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.I.; Pichna, B.A.; Shi, Y.; Bowes, A.J.; Werstuck, G.H. Evidence Supporting a Role for Endoplasmic Reticulum Stress in the Development of Atherosclerosis in a Hyperglycaemic Mouse Model. Antioxid. Redox Signal. 2009, 11, 2289–2298. [Google Scholar] [CrossRef] [PubMed]
- Rösen, P.; Nawroth, P.P.; King, G.; Möller, W.; Tritschler, H.J.; Packer, L. The Role of Oxidative Stress in the Onset and Progression of Diabetes and Its Complications: A Summary of a Congress Series Sponsored by UNESCO-MCBN, the American Diabetes Association and the German Diabetes Society. Diabetes/Metab. Res. Rev. 2001, 17, 189–212. [Google Scholar] [CrossRef] [PubMed]
- Zeiher, A.M.; Fisslthaler, B.; Schray-Utz, B.; Busse, R. Nitric Oxide Modulates the Expression of Monocyte Chemoattractant Protein 1 in Cultured Human Endothelial Cells. Circ. Res. 1995, 76, 980–986. [Google Scholar] [CrossRef]
- Christlieb, A.R.; Janka, H.U.; Kraus, B.; Gleason, R.E.; Icasas-Cabral, E.A.; Aiello, L.M.; Cabral, B.V.; Solano, A. Vascular Reactivity to Angiotensin II and to Norepinephrine in Diabetic Subjects. Diabetes 1976, 25, 268–274. [Google Scholar] [CrossRef]
- Nugent, A.G.; McGurk, C.; Hayes, J.R.; Johnston, G.D. Impaired Vasoconstriction to Endothelin 1 in Patients with NIDDM. Diabetes 1996, 45, 105–107. [Google Scholar] [CrossRef][Green Version]
- Tesfamariam, B.; Cohen, R.A. Enhanced Adrenergic Neurotransmission in Diabetic Rabbit Carotid Artery. Cardiovasc. Res. 1995, 29, 549–554. [Google Scholar] [CrossRef]
- McDaid, E.A.; Monaghan, B.; Parker, A.I.; Hayes, J.R.; Allen, J.A. Peripheral Autonomic Impairment in Patients Newly Diagnosed with Type II Diabetes. Diabetes Care 1994, 17, 1422–1427. [Google Scholar] [CrossRef]
- Fukumoto, H.; Naito, Z.; Asano, G.; Aramaki, T. Immunohistochemical and Morphometric Evaluations of Coronary Atherosclerotic Plaques Associated with Myocardial Infarction and Diabetes Mellitus. J. Atheroscler. Thromb. 1998, 5, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, S.; Oinuma, T.; Yamada, T. A Comparative Study of Cultured Smooth Muscle Cell Proliferation and Injury, Utilizing Glycated Low Density Lipoproteins with Slight Oxidation, Auto-Oxidation, or Extensive Oxidation. J. Atheroscler. Thromb. 2000, 7, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Assert, R.; Scherk, G.; Bumbure, A.; Pirags, V.; Schatz, H.; Pfeiffer, A.F. Regulation of Protein Kinase C by Short Term Hyperglycemia in Human Platelets in Vivo and in Vitro. Diabetologia 2001, 44, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Vinik, A.I.; Erbas, T.; Park, T.S.; Nolan, R.; Pittenger, G.L. Platelet Dysfunction in Type 2 Diabetes. Diabetes Care 2001, 24, 1476–1485. [Google Scholar] [CrossRef]
- Carr, M.E. Diabetes Mellitus: A Hypercoagulable State. J. Diabetes Complicat. 2001, 15, 44–54. [Google Scholar] [CrossRef]
- Nordt, T.K.; Bode, C. Impaired Endogenous Fibrinolysis in Diabetes Mellitus: Mechanisms and Therapeutic Approaches. Semin. Thromb. Hemost. 2000, 26, 495–501. [Google Scholar] [CrossRef]
- Joshi, M.B.; Lad, A.; Prasad, A.S.B.; Balakrishnan, A.; Ramachandra, L.; Satyamoorthy, K. High Glucose Modulates IL-6 Mediated Immune Homeostasis through Impeding Neutrophil Extracellular Trap Formation. FEBS Lett. 2013, 587, 2241–2246. [Google Scholar] [CrossRef]
- Ye, Y.; Zeng, Z.; Jin, T.; Zhang, H.; Xiong, X.; Gu, L. The Role of High Mobility Group Box 1 in Ischemic Stroke. Front. Cell. Neurosci. 2019, 13, 127. [Google Scholar] [CrossRef]
- Ogurtsova, K.; da Rocha Fernandes, J.D.; Huang, Y.; Linnenkamp, U.; Guariguata, L.; Cho, N.H.; Cavan, D.; Shaw, J.E.; Makaroff, L.E. IDF Diabetes Atlas: Global Estimates for the Prevalence of Diabetes for 2015 and 2040. Diabetes Res. Clin. Pract. 2017, 128, 40–50. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart Disease and Stroke Statistics—2018 Update: A Report From the American Heart Association. Circulation 2018, 137, e67–e492. [Google Scholar] [CrossRef]
- Bertoni, A.G.; Krop, J.S.; Anderson, G.F.; Brancati, F.L. Diabetes-Related Morbidity and Mortality in a National Sample of U.S. Elders. Diabetes Care 2002, 25, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Katakura, M.; Naka, M.; Kondo, T.; Nishii, N.; Komatsu, M.; Sato, Y.; Yamauchi, K.; Hiramatsu, K.; Ikeda, M.; Aizawa, T.; et al. Prospective Analysis of Mortality, Morbidity, and Risk Factors in Elderly Diabetic Subjects: Nagano Study. Diabetes Care 2003, 26, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Kannel, W.B.; McGee, D.L. Diabetes and Cardiovascular Disease. The Framingham Study. JAMA 1979, 241, 2035–2038. [Google Scholar] [CrossRef]
- Kissela, B.M.; Khoury, J.; Kleindorfer, D.; Woo, D.; Schneider, A.; Alwell, K.; Miller, R.; Ewing, I.; Moomaw, C.J.; Szaflarski, J.P.; et al. Epidemiology of Ischemic Stroke in Patients with Diabetes: The Greater Cincinnati/Northern Kentucky Stroke Study. Diabetes Care 2005, 28, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Almdal, T.; Scharling, H.; Jensen, J.S.; Vestergaard, H. The Independent Effect of Type 2 Diabetes Mellitus on Ischemic Heart Disease, Stroke, and Death: A Population-Based Study of 13,000 Men and Women with 20 Years of Follow-Up. Arch. Intern. Med. 2004, 164, 1422–1426. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Pinto, A.; Salemi, G.; Di Raimondo, D.; Di Sciacca, R.; Fernandez, P.; Ragonese, P.; Savettieri, G.; Licata, G. Diabetic and Non-Diabetic Subjects with Ischemic Stroke: Differences, Subtype Distribution and Outcome. Nutr. Metab. Cardiovasc. Dis. 2008, 18, 152–157. [Google Scholar] [CrossRef]
- Mulnier, H.E.; Seaman, H.E.; Raleigh, V.S.; Soedamah-Muthu, S.S.; Colhoun, H.M.; Lawrenson, R.A.; De Vries, C.S. Risk of Stroke in People with Type 2 Diabetes in the UK: A Study Using the General Practice Research Database. Diabetologia 2006, 49, 2859–2865. [Google Scholar] [CrossRef]
- Selvin, E.; Coresh, J.; Shahar, E.; Zhang, L.; Steffes, M.; Sharrett, A.R. Glycaemia (Haemoglobin A1c) and Incident Ischaemic Stroke: The Atherosclerosis Risk in Communities (ARIC) Study. Lancet Neurol. 2005, 4, 821–826. [Google Scholar] [CrossRef]
- Guerrero-Romero, F.; Rodríguez-Morán, M. Proteinuria Is an Independent Risk Factor for Ischemic Stroke in Non-Insulin-Dependent Diabetes Mellitus. Stroke 1999, 30, 1787–1791. [Google Scholar] [CrossRef]
- Cholesterol Treatment Trialists’ (CTT) Collaborators; Kearney, P.M.; Blackwell, L.; Collins, R.; Keech, A.; Simes, J.; Peto, R.; Armitage, J.; Baigent, C. Efficacy of Cholesterol-Lowering Therapy in 18,686 People with Diabetes in 14 Randomised Trials of Statins: A Meta-Analysis. Lancet 2008, 371, 117–125. [Google Scholar] [CrossRef]
- UK Prospective Diabetes Study Group. Tight Blood Pressure Control and Risk of Macrovascular and Microvascular Complications in Type 2 Diabetes: UKPDS 38. UK Prospective Diabetes Study Group. BMJ 1998, 317, 703–713. [Google Scholar] [CrossRef]
- PROGRESS Collaborative Group Randomised Trial of a Perindopril-Based Blood-Pressure-Lowering Regimen among 6105 Individuals with Previous Stroke or Transient Ischaemic Attack. Lancet 2001, 358, 1033–1041. [CrossRef]
- Lichtman, J.H.; Krumholz, H.M.; Wang, Y.; Radford, M.J.; Brass, L.M. Risk and Predictors of Stroke after Myocardial Infarction among the Elderly: Results from the Cooperative Cardiovascular Project. Circulation 2002, 105, 1082–1087. [Google Scholar] [CrossRef] [PubMed]
- D’Ancona, G.; de Ibarra, J.I.S.; Baillot, R.; Mathieu, P.; Doyle, D.; Metras, J.; Desaulniers, D.; Dagenais, F. Determinants of Stroke after Coronary Artery Bypass Grafting. Eur. J. Cardio-Thorac. Surg. 2003, 24, 552–556. [Google Scholar] [CrossRef]
- Anderson, C.S.; Carter, K.N.; Hackett, M.L.; Feigin, V.; Barber, P.A.; Broad, J.B.; Bonita, R.; Auckland Regional Community Stroke (ARCOS) Study Group. Trends in Stroke Incidence in Auckland, New Zealand, during 1981 to 2003. Stroke 2005, 36, 2087–2093. [Google Scholar] [CrossRef]
- Benatru, I.; Rouaud, O.; Durier, J.; Contegal, F.; Couvreur, G.; Bejot, Y.; Osseby, G.V.; Ben Salem, D.; Ricolfi, F.; Moreau, T.; et al. Stable Stroke Incidence Rates but Improved Case-Fatality in Dijon, France, from 1985 to 2004. Stroke 2006, 37, 1674–1679. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Coull, A.J.; Giles, M.F.; Howard, S.C.; Silver, L.E.; Bull, L.M.; Gutnikov, S.A.; Edwards, P.; Mant, D.; Sackley, C.M.; et al. Change in Stroke Incidence, Mortality, Case-Fatality, Severity, and Risk Factors in Oxfordshire, UK from 1981 to 2004 (Oxford Vascular Study). Lancet 2004, 363, 1925–1933. [Google Scholar] [CrossRef]
- Gray, C.S.; Scott, J.F.; French, J.M.; Alberti, K.G.M.M.; O’Connell, J.E. Prevalence and Prediction of Unrecognised Diabetes Mellitus and Impaired Glucose Tolerance Following Acute Stroke. Age Ageing 2004, 33, 71–77. [Google Scholar] [CrossRef]
- Karapanayiotides, T.; Piechowski-Jozwiak, B.; van Melle, G.; Bogousslavsky, J.; Devuyst, G. Stroke Patterns, Etiology, and Prognosis in Patients with Diabetes Mellitus. Neurology 2004, 62, 1558–1562. [Google Scholar] [CrossRef]
- Jackson, C.; Sudlow, C. Are Lacunar Strokes Really Different? A Systematic Review of Differences in Risk Factor Profiles between Lacunar and Nonlacunar Infarcts. Stroke 2005, 36, 891–901. [Google Scholar] [CrossRef]
- Schulz, U.; Rothwell, P. Differences in Vascular Risk Factors Between Etiological Subtypes of Ischemic Stroke. Stroke 2003, 34, 2050–2059. [Google Scholar] [CrossRef] [PubMed]
- Manolio, T.A.; Kronmal, R.A.; Burke, G.L.; O’Leary, D.H.; Price, T.R. Short-Term Predictors of Incident Stroke in Older Adults. Stroke 1996, 27, 1479–1486. [Google Scholar] [CrossRef] [PubMed]
- Giles, W.H.; Kittner, S.J.; Hebel, J.R.; Losonczy, K.G.; Sherwin, R.W. Determinants of Black-White Differences in the Risk of Cerebral Infarction. The National Health and Nutrition Examination Survey Epidemiologic Follow-up Study. Arch. Intern. Med. 1995, 155, 1319–1324. [Google Scholar] [CrossRef] [PubMed]
- Megherbi, S.-E.; Milan, C.; Minier, D.; Couvreur, G.; Osseby, G.-V.; Tilling, K.; Di Carlo, A.; Inzitari, D.; Wolfe, C.D.A.; Moreau, T.; et al. Association between Diabetes and Stroke Subtype on Survival and Functional Outcome 3 Months after Stroke: Data from the European BIOMED Stroke Project. Stroke 2003, 34, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Scott, R.A.; Traylor, M.; Langenberg, C.C.; Hindy, G.; Melander, O.; Orho-Melander, M.; Seshadri, S.; Wareham, N.J.; Markus, H.S.; et al. Type 2 Diabetes, Glucose, Insulin, BMI, and Ischemic Stroke Subtypes. Neurology 2017, 89, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Roper, N.A.; Bilous, R.W.; Kelly, W.F.; Unwin, N.C.; Connolly, V.M. Excess Mortality in a Population with Diabetes and the Impact of Material Deprivation: Longitudinal, Population Based Study. BMJ 2001, 322, 1389–1393. [Google Scholar] [CrossRef]
- Pinto, A.; Tuttolomondo, A.; Di Raimondo, D.; Fernandez, P.; La Placa, S.; Di Gati, M.; Licata, G. Cardiovascular Risk Profile and Morbidity in Subjects Affected by Type 2 Diabetes Mellitus with and without Diabetic Foot. Metabolism 2008, 57, 676–682. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; Casuccio, A.; Guercio, G.; Maida, C.; Del Cuore, A.; Di Raimondo, D.; Simonetta, I.; Di Bona, D.; Pecoraro, R.; Della Corte, V.; et al. Arterial Stiffness, Endothelial and Cognitive Function in Subjects with Type 2 Diabetes in Accordance with Absence or Presence of Diabetic Foot Syndrome. Cardiovasc. Diabetol. 2017, 16, 2. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; Del Cuore, A.; La Malfa, A.; Casuccio, A.; Daidone, M.; Maida, C.D.; Di Raimondo, D.; Di Chiara, T.; Puleo, M.G.; Norrito, R.; et al. Assessment of Heart Rate Variability (HRV) in Subjects with Type 2 Diabetes Mellitus with and without Diabetic Foot: Correlations with Endothelial Dysfunction Indices and Markers of Adipo-Inflammatory Dysfunction. Cardiovasc. Diabetol. 2021, 20, 142. [Google Scholar] [CrossRef]
- Wardlaw, J.M.; Smith, E.E.; Biessels, G.J.; Cordonnier, C.; Fazekas, F.; Frayne, R.; Lindley, R.I.; O’Brien, J.T.; Barkhof, F.; Benavente, O.R.; et al. Neuroimaging Standards for Research into Small Vessel Disease and Its Contribution to Ageing and Neurodegeneration. Lancet Neurol. 2013, 12, 822–838. [Google Scholar] [CrossRef]
- Martinez-Majander, N.; Gordin, D.; Joutsi-Korhonen, L.; Salopuro, T.; Adeshara, K.; Sibolt, G.; Curtze, S.; Pirinen, J.; Liebkind, R.; Soinne, L.; et al. Endothelial Dysfunction Is Associated With Early-Onset Cryptogenic Ischemic Stroke in Men and With Increasing Age. J. Am. Heart Assoc. 2021, 10, e020838. [Google Scholar] [CrossRef] [PubMed]
- Shchepankevich, L.A.; Vostrikova, E.V.; Pilipenko, P.I.; Iarmoshchuk, A.V.; Akhundova, L.E.; Miasnikova, N.G.; Fedorova, K.O.; Kononova, E.A. Endothelial dysfunction in ischemic strokes in diabetes mellitus. Zhurnal Nevrol. Psikhiatrii Im. SS Korsakova 2011, 111, 28–30. [Google Scholar]
- Tuttolomondo, A.; Casuccio, A.; Della Corte, V.; Maida, C.; Pecoraro, R.; Di Raimondo, D.; Vassallo, V.; Simonetta, I.; Arnao, V.; Pinto, A. Endothelial Function and Arterial Stiffness Indexes in Subjects with Acute Ischemic Stroke: Relationship with TOAST Subtype. Atherosclerosis 2017, 256, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.; Hunt, B.J.; O’Sullivan, M.; Parmar, K.; Bamford, J.M.; Briley, D.; Brown, M.M.; Thomas, D.J.; Markus, H.S. Markers of Endothelial Dysfunction in Lacunar Infarction and Ischaemic Leukoaraiosis. Brain 2003, 126, 424–432. [Google Scholar] [CrossRef]
- Caplan, L.R. Intracranial Branch Atheromatous Disease: A Neglected, Understudied, and Underused Concept. Neurology 1989, 39, 1246–1250. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Ohara, T.; Hamanaka, M.; Hosomi, A.; Tamura, A.; Akiguchi, I. Characteristics of Intracranial Branch Atheromatous Disease and Its Association with Progressive Motor Deficits. J. Neurol. Sci. 2011, 304, 78–82. [Google Scholar] [CrossRef]
- Cordonnier, C.; Salman, R.A.-S.; Wardlaw, J. Spontaneous Brain Microbleeds: Systematic Review, Subgroup Analyses and Standards for Study Design and Reporting. Brain 2007, 130, 1988–2003. [Google Scholar] [CrossRef]
- Wang, D.-Q.; Wang, L.; Wei, M.-M.; Xia, X.-S.; Tian, X.-L.; Cui, X.-H.; Li, X. Relationship Between Type 2 Diabetes and White Matter Hyperintensity: A Systematic Review. Front. Endocrinol. 2020, 11, 595962. [Google Scholar] [CrossRef]
- Kato, H.; Izumiyama, M.; Izumiyama, K.; Takahashi, A.; Itoyama, Y. Silent Cerebral Microbleeds on T2*-Weighted MRI: Correlation with Stroke Subtype, Stroke Recurrence, and Leukoaraiosis. Stroke 2002, 33, 1536–1540. [Google Scholar] [CrossRef]
- Kuller, L.H.; Longstreth, W.T.; Arnold, A.M.; Bernick, C.; Bryan, R.N.; Beauchamp, N.J.; Cardiovascular Health Study Collaborative Research Group. White Matter Hyperintensity on Cranial Magnetic Resonance Imaging: A Predictor of Stroke. Stroke 2004, 35, 1821–1825. [Google Scholar] [CrossRef]
- Candelise, L.; Landi, G.; Orazio, E.N.; Boccardi, E. Prognostic Significance of Hyperglycemia in Acute Stroke. Arch. Neurol. 1985, 42, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, H.; Nakayama, H.; Raaschou, H.O.; Olsen, T.S. Stroke in Patients with Diabetes. The Copenhagen Stroke Study. Stroke 1994, 25, 1977–1984. [Google Scholar] [CrossRef] [PubMed]
- Kiers, L.; Davis, S.M.; Larkins, R.; Hopper, J.; Tress, B.; Rossiter, S.C.; Carlin, J.; Ratnaike, S. Stroke Topography and Outcome in Relation to Hyperglycemia and Diabetes. J. Neurol. Neurosurg. Psychiatry 1992, 55, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Capes, S.E.; Hunt, D.; Malmberg, K.; Pathak, P.; Gerstein, H.C. Stress Hyperglycemia and Prognosis of Stroke in Nondiabetic and Diabetic Patients: A Systematic Overview. Stroke 2001, 32, 2426–2432. [Google Scholar] [CrossRef]
- Tracey, F.; Crawford, V.L.S.; Lawson, J.T.; Buchanan, K.D.; Stout, R.W. Hyperglycemia and Mortality from Acute Stroke. QJM Int. J. Med. 1993, 86, 439–446. [Google Scholar]
- Barth, E.; Albuszies, G.; Baumgart, K.; Matejovic, M.; Wachter, U.; Vogt, J.; Radermacher, P.; Calzia, E. Glucose Metabolism and Catecholamines. Crit. Care Med. 2007, 35, S508–S518. [Google Scholar] [CrossRef]
- Seematter, G.; Binnert, C.; Martin, J.-L.; Tappy, L. Relationship between Stress, Inflammation and Metabolism. Curr. Opin. Clin. Nutr. Metab. Care 2004, 7, 169–173. [Google Scholar] [CrossRef]
- Feibel, J.H.; Hardy, P.M.; Campbell, R.G.; Goldstein, M.N.; Joynt, R.J. Prognostic Value of the Stress Response Following Stroke. JAMA 1977, 238, 1374–1376. [Google Scholar] [CrossRef]
- O’Neill, P.A.; Davies, I.; Fullerton, K.J.; Bennett, D. Stress Hormone and Blood Glucose Response Following Acute Stroke in the Elderly. Stroke 1991, 22, 842–847. [Google Scholar] [CrossRef]
- Vila, N.; Castillo, J.; Dávalos, A.; Chamorro, A. Proinflammatory Cytokines and Early Neurological Worsening in Ischemic Stroke. Stroke 2000, 31, 2325–2329. [Google Scholar] [CrossRef]
- Chrousos, G.P. The Hypothalamic-Pituitary-Adrenal Axis and Immune-Mediated Inflammation. N. Engl. J. Med. 1995, 332, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Plomgaard, P.; Bouzakri, K.; Krogh-Madsen, R.; Mittendorfer, B.; Zierath, J.R.; Pedersen, B.K. Tumor Necrosis Factor-Alpha Induces Skeletal Muscle Insulin Resistance in Healthy Human Subjects via Inhibition of Akt Substrate 160 Phosphorylation. Diabetes 2005, 54, 2939–2945. [Google Scholar] [CrossRef] [PubMed]
- Rask-Madsen, C.; Domínguez, H.; Ihlemann, N.; Hermann, T.; Køber, L.; Torp-Pedersen, C. Tumor Necrosis Factor-Alpha Inhibits Insulin’s Stimulating Effect on Glucose Uptake and Endothelium-Dependent Vasodilation in Humans. Circulation 2003, 108, 1815–1821. [Google Scholar] [CrossRef] [PubMed]
- Dhindsa, S.; Tripathy, D.; Mohanty, P.; Ghanim, H.; Syed, T.; Aljada, A.; Dandona, P. Differential Effects of Glucose and Alcohol on Reactive Oxygen Species Generation and Intranuclear Nuclear Factor-KappaB in Mononuclear Cells. Metabolism 2004, 53, 330–334. [Google Scholar] [CrossRef]
- Kawai, N.; Keep, R.F.; Betz, A.L.; Nagao, S. Hyperglycemia Induces Progressive Changes in the Cerebral Microvasculature and Blood-Brain Barrier Transport during Focal Cerebral Ischemia. In Intracranial Pressure and Neuromonitoring in Brain Injury; Springer: Vienna, Austria, 1998; Volume 71, pp. 219–221. [Google Scholar] [CrossRef]
- Li, P.-A.; Shuaib, A.; Miyashita, H.; He, Q.-P.; Siesjö, B.K. Hyperglycemia Enhances Extracellular Glutamate Accumulation in Rats Subjected to Forebrain Ischemia. Stroke 2000, 31, 183–192. [Google Scholar] [CrossRef]
- Folbergrová, J.; Memezawa, H.; Smith, M.L.; Siesjö, B.K. Focal and Perifocal Changes in Tissue Energy State during Middle Cerebral Artery Occlusion in Normo- and Hyperglycemic Rats. J. Cereb. Blood Flow Metab. 1992, 12, 25–33. [Google Scholar] [CrossRef]
- Barnes, P.J.; Karin, M. Nuclear Factor-KappaB—A Pivotal Transcription Factor in Chronic Inflammatory Diseases. N. Engl. J. Med. 1997, 336, 1066–1071. [Google Scholar] [CrossRef]
- Nurmi, A.; Lindsberg, P.J.; Koistinaho, M.; Zhang, W.; Juettler, E.; Karjalainen-Lindsberg, M.-L.; Weih, F.; Frank, N.; Schwaninger, M.; Koistinaho, J. Nuclear Factor-ΚB Contributes to Infarction After Permanent Focal Ischemia. Stroke 2004, 35, 987–991. [Google Scholar] [CrossRef]
- Schneider, A.; Martin-Villalba, A.; Weih, F.; Vogel, J.; Wirth, T.; Schwaninger, M. NF-KappaB Is Activated and Promotes Cell Death in Focal Cerebral Ischemia. Nat. Med. 1999, 5, 554–559. [Google Scholar] [CrossRef]
- Aljada, A.; Ghanim, H.; Mohanty, P.; Syed, T.; Bandyopadhyay, A.; Dandona, P. Glucose Intake Induces an Increase in Activator Protein 1 and Early Growth Response 1 Binding Activities, in the Expression of Tissue Factor and Matrix Metalloproteinase in Mononuclear Cells, and in Plasma Tissue Factor and Matrix Metalloproteinase Concentrations. Am. J. Clin. Nutr. 2004, 80, 51–57. [Google Scholar] [CrossRef]
- Berger, L.; Hakim, A.M. The Association of Hyperglycemia with Cerebral Edema in Stroke. Stroke 1986, 17, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Gursoy-Ozdemir, Y.; Qiu, J.; Matsuoka, N.; Bolay, H.; Bermpohl, D.; Jin, H.; Wang, X.; Rosenberg, G.A.; Lo, E.H.; Moskowitz, M.A. Cortical Spreading Depression Activates and Upregulates MMP-9. J. Clin. Investig. 2004, 113, 1447–1455. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, P.; Hamouda, W.; Garg, R.; Aljada, A.; Ghanim, H.; Dandona, P. Glucose Challenge Stimulates Reactive Oxygen Species (ROS) Generation by Leucocytes. J. Clin. Endocrinol. Metab. 2000, 85, 2970–2973. [Google Scholar] [CrossRef] [PubMed]
- Dandona, P.; James, I.M.; Newbury, P.A.; Woollard, M.L.; Beckett, A.G. Cerebral Blood Flow in Diabetes Mellitus: Evidence of Abnormal Cerebrovascular Reactivity. Br. Med. J. 1978, 2, 325–326. [Google Scholar] [CrossRef]
- Duckrow, R.B.; Beard, D.C.; Brennan, R.W. Regional Cerebral Blood Flow Decreases during Chronic and Acute Hyperglycemia. Stroke 1987, 18, 52–58. [Google Scholar] [CrossRef]
- Araki, N.; Greenberg, J.H.; Sladky, J.T.; Uematsu, D.; Karp, A.; Reivich, M. The Effect of Hyperglycemia on Intracellular Calcium in Stroke. J. Cereb. Blood Flow Metab. 1992, 12, 469–476. [Google Scholar] [CrossRef]
- Chew, W.; Kucharczyk, J.; Moseley, M.; Derugin, N.; Norman, D. Hyperglycemia Augments Ischemic Brain Injury: In Vivo MR Imaging/Spectroscopic Study with Nicardipine in Cats with Occluded Middle Cerebral Arteries. AJNR Am. J. Neuroradiol. 1991, 12, 603–609. [Google Scholar]
- Anderson, R.E.; Tan, W.K.; Martin, H.S.; Meyer, F.B. Effects of Glucose and PaO2 Modulation on Cortical Intracellular Acidosis, NADH Redox State, and Infarction in the Ischemic Penumbra. Stroke 1999, 30, 160–170. [Google Scholar] [CrossRef]
- Hamilton, M.G.; Tranmer, B.I.; Auer, R.N. Insulin Reduction of Cerebral Infarction Due to Transient Focal Ischemia. J. Neurosurg. 1995, 82, 262–268. [Google Scholar] [CrossRef]
- Voll, C.L.; Auer, R.N. Insulin Attenuates Ischemic Brain Damage Independent of Its Hypoglycemic Effect. J. Cereb. Blood Flow Metab. 1991, 11, 1006–1014. [Google Scholar] [CrossRef]
- Aljada, A.; Ghanim, H.; Saadeh, R.; Dandona, P. Insulin Inhibits NFkappaB and MCP-1 Expression in Human Aortic Endothelial Cells. J. Clin. Endocrinol. Metab. 2001, 86, 450–453. [Google Scholar] [CrossRef]
- Dandona, P.; Aljada, A.; Mohanty, P.; Ghanim, H.; Hamouda, W.; Assian, E.; Ahmad, S. Insulin Inhibits Intranuclear Nuclear Factor KappaB and Stimulates IkappaB in Mononuclear Cells in Obese Subjects: Evidence for an Anti-Inflammatory Effect? J. Clin. Endocrinol. Metab. 2001, 86, 3257–3265. [Google Scholar] [CrossRef] [PubMed]
- Aljada, A.; Ghanim, H.; Mohanty, P.; Kapur, N.; Dandona, P. Insulin Inhibits the Pro-Inflammatory Transcription Factor Early Growth Response Gene-1 (Egr)-1 Expression in Mononuclear Cells (MNC) and Reduces Plasma Tissue Factor (TF) and Plasminogen Activator Inhibitor-1 (PAI-1) Concentrations. J. Clin. Endocrinol. Metab. 2002, 87, 1419–1422. [Google Scholar] [CrossRef] [PubMed]
- Dandona, P.; Aljada, A.; Mohanty, P.; Ghanim, H.; Bandyopadhyay, A.; Chaudhuri, A. Insulin Suppresses Plasma Concentration of Vascular Endothelial Growth Factor and Matrix Metalloproteinase-9. Diabetes Care 2003, 26, 3310–3314. [Google Scholar] [CrossRef] [PubMed]
- Mikhailidis, D.P.; Mikhailidis, A.M.; Barradas, M.A.; Dandona, P. Effect of Nonesterified Fatty Acids on the Stability of Prostacyclin Activity. Metabolism 1983, 32, 717–721. [Google Scholar] [CrossRef]
- Dandona, P. Endothelium, Inflammation, and Diabetes. Curr. Diabetes Rep. 2002, 2, 311–315. [Google Scholar] [CrossRef]
- Boulton, A.J.M.; Vileikyte, L.; Ragnarson-Tennvall, G.; Apelqvist, J. The Global Burden of Diabetic Foot Disease. Lancet 2005, 366, 1719–1724. [Google Scholar] [CrossRef]
- Jeffcoate, W.J.; Game, F.; Cavanagh, P.R. The Role of Proinflammatory Cytokines in the Cause of Neuropathic Osteoarthropathy (Acute Charcot Foot) in Diabetes. Lancet 2005, 366, 2058–2061. [Google Scholar] [CrossRef]
- American Diabetes Association. 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes—2020. Diabetes Care 2020, 43, S98–S110. [Google Scholar] [CrossRef]
- Boulton, A.J.M.; Armstrong, D.G.; Albert, S.F.; Frykberg, R.G.; Hellman, R.; Kirkman, M.S.; Lavery, L.A.; Lemaster, J.W.; Mills, J.L.; Mueller, M.J.; et al. Comprehensive Foot Examination and Risk Assessment: A Report of the Task Force of the Foot Care Interest Group of the American Diabetes Association, with Endorsement by the American Association of Clinical Endocrinologists. Diabetes Care 2008, 31, 1679–1685. [Google Scholar] [CrossRef]
- CDC. National Diabetes Statistics Report 2020. Estimates of Diabetes and Its Burden in the United States; CDC: Atlanta, GA, USA, 2020; p. 32.
- Pecoraro, R.E.; Reiber, G.E.; Burgess, E.M. Pathways to Diabetic Limb Amputation: Basis for Prevention. Diabetes Care 1990, 13, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Abbott, C.A.; Vileikyte, L.; Williamson, S.; Carrington, A.L.; Boulton, A.J. Multicenter Study of the Incidence of and Predictive Risk Factors for Diabetic Neuropathic Foot Ulceration. Diabetes Care 1998, 21, 1071–1075. [Google Scholar] [CrossRef] [PubMed]
- Moss, S.E.; Klein, R.; Klein, B.E. The Prevalence and Incidence of Lower Extremity Amputation in a Diabetic Population. Arch. Intern. Med. 1992, 152, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, S.D.; Newton, K.; Blough, D.; McCulloch, D.K.; Sandhu, N.; Reiber, G.E.; Wagner, E.H. Incidence, Outcomes, and Cost of Foot Ulcers in Patients with Diabetes. Diabetes Care 1999, 22, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Faris, I. The Management of the Diabetic Foot; Churchill Livingstone: London, UK, 1991. [Google Scholar]
- Reiber, G.E.; Vileikyte, L.; Boyko, E.J.; del Aguila, M.; Smith, D.G.; Lavery, L.A.; Boulton, A.J. Causal Pathways for Incident Lower-Extremity Ulcers in Patients with Diabetes from Two Settings. Diabetes Care 1999, 22, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Pascarelli, E.F.; Bertrand, C.A.; Lopez, M. Comparison of blood pressures in the arms and legs. N. Engl. J. Med. 1964, 270, 693–698. [Google Scholar] [CrossRef] [PubMed]
- O’Hare, A.M.; Katz, R.; Shlipak, M.G.; Cushman, M.; Newman, A.B. Mortality and Cardiovascular Risk across the Ankle-Arm Index Spectrum: Results from the Cardiovascular Health Study. Circulation 2006, 113, 388–393. [Google Scholar] [CrossRef]
- Daousi, C.; MacFarlane, I.A.; Woodward, A.; Nurmikko, T.J.; Bundred, P.E.; Benbow, S.J. Chronic Painful Peripheral Neuropathy in an Urban Community: A Controlled Comparison of People with and without Diabetes. Diabet. Med. 2004, 21, 976–982. [Google Scholar] [CrossRef]
- Astasio-Picado, Á.; Cobos-Moreno, P.; Gómez-Martín, B. Self-Care Planning and Sanitary Education in the Prevention of the Diabetic Foot. Appl. Sci. 2021, 11, 7281. [Google Scholar] [CrossRef]
- Clinically Relevant Experimental Rodent Models of Diabetic Foot Ulcer-PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/35089527/ (accessed on 11 February 2022).
- Jude, E.B.; Oyibo, S.O.; Chalmers, N.; Boulton, A.J. Peripheral Arterial Disease in Diabetic and Nondiabetic Patients: A Comparison of Severity and Outcome. Diabetes Care 2001, 24, 1433–1437. [Google Scholar] [CrossRef]
- Pinto, A.; Tuttolomondo, A.; Di Raimondo, D.; La Placa, S.; Di Sciacca, R.; Fernandez, P.; Di Gati, M.; Raffa, A.; Licata, G. Ischemic Stroke in Patients with Diabetic Foot. Int. Angiol. 2007, 26, 266–269. [Google Scholar] [PubMed]
- Tuttle, H.A.; Davis-Gorman, G.; Goldman, S.; Copeland, J.G.; McDonagh, P.F. Proinflammatory Cytokines Are Increased in Type 2 Diabetic Women with Cardiovascular Disease. J. Diabetes Complicat. 2004, 18, 343–351. [Google Scholar] [CrossRef]
- Weigelt, C.; Rose, B.; Poschen, U.; Ziegler, D.; Friese, G.; Kempf, K.; Koenig, W.; Martin, S.; Herder, C. Immune Mediators in Patients with Acute Diabetic Foot Syndrome. Diabetes Care 2009, 32, 1491–1496. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; La Placa, S.; Di Raimondo, D.; Bellia, C.; Caruso, A.; Sasso, B.L.; Guercio, G.; Diana, G.; Ciaccio, M.; Licata, G.; et al. Adiponectin, Resistin and IL-6 Plasma Levels in Subjects with Diabetic Foot and Possible Correlations with Clinical Variables and Cardiovascular Co-Morbidity. Cardiovasc. Diabetol. 2010, 9, 50. [Google Scholar] [CrossRef]
- Osawa, H.; Doi, Y.; Makino, H.; Ninomiya, T.; Yonemoto, K.; Kawamura, R.; Hata, J.; Tanizaki, Y.; Iida, M.; Kiyohara, Y. Diabetes and Hypertension Markedly Increased the Risk of Ischemic Stroke Associated with High Serum Resistin Concentration in a General Japanese Population: The Hisayama Study. Cardiovasc. Diabetol. 2009, 8, 60. [Google Scholar] [CrossRef]
- Reilly, M.P.; Lehrke, M.; Wolfe, M.L.; Rohatgi, A.; Lazar, M.A.; Rader, D.J. Resistin Is an Inflammatory Marker of Atherosclerosis in Humans. Circulation 2005, 111, 932–939. [Google Scholar] [CrossRef]
- Hotta, K.; Funahashi, T.; Arita, Y.; Takahashi, M.; Matsuda, M.; Okamoto, Y.; Iwahashi, H.; Kuriyama, H.; Ouchi, N.; Maeda, K.; et al. Plasma Concentrations of a Novel, Adipose-Specific Protein, Adiponectin, in Type 2 Diabetic Patients. Arter. Thromb. Vasc. Biol. 2000, 20, 1595–1599. [Google Scholar] [CrossRef]
- Kumada, M.; Kihara, S.; Sumitsuji, S.; Kawamoto, T.; Matsumoto, S.; Ouchi, N.; Arita, Y.; Okamoto, Y.; Shimomura, I.; Hiraoka, H.; et al. Association of Hypoadiponectinemia with Coronary Artery Disease in Men. Arter. Thromb. Vasc. Biol. 2003, 23, 85–89. [Google Scholar] [CrossRef]
- Baratta, R.; Amato, S.; Degano, C.; Farina, M.G.; Patanè, G.; Vigneri, R.; Frittitta, L. Adiponectin Relationship with Lipid Metabolism Is Independent of Body Fat Mass: Evidence from Both Cross-Sectional and Intervention Studies. J. Clin. Endocrinol. Metab. 2004, 89, 2665–2671. [Google Scholar] [CrossRef]
- Albanese, A.; Tuttolomondo, A.; Anile, C.; Sabatino, G.; Pompucci, A.; Pinto, A.; Licata, G.; Mangiola, A. Spontaneous chronic subdural hematomas in young adults with a deficiency in coagulation factor XIII. Report of three cases. J. Neurosurg. 2005, 102, 1130–1132. [Google Scholar] [CrossRef]
- Della Corte, V.; Tuttolomondo, A.; Pecoraro, R.; Di Raimondo, D.; Vassallo, V.; Pinto, A. Inflammation, Endothelial Dysfunction and Arterial Stiffness as Therapeutic Targets in Cardiovascular Medicine. Curr. Pharm. Des. 2016, 22, 4658–4668. [Google Scholar] [CrossRef] [PubMed]
- Zanoli, L.; Boutouyrie, P.; Fatuzzo, P.; Granata, A.; Lentini, P.; Oztürk, K.; Cappello, M.; Theocharidou, E.; Tuttolomondo, A.; Pinto, A.; et al. Inflammation and Aortic Stiffness: An Individual Participant Data Meta-Analysis in Patients With Inflammatory Bowel Disease. J. Am. Heart Assoc. 2017, 6, e007003. [Google Scholar] [CrossRef] [PubMed]
- Basili, S.; Raparelli, V.; Napoleone, L.; Talerico, G.; Corazza, G.R.; Perticone, F.; Sacerdoti, D.; Andriulli, A.; Licata, A.; Pietrangelo, A.; et al. Platelet Count Does Not Predict Bleeding in Cirrhotic Patients: Results from the PRO-LIVER Study. Am. J. Gastroenterol. 2018, 113, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Siragusa, S.; Malato, A.; Saccullo, G.; Iorio, A.; Di Ianni, M.; Caracciolo, C.; Coco, L.L.; Raso, S.; Santoro, M.; Guarneri, F.P.; et al. Residual vein thrombosis for assessing duration of anticoagulation after unprovoked deep vein thrombosis of the lower limbs: The extended DACUS study. Am. J. Hematol. 2011, 86, 914–917. [Google Scholar] [CrossRef] [PubMed]
- Di Raimondo, D.; Tuttolomondo, A.; Buttà, C.; Casuccio, A.; Giarrusso, L.; Miceli, G.; Licata, G.; Pinto, A. Metabolic and anti-inflammatory effects of a home-based programme of aerobic physical exercise. Int. J. Clin. Pract. 2013, 67, 1247–1253. [Google Scholar] [CrossRef]
- Pinto, A.; Tuttolomondo, A.; Di Raimondo, D.; Fernandez, P.; Licata, G. Risk factors profile and clinical outcome of ischemic stroke patients admitted in a Department of Internal Medicine and classified by TOAST classification. Int. Angiol. 2006, 25, 261–267. [Google Scholar]
- Tuttolomondo, A.; Pinto, A.; Corrao, S.; Di Raimondo, D.; Fernandez, P.; Di Sciacca, R.; Arnao, V.; Licata, G. Immuno-inflammatory and thrombotic/fibrinolytic variables associated with acute ischemic stroke diagnosis. Atherosclerosis 2009, 203, 503–508. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; Simonetta, I.; Daidone, M.; Mogavero, A.; Ortello, A.; Pinto, A. Metabolic and Vascular Effect of the Mediterranean Diet. Int. J. Mol. Sci. 2019, 20, 4716. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Main Author | Study Design | Results | Ref. |
|---|---|---|---|
| Framingham study | Cohort study. 20 years of cohort surveillance | 2.5-fold incidence of ischemic stroke in diabetic men and a 3.6-fold one in diabetic women. | [74] |
| Tuttolomondo | Case-control prospective study | Diabetes was associated with lacunar ischemic stroke subtype, with a record of hypertension, and a better Scandinavian Stroke Scale score at admission. The association of diabetes with lacunar stroke remained significant even after adjustment for hypertension or large artery atherosclerotic and cardioembolic stroke subtypes. | [77] |
| Karapanayiotides | Case-control study | Diabetes was associated with a higher relative frequency of small-vessel and large-artery disease, a lower relative prevalence of intracerebral hemorrhage, a higher relative prevalence of subcortical infarction. | [90] |
| Manolio | Prospective study | Odds ratio (OR) of 2.12 in those who have diabetes after an adjustment for other risk factors. | [93] |
| Giles | Prospective study | Odds ratio (OR) of 2.47 in those who have diabetes after an adjustment for other risk factors. | [94] |
| Megherbi | Prospective study | Diabetic patients, compared with those without diabetes, were more likely to have limb weakness, dysarthria, ischemic stroke, and lacunar cerebral infarction. | [95] |
| Larsson | Mendelian randomization (MR) analysis | MR analysis showed associations between genetically predicted T2D (type 2 mellitus diabetes) and large artery stroke (OR 1.28) and small vessel stroke (OR 1.21) but not cardioembolic stroke. | [96] |
| Roper | Longitudinal, population-based study | All-cause standardized mortality ratios for type 2 diabetes were 160 (147 to 174) in women and 141 (130 to 152) in men. Cause-specific standardized mortality ratios were increased for ischemic heart disease, cerebrovascular disease, and renal disease. | [97] |
| Pinto | Case-control prospective study | Higher prevalence of major cardiovascular risk factors (such as dyslipidemia), of asymptomatic markers of cardiovascular disease (CVD), and a higher prevalence and incidence of previous and new-onset vascular events (coronary artery disease, transient ischemic attack/ischemic stroke, diabetic retinopathy) in diabetic patients with foot complications. | [98] |
| Tuttolomondo | Case-control study | Patients with DFS (diabetic foot syndrome) showed higher mean values of PWV (pulse wave velocity), lower mean values of RHI (reactive hyperemia index), and lower mean MMSE (mini-mental state examination). | [99] |
| Tuttolomondo | Case-control study | DFS patients show a higher degree of activation of the parasympathetic system than diabetic controls and a higher degree of vascular impairment, as indicated by lower RHI values. | [100] |
| Subtypes of SVD Neuroradiological Aspect | |
|---|---|
| Recent small subcortical infarct | • Recent infarction in one perforating arteriole and its territory • Increased DWI, FLAIR, T2-weighted signal • Decreased T1-weighted signal • Iso-intense T2-weighted GRE signal |
| WMHs | • Increase intensity or hyperintensity on T2-weighted, T2 weighted GRE and FLAIR signal • Iso-intense on DWI and T1-weighted signal • Decrease intensity or hypointense on T1-weighted signal |
| Lacune | • Round or ovoid fluid filed cavity mostly in subcortical region • Hyperintensity on T2-weighted signal • Decreased signal in FLAIR and T1-weighted images • Signal similar to CSF • Decreased or iso-intense signal on DWI |
| Perivascular space | • Fluid-filled spaces that follow the typical course of a vessel as it goes through gray or white matter similar signal intensity with CSF • Decrease FLAIR and T1-weighted signal • IncreasedT2-weighted-signal • DW1 and T2- weighted GRE signal seems iso-intense |
| Cerebral microbleed | • Small, rounded areas of signal void • Iso-intense DWI, FLAIR, 12- and-weighted signal • Best seen in T21 weighted GRE with decreased signal |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maida, C.D.; Daidone, M.; Pacinella, G.; Norrito, R.L.; Pinto, A.; Tuttolomondo, A. Diabetes and Ischemic Stroke: An Old and New Relationship an Overview of the Close Interaction between These Diseases. Int. J. Mol. Sci. 2022, 23, 2397. https://doi.org/10.3390/ijms23042397
Maida CD, Daidone M, Pacinella G, Norrito RL, Pinto A, Tuttolomondo A. Diabetes and Ischemic Stroke: An Old and New Relationship an Overview of the Close Interaction between These Diseases. International Journal of Molecular Sciences. 2022; 23(4):2397. https://doi.org/10.3390/ijms23042397
Chicago/Turabian StyleMaida, Carlo Domenico, Mario Daidone, Gaetano Pacinella, Rosario Luca Norrito, Antonio Pinto, and Antonino Tuttolomondo. 2022. "Diabetes and Ischemic Stroke: An Old and New Relationship an Overview of the Close Interaction between These Diseases" International Journal of Molecular Sciences 23, no. 4: 2397. https://doi.org/10.3390/ijms23042397
APA StyleMaida, C. D., Daidone, M., Pacinella, G., Norrito, R. L., Pinto, A., & Tuttolomondo, A. (2022). Diabetes and Ischemic Stroke: An Old and New Relationship an Overview of the Close Interaction between These Diseases. International Journal of Molecular Sciences, 23(4), 2397. https://doi.org/10.3390/ijms23042397