
Antibody-Drug Conjugate Targeting c-Kit for the Treatment of Small Cell Lung Cancer


Abstract
:1. Introduction
2. Results
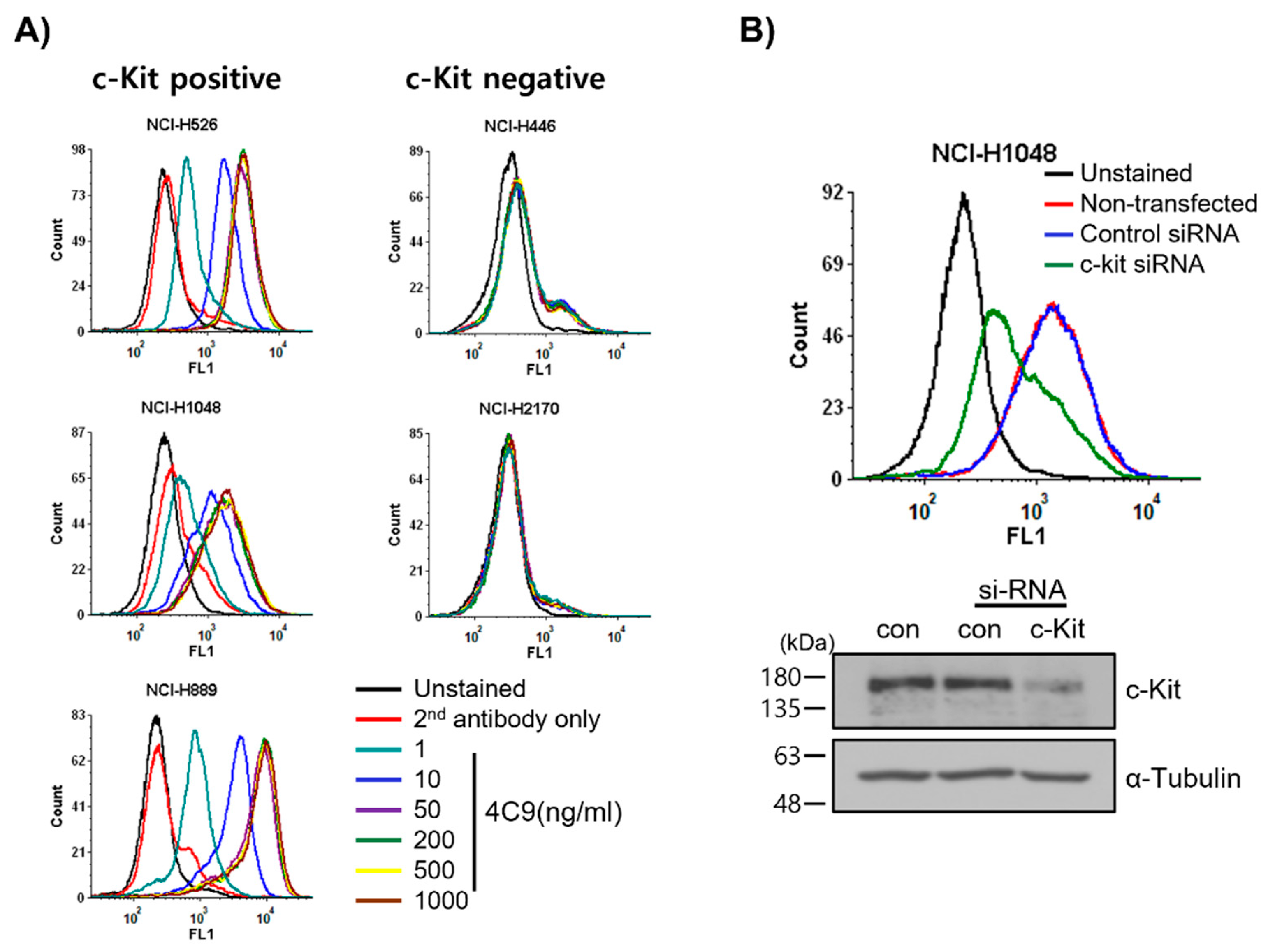
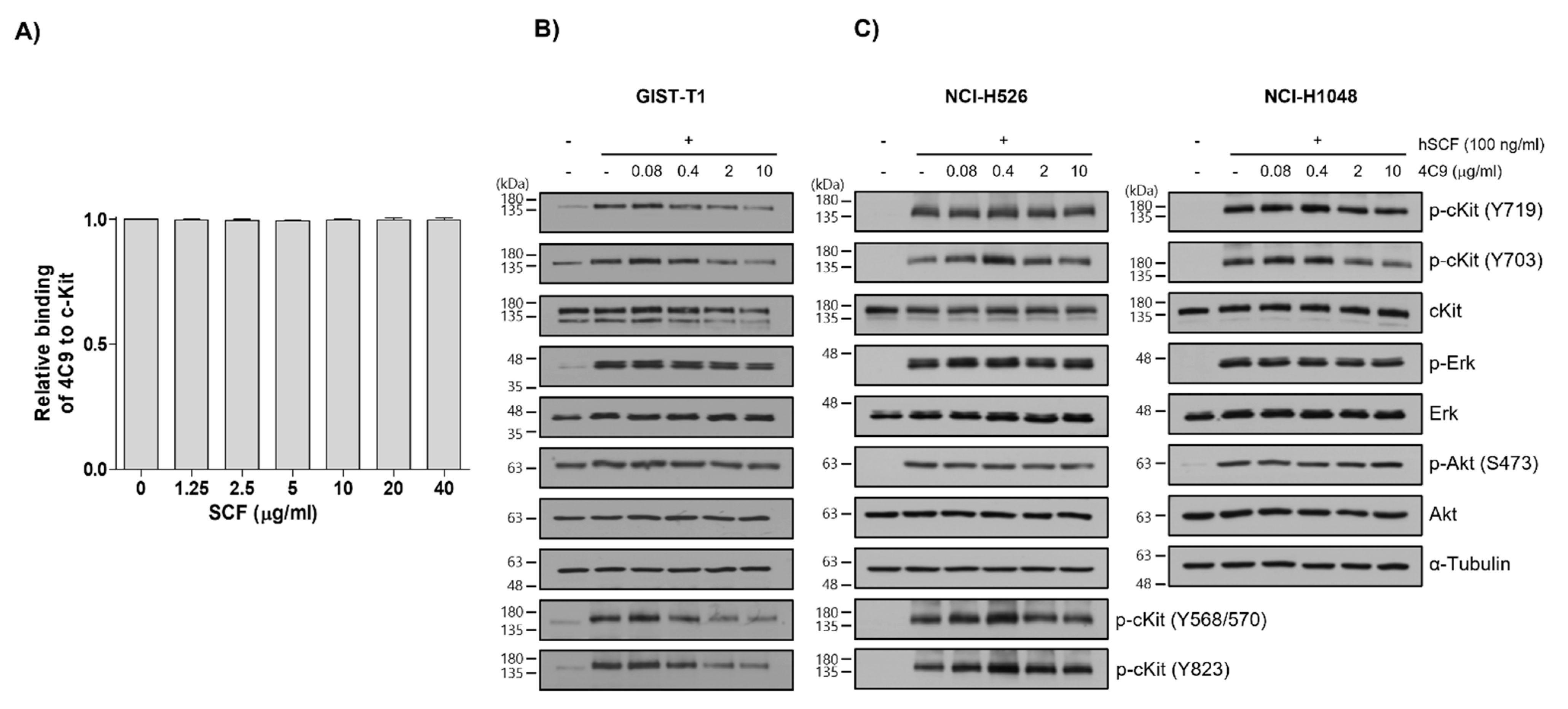
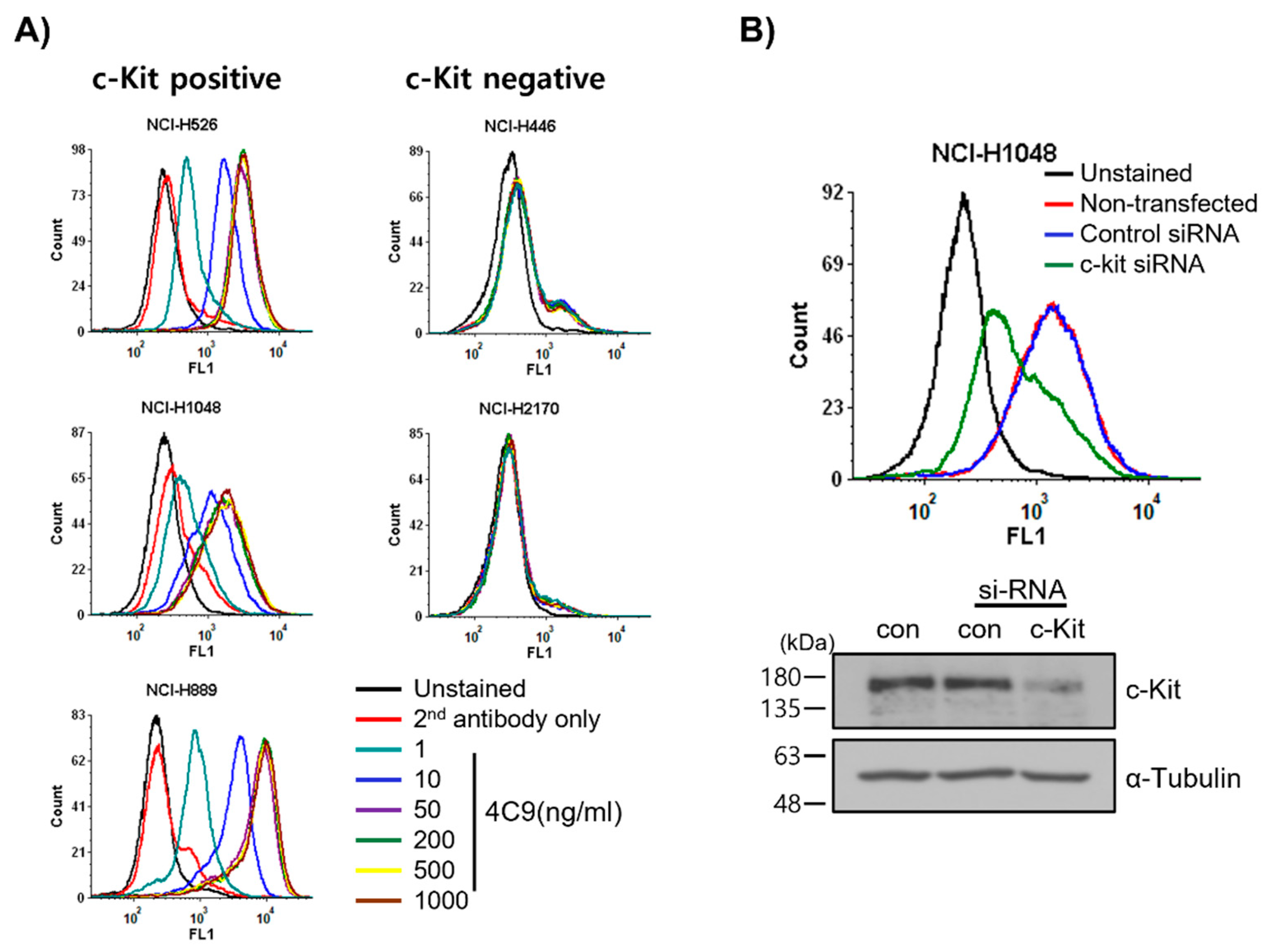
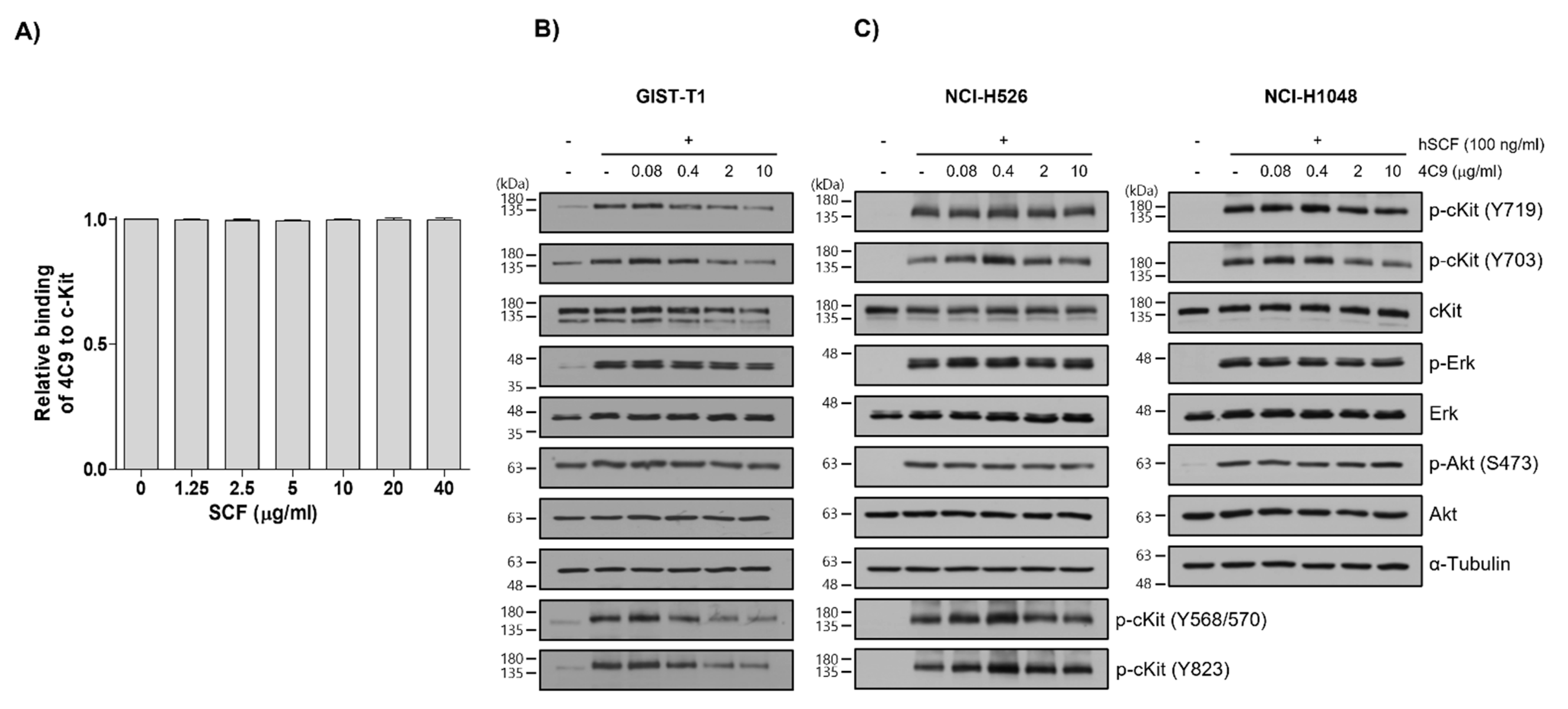
2.1. 4C9 Antibody Specifically Binds to c-Kit
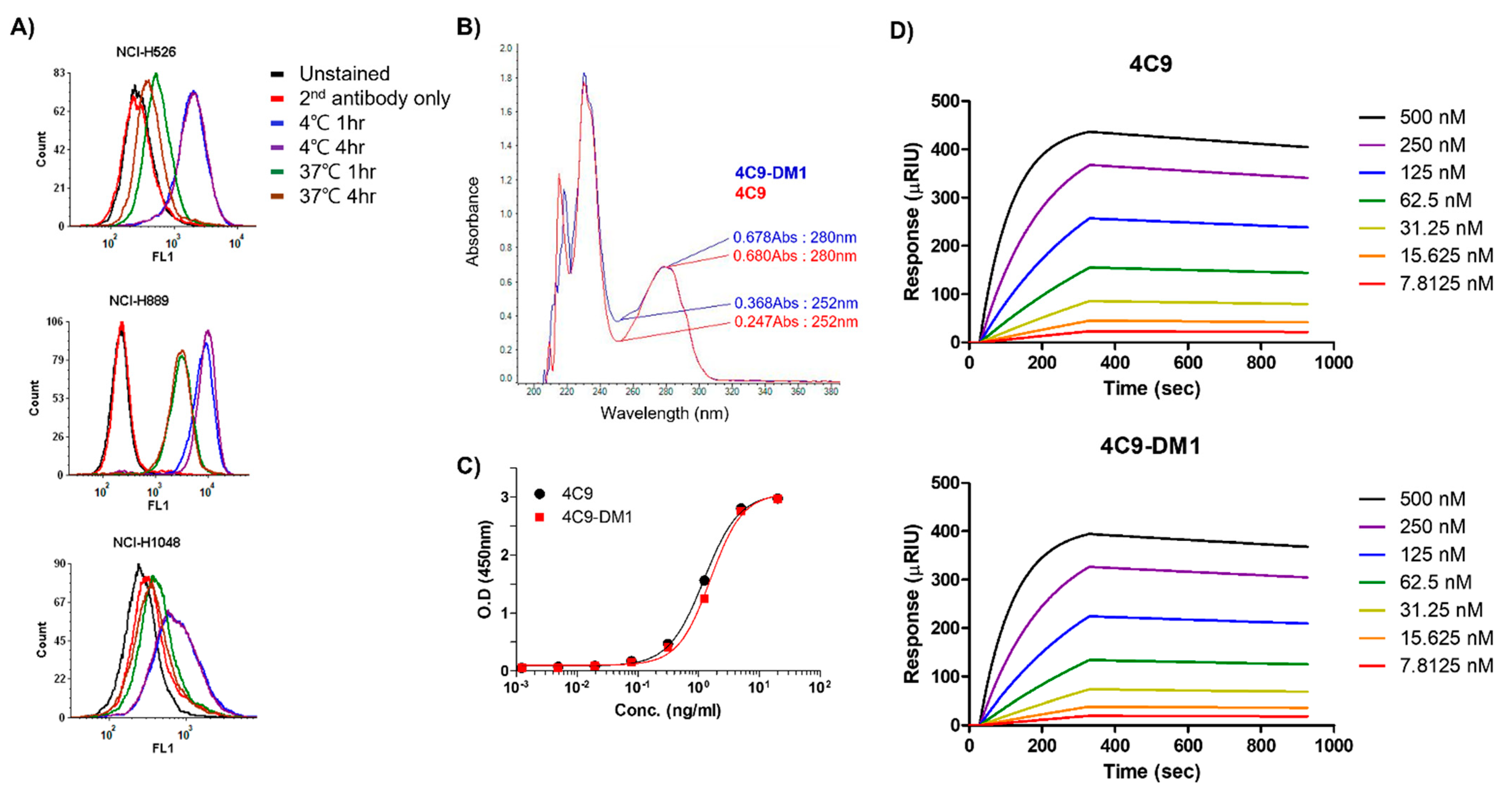
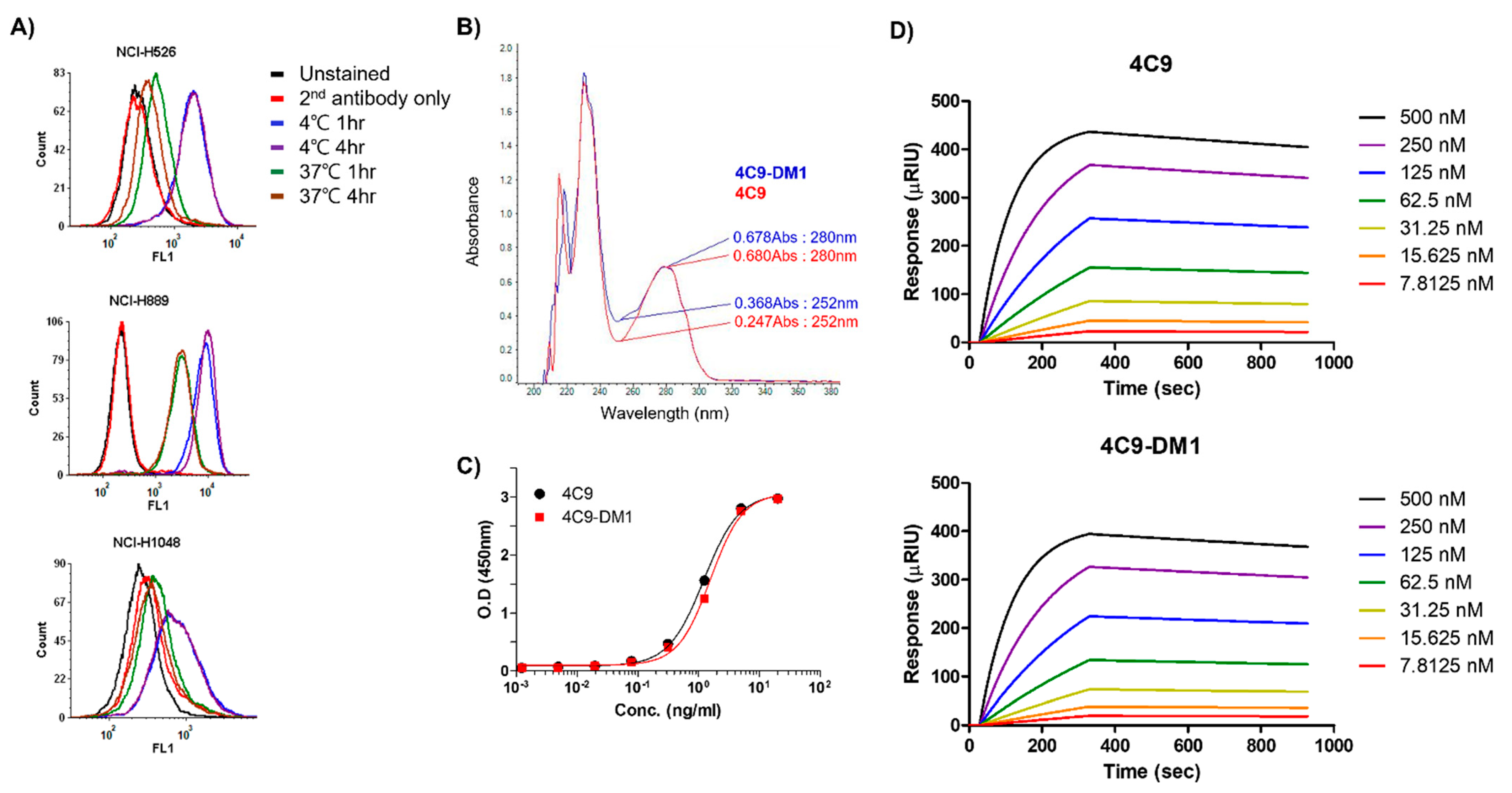
2.2. Generation and Characterization of ADC (4C9-DM1)
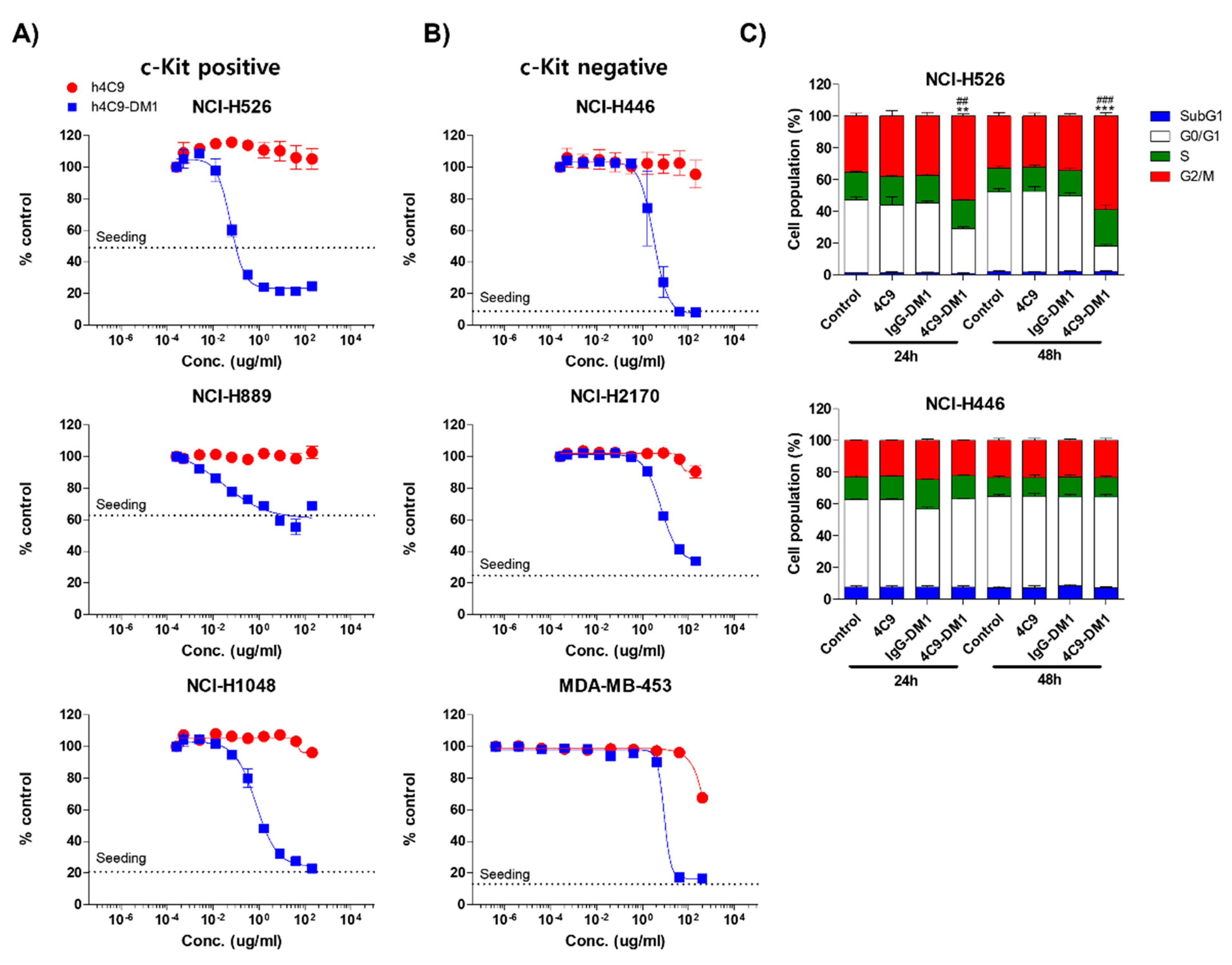
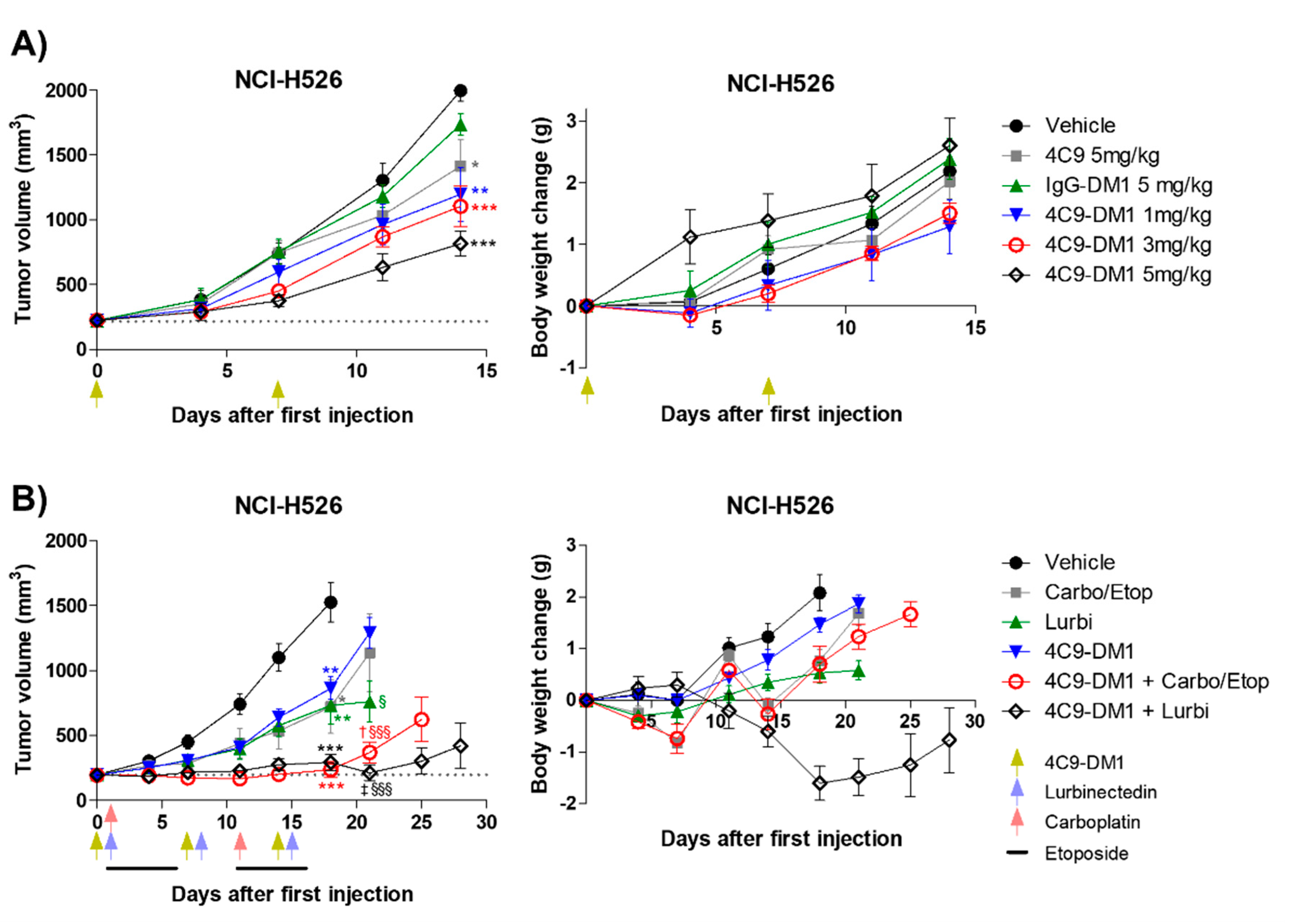
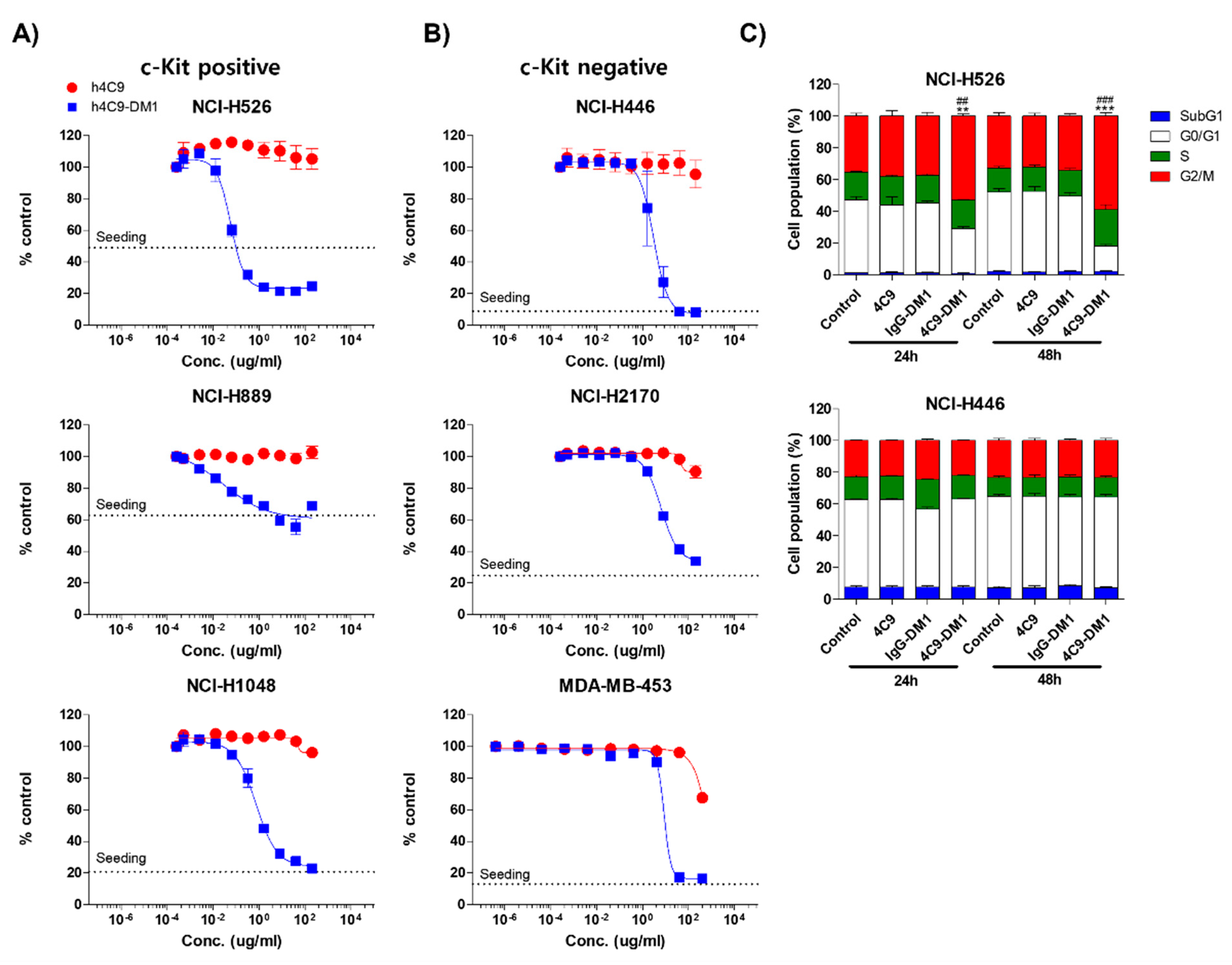
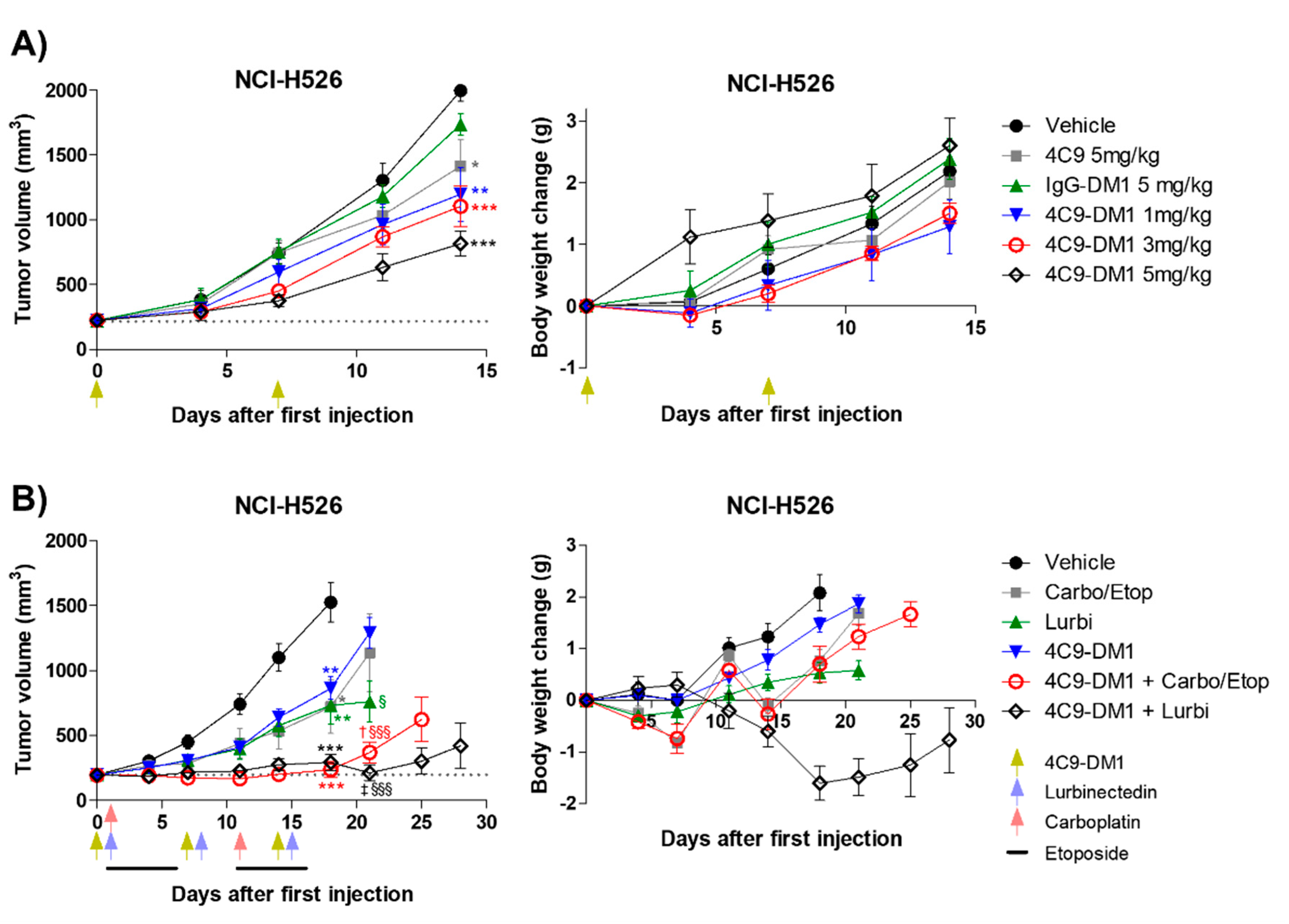
2.3. 4C9-DM1 Exhibits Antitumor Activity In Vitro and In Vivo
3. Discussion
4. Materials and Methods
4.1. Generation and Production of 4C9 Antibody
4.2. Cell Lines and Culture
4.3. Flow Cytometry
4.4. Western Blot Analysis
4.5. si-RNA Study
4.6. ELISA
4.7. Internalization Assay
4.8. Generation of ADC
- εD280 = molar extinction coefficient of SMCC-DM1 at 280 nm.
- εAb280 = molar extinction coefficient of 4C9 at 280 nm.
- εD252 = molar extinction coefficient of SMCC-DM1 at 252 nm.
- εAb252 = molar extinction coefficient of 4C9 at 252 nm.
- CD = molar concentration of SMCC-DM1.
- CAb = molar concentration of 4C9.
4.9. In Vitro Cytotoxicity Test
4.10. In Vivo Studies Using Xenograft Mouse Model
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| SCLC | Small cell lung cancer |
| ADC | Antibody-drug conjugate |
| TGI | Tumor growth inhibition |
| NSCLC | Non-small cell lung cancer |
| EGFR | Epidermal growth factor receptor |
| mTOR | Mammalian target of rapamycin |
| PI3K | Phosphoinositide 3-kinase |
| SCF | Stem cell factor |
| GIST | Gastrointestinal stromal tumor |
| SMCC | N-succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate |
| DAR | Drug-antibody ratio |
| IC50 | Half-maximum inhibitory concentration |
| Ppp1r3b | Protein phosphatase 1 regulatory subunit 3B |
| PD-L1 | Programmed death-ligand 1 |
References
- Blandin Knight, S.; Crosbie, P.A.; Balata, H.; Chudziak, J.; Hussell, T.; Dive, C. Progress and prospects of early detection in lung cancer. Open Biol. 2017, 7, 170070. [Google Scholar] [CrossRef] [Green Version]
- Elias, A.D. Small cell lung cancer: State-of-the-art therapy in 1996. Chest 1997, 112, 251S–258S. [Google Scholar] [CrossRef]
- Dores, G.M.; Qubaiah, O.; Mody, A.; Ghabach, B.; Devesa, S.S. A population-based study of incidence and patient survival of small cell carcinoma in the United States, 1992–2010. BMC Cancer 2015, 15, 185. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Tang, J.; Sun, T.; Zheng, X.; Li, J.; Sun, H.; Zhou, X.; Zhou, C.; Zhang, H.; Cheng, Z.; et al. Survival changes in patients with small cell lung cancer and disparities between different sexes, socioeconomic statuses and ages. Sci. Rep. 2017, 7, 1339. [Google Scholar] [CrossRef]
- Salgia, R.; Skarin, A.T. Molecular abnormalities in lung cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1998, 16, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Rozengurt, E. Autocrine loops, signal transduction, and cell cycle abnormalities in the molecular biology of lung cancer. Curr. Opin. Oncol. 1999, 11, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Wojtalla, A.; Fischer, B.; Kotelevets, N.; Mauri, F.A.; Sobek, J.; Rehrauer, H.; Wotzkow, C.; Tschan, M.P.; Seckl, M.J.; Zangemeister-Wittke, U.; et al. Targeting the phosphoinositide 3-kinase p110-alpha isoform impairs cell proliferation, survival, and tumor growth in small cell lung cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.H.; Girard, L.; Giacomini, C.P.; Wang, P.; Hernandez-Boussard, T.; Tibshirani, R.; Minna, J.D.; Pollack, J.R. Combined microarray analysis of small cell lung cancer reveals altered apoptotic balance and distinct expression signatures of MYC family gene amplification. Oncogene 2006, 25, 130–138. [Google Scholar] [CrossRef] [Green Version]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretic, L.; Kong, G.; Leenders, F.; Lu, X.; Fernandez-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef]
- Carvajal, L.A.; Manfredi, J.J. Another fork in the road--life or death decisions by the tumour suppressor p53. EMBO Rep. 2013, 14, 414–421. [Google Scholar] [CrossRef] [Green Version]
- Gazdar, A.F.; Bunn, P.A.; Minna, J.D. Small-cell lung cancer: What we know, what we need to know and the path forward. Nat. Rev. Cancer 2017, 17, 765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, B.W.; Glisson, B.S.; Truong, M.T.; Erasmus, J.J. Small cell lung carcinoma: Staging, imaging, and treatment considerations. Radiographics 2014, 34, 1707–1721. [Google Scholar] [CrossRef] [PubMed]
- Pleasance, E.D.; Stephens, P.J.; O’Meara, S.; McBride, D.J.; Meynert, A.; Jones, D.; Lin, M.L.; Beare, D.; Lau, K.W.; Greenman, C.; et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature 2010, 463, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Ju, Y.S.; Haase, K.; Van Loo, P.; Martincorena, I.; Nik-Zainal, S.; Totoki, Y.; Fujimoto, A.; Nakagawa, H.; Shibata, T.; et al. Mutational signatures associated with tobacco smoking in human cancer. Science 2016, 354, 618–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarden, Y.; Kuang, W.J.; Yang-Feng, T.; Coussens, L.; Munemitsu, S.; Dull, T.J.; Chen, E.; Schlessinger, J.; Francke, U.; Ullrich, A. Human proto-oncogene c-kit: A new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO J. 1987, 6, 3341–3351. [Google Scholar] [CrossRef] [PubMed]
- Geissler, E.N.; Ryan, M.A.; Housman, D.E. The dominant-white spotting (W) locus of the mouse encodes the c-kit proto-oncogene. Cell 1988, 55, 185–192. [Google Scholar] [CrossRef]
- Ashman, L.K. The biology of stem cell factor and its receptor C-kit. Int. J. Biochem. Cell Biol. 1999, 31, 1037–1051. [Google Scholar] [CrossRef]
- Vose, J.M.; Armitage, J.O. Clinical applications of hematopoietic growth factors. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1995, 13, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Lennartsson, J.; Ronnstrand, L. Stem cell factor receptor/c-Kit: From basic science to clinical implications. Physiol. Rev. 2012, 92, 1619–1649. [Google Scholar] [CrossRef] [Green Version]
- Bougherara, H.; Subra, F.; Crepin, R.; Tauc, P.; Auclair, C.; Poul, M.A. The aberrant localization of oncogenic kit tyrosine kinase receptor mutants is reversed on specific inhibitory treatment. Mol. Cancer Res. MCR 2009, 7, 1525–1533. [Google Scholar] [CrossRef] [Green Version]
- Krystal, G.W.; Hines, S.J.; Organ, C.P. Autocrine growth of small cell lung cancer mediated by coexpression of c-kit and stem cell factor. Cancer Res. 1996, 56, 370–376. [Google Scholar]
- Hibi, K.; Takahashi, T.; Sekido, Y.; Ueda, R.; Hida, T.; Ariyoshi, Y.; Takagi, H.; Takahashi, T. Coexpression of the stem cell factor and the c-kit genes in small-cell lung cancer. Oncogene 1991, 6, 2291–2296. [Google Scholar] [PubMed]
- Sekido, Y.; Obata, Y.; Ueda, R.; Hida, T.; Suyama, M.; Shimokata, K.; Ariyoshi, Y.; Takahashi, T. Preferential expression of c-kit protooncogene transcripts in small cell lung cancer. Cancer Res. 1991, 51, 2416–2419. [Google Scholar] [PubMed]
- Hida, T.; Ueda, R.; Sekido, Y.; Hibi, K.; Matsuda, R.; Ariyoshi, Y.; Sugiura, T.; Takahashi, T.; Takahashi, T. Ectopic expression of c-kit in small-cell lung cancer. Int. J. Cancer. Suppl. = J. Int. Du Cancer. Suppl. 1994, 8, 108–109. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Qian, S.X. Clinical efficacy and safety of imatinib in the management of Ph(+) chronic myeloid or acute lymphoblastic leukemia in Chinese patients. OncoTargets Ther. 2014, 7, 395–404. [Google Scholar] [CrossRef] [Green Version]
- Lopes, L.F.; Bacchi, C.E. Imatinib treatment for gastrointestinal stromal tumour (GIST). J. Cell. Mol. Med. 2010, 14, 42–50. [Google Scholar] [CrossRef] [Green Version]
- Helbig, G. Imatinib for the treatment of hypereosinophilic syndromes. Expert Rev. Clin. Immunol. 2018, 14, 163–170. [Google Scholar] [CrossRef]
- Sacha, T. Imatinib in chronic myeloid leukemia: An overview. Mediterr. J. Hematol. Infect. Dis. 2014, 6, e2014007. [Google Scholar] [CrossRef]
- Krystal, G.W.; Honsawek, S.; Litz, J.; Buchdunger, E. The selective tyrosine kinase inhibitor STI571 inhibits small cell lung cancer growth. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2000, 6, 3319–3326. [Google Scholar]
- Wang, W.L.; Healy, M.E.; Sattler, M.; Verma, S.; Lin, J.; Maulik, G.; Stiles, C.D.; Griffin, J.D.; Johnson, B.E.; Salgia, R. Growth inhibition and modulation of kinase pathways of small cell lung cancer cell lines by the novel tyrosine kinase inhibitor STI 571. Oncogene 2000, 19, 3521–3528. [Google Scholar] [CrossRef]
- Soria, J.C.; Johnson, B.E.; Chevalier, T.L. Imatinib in small cell lung cancer. Lung Cancer 2003, 41 (Suppl. S1), S49–S53. [Google Scholar] [CrossRef]
- Johnson, B.E.; Fischer, T.; Fischer, B.; Dunlop, D.; Rischin, D.; Silberman, S.; Kowalski, M.O.; Sayles, D.; Dimitrijevic, S.; Fletcher, C.; et al. Phase II study of imatinib in patients with small cell lung cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 5880–5887. [Google Scholar]
- Dy, G.K.; Miller, A.A.; Mandrekar, S.J.; Aubry, M.C.; Langdon, R.M., Jr.; Morton, R.F.; Schild, S.E.; Jett, J.R.; Adjei, A.A. A phase II trial of imatinib (ST1571) in patients with c-kit expressing relapsed small-cell lung cancer: A CALGB and NCCTG study. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2005, 16, 1811–1816. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.O.; Kim, K.H.; Baek, E.J.; Park, B.; So, M.K.; Ko, B.J.; Ko, H.J.; Park, S.G. A novel anti-c-Kit antibody-drug conjugate to treat wild-type and activating-mutant c-Kit-positive tumors. Mol. Oncol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Wagh, A.; Song, H.; Zeng, M.; Tao, L.; Das, T.K. Challenges and new frontiers in analytical characterization of antibody-drug conjugates. mAbs 2018, 10, 222–243. [Google Scholar] [CrossRef] [PubMed]
- Tedeschini, T.; Campara, B.; Grigoletto, A.; Bellini, M.; Salvalaio, M.; Matsuno, Y.; Suzuki, A.; Yoshioka, H.; Pasut, G. Polyethylene glycol-based linkers as hydrophilicity reservoir for antibody-drug conjugates. J. Control. Release Off. J. Control. Release Soc. 2021, 337, 431–447. [Google Scholar] [CrossRef]
- Singh, S.; Jaigirdar, A.A.; Mulkey, F.; Cheng, J.; Hamed, S.S.; Li, Y.; Liu, J.; Zhao, H.; Goheer, A.; Helms, W.S.; et al. FDA Approval Summary: Lurbinectedin for the Treatment of Metastatic Small Cell Lung Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 2378–2382. [Google Scholar] [CrossRef]
- Farago, A.F.; Keane, F.K. Current standards for clinical management of small cell lung cancer. Transl. Lung Cancer Res. 2018, 7, 69–79. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.; Salgia, R. Managing Patients With Relapsed Small-Cell Lung Cancer. J. Oncol. Pract. 2018, 14, 359–366. [Google Scholar] [CrossRef]
- Mehta, M.B.; Shewale, S.V.; Sequeira, R.N.; Millar, J.S.; Hand, N.J.; Rader, D.J. Hepatic protein phosphatase 1 regulatory subunit 3B (Ppp1r3b) promotes hepatic glycogen synthesis and thereby regulates fasting energy homeostasis. J. Biol. Chem. 2017, 292, 10444–10454. [Google Scholar] [CrossRef] [Green Version]
- Yuzawa, S.; Opatowsky, Y.; Zhang, Z.; Mandiyan, V.; Lax, I.; Schlessinger, J. Structural basis for activation of the receptor tyrosine kinase KIT by stem cell factor. Cell 2007, 130, 323–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masson, K.; Heiss, E.; Band, H.; Ronnstrand, L. Direct binding of Cbl to Tyr568 and Tyr936 of the stem cell factor receptor/c-Kit is required for ligand-induced ubiquitination, internalization and degradation. Biochem. J. 2006, 399, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Bayle, J.; Letard, S.; Frank, R.; Dubreuil, P.; De Sepulveda, P. Suppressor of cytokine signaling 6 associates with KIT and regulates KIT receptor signaling. J. Biol. Chem. 2004, 279, 12249–12259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Sun, Y.; Li, J.; Zhao, X.; Shi, X.; Gong, T.; Pan, S.; Zheng, Z.; Zhang, X. SOCS6 promotes radiosensitivity and decreases cancer cell stemness in esophageal squamous cell carcinoma by regulating c-Kit ubiquitylation. Cancer Cell Int. 2021, 21, 165. [Google Scholar] [CrossRef]
- Tsurutani, J.; West, K.A.; Sayyah, J.; Gills, J.J.; Dennis, P.A. Inhibition of the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin pathway but not the MEK/ERK pathway attenuates laminin-mediated small cell lung cancer cellular survival and resistance to imatinib mesylate or chemotherapy. Cancer Res. 2005, 65, 8423–8432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poruchynsky, M.S.; Komlodi-Pasztor, E.; Trostel, S.; Wilkerson, J.; Regairaz, M.; Pommier, Y.; Zhang, X.; Kumar Maity, T.; Robey, R.; Burotto, M.; et al. Microtubule-targeting agents augment the toxicity of DNA-damaging agents by disrupting intracellular trafficking of DNA repair proteins. Proc. Natl. Acad. Sci. USA 2015, 112, 1571–1576. [Google Scholar] [CrossRef] [Green Version]
- Kauffmann-Guerrero, D.; Huber, R.M. Orphan Drugs in Development for the Treatment of Small-Cell Lung Cancer: Emerging Data on Lurbinectedin. Lung Cancer 2020, 11, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Pavan, A.; Attili, I.; Pasello, G.; Guarneri, V.; Conte, P.F.; Bonanno, L. Immunotherapy in small-cell lung cancer: From molecular promises to clinical challenges. J. Immunother. Cancer 2019, 7, 205. [Google Scholar] [CrossRef] [Green Version]
- Akinleye, A.; Rasool, Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J. Hematol. Oncol. 2019, 12, 92. [Google Scholar] [CrossRef] [Green Version]
- Schultheis, A.M.; Scheel, A.H.; Ozretic, L.; George, J.; Thomas, R.K.; Hagemann, T.; Zander, T.; Wolf, J.; Buettner, R. PD-L1 expression in small cell neuroendocrine carcinomas. Eur. J. Cancer 2015, 51, 421–426. [Google Scholar] [CrossRef] [Green Version]
- Acheampong, E.; Abed, A.; Morici, M.; Bowyer, S.; Amanuel, B.; Lin, W.; Millward, M.; Gray, E.S. Tumour PD-L1 Expression in Small-Cell Lung Cancer: A Systematic Review and Meta-Analysis. Cells 2020, 9, 2393. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.O.; Kim, H.N.; Kim, K.H.; Baek, E.J.; Park, J.Y.; Ha, K.; Heo, D.R.; Seo, M.D.; Park, S.G. Development and characterization of a fully human antibody targeting SCF/c-kit signaling. Int. J. Biol. Macromol. 2020, 159, 66–78. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| c-Kit Expression | Tissue Type | Cell Line | 4C9-DM1 * | SMCC-DM1 |
|---|---|---|---|---|
| c-Kit positive | SCLC | NCI-H526 | 0.158 | 12.23 |
| NCI-H889 | 0.323 | 11.62 | ||
| NCI-H1048 | 4.08 | 30.45 | ||
| c-Kit negative | SCLC | NCI-H446 | 16.58 | 6.97 |
| NCI-H2170 | 35.5 | 20.17 | ||
| Breast cancer | MDA-MB-453 | 47.63 | 9.763 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, K.-H.; Kim, J.-O.; Park, J.-Y.; Seo, M.-D.; Park, S.G. Antibody-Drug Conjugate Targeting c-Kit for the Treatment of Small Cell Lung Cancer. Int. J. Mol. Sci. 2022, 23, 2264. https://doi.org/10.3390/ijms23042264
Kim K-H, Kim J-O, Park J-Y, Seo M-D, Park SG. Antibody-Drug Conjugate Targeting c-Kit for the Treatment of Small Cell Lung Cancer. International Journal of Molecular Sciences. 2022; 23(4):2264. https://doi.org/10.3390/ijms23042264
Chicago/Turabian StyleKim, Kwang-Hyeok, Jin-Ock Kim, Jeong-Yang Park, Min-Duk Seo, and Sang Gyu Park. 2022. "Antibody-Drug Conjugate Targeting c-Kit for the Treatment of Small Cell Lung Cancer" International Journal of Molecular Sciences 23, no. 4: 2264. https://doi.org/10.3390/ijms23042264
APA StyleKim, K.-H., Kim, J.-O., Park, J.-Y., Seo, M.-D., & Park, S. G. (2022). Antibody-Drug Conjugate Targeting c-Kit for the Treatment of Small Cell Lung Cancer. International Journal of Molecular Sciences, 23(4), 2264. https://doi.org/10.3390/ijms23042264