
The Involvement of RAGE and Its Ligands during Progression of ALS in SOD1 G93A Transgenic Mice

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
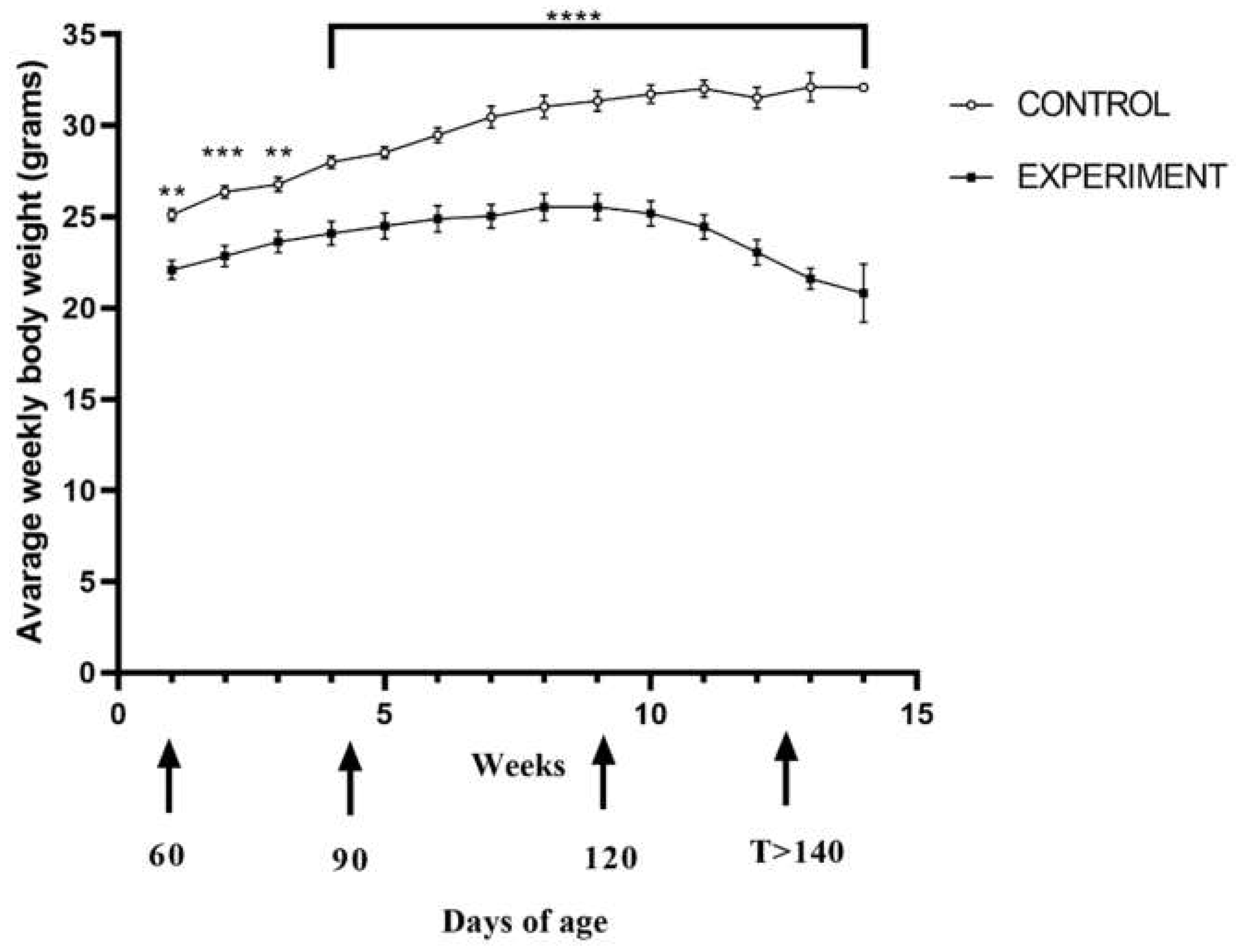
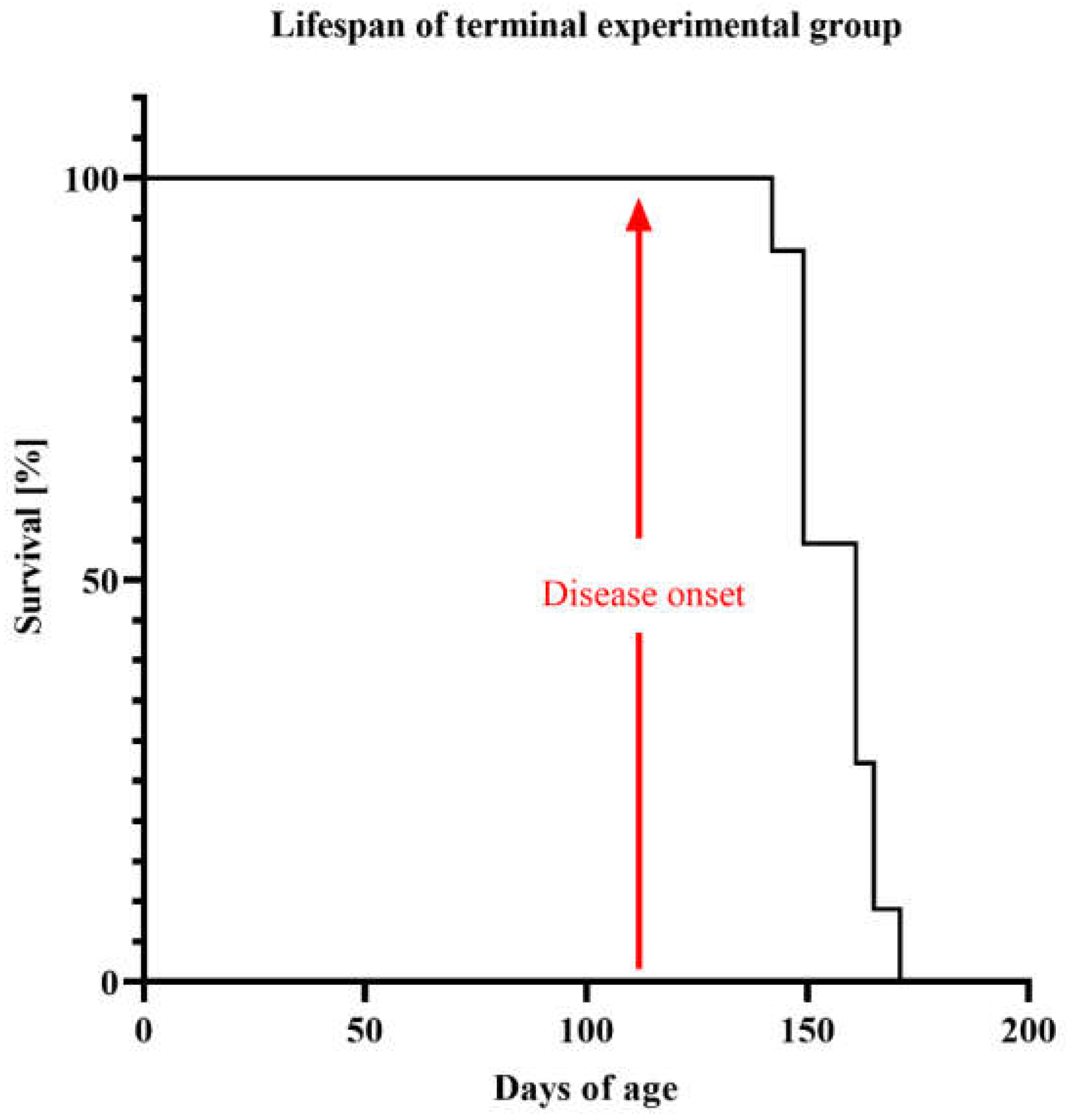
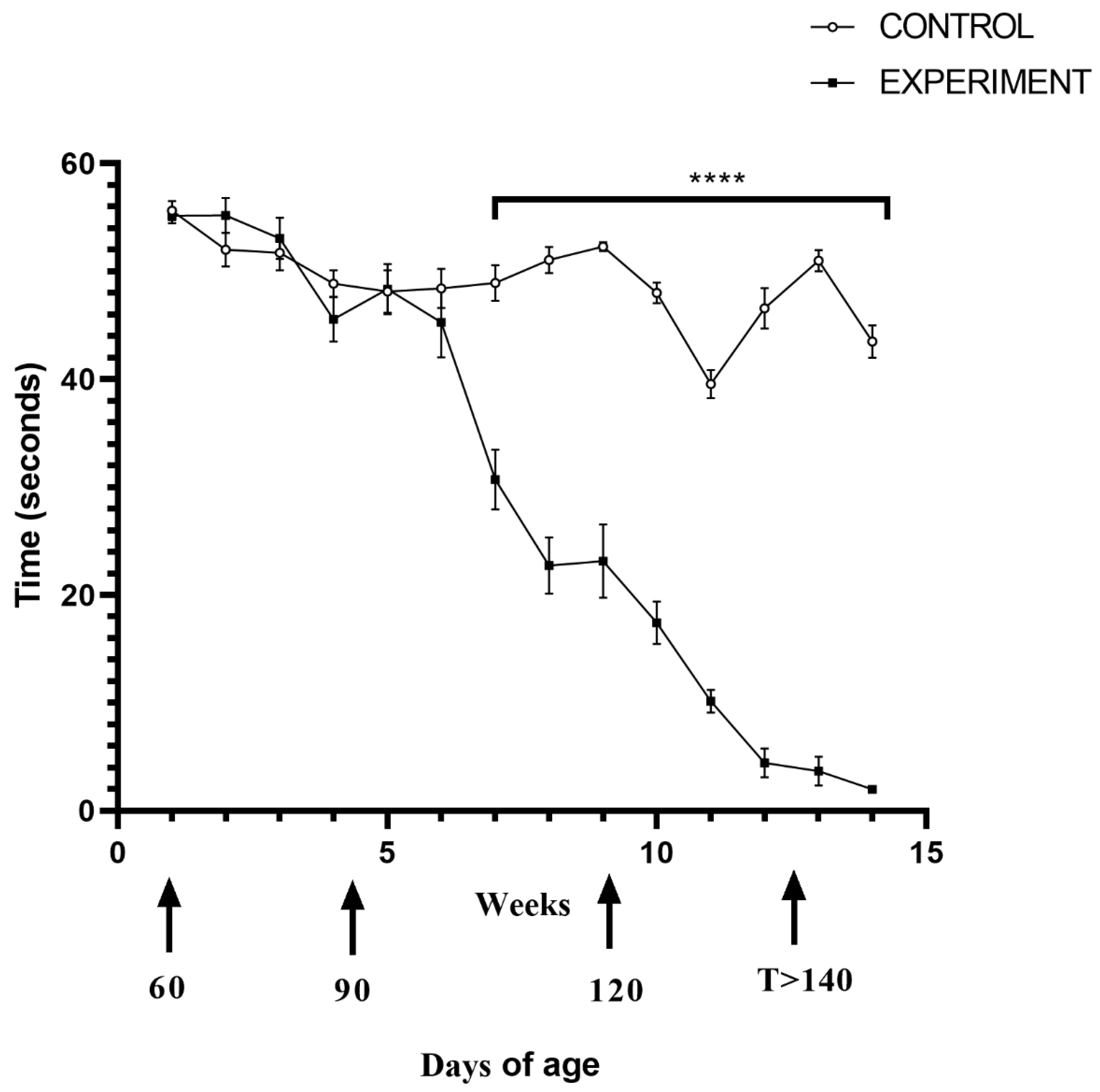
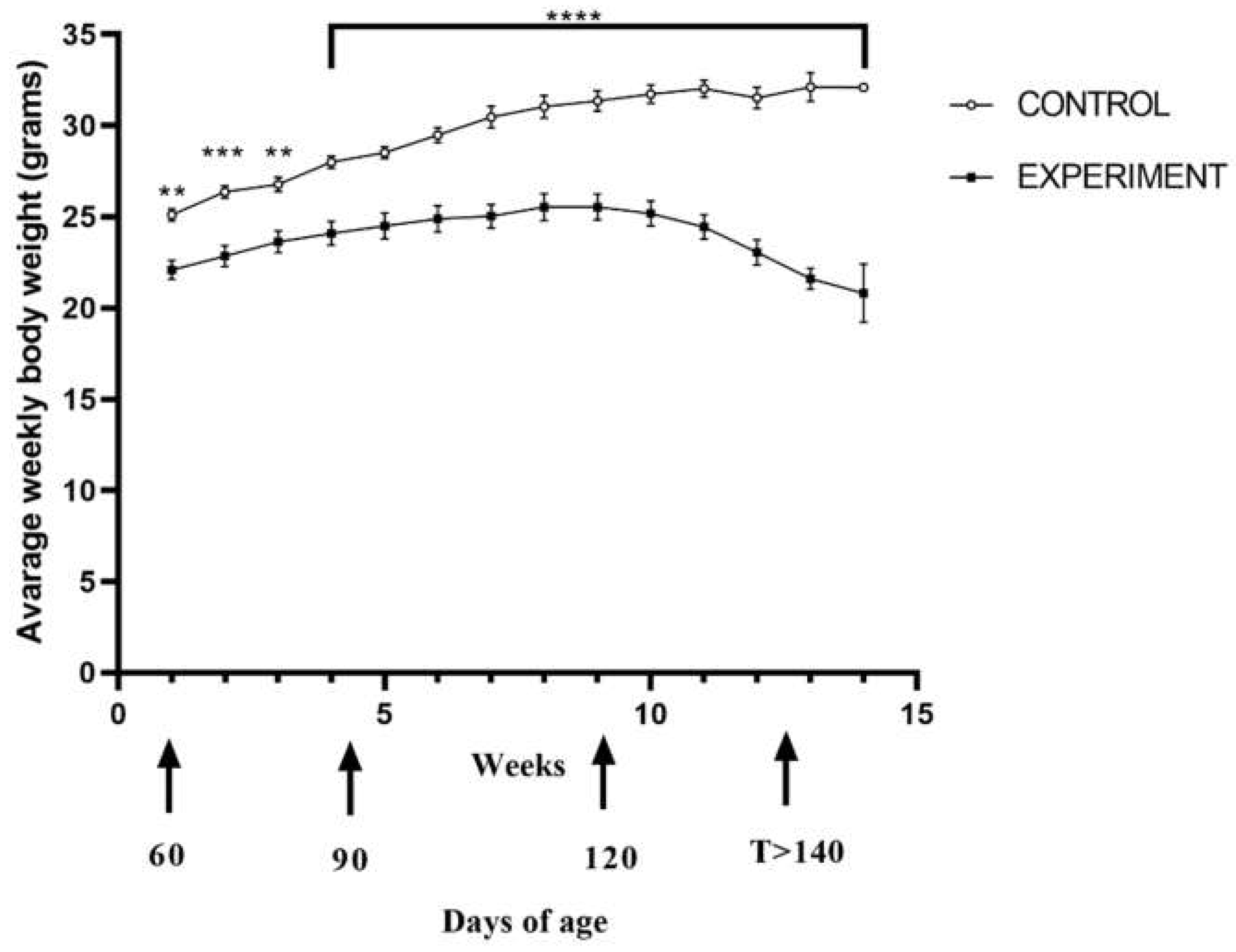
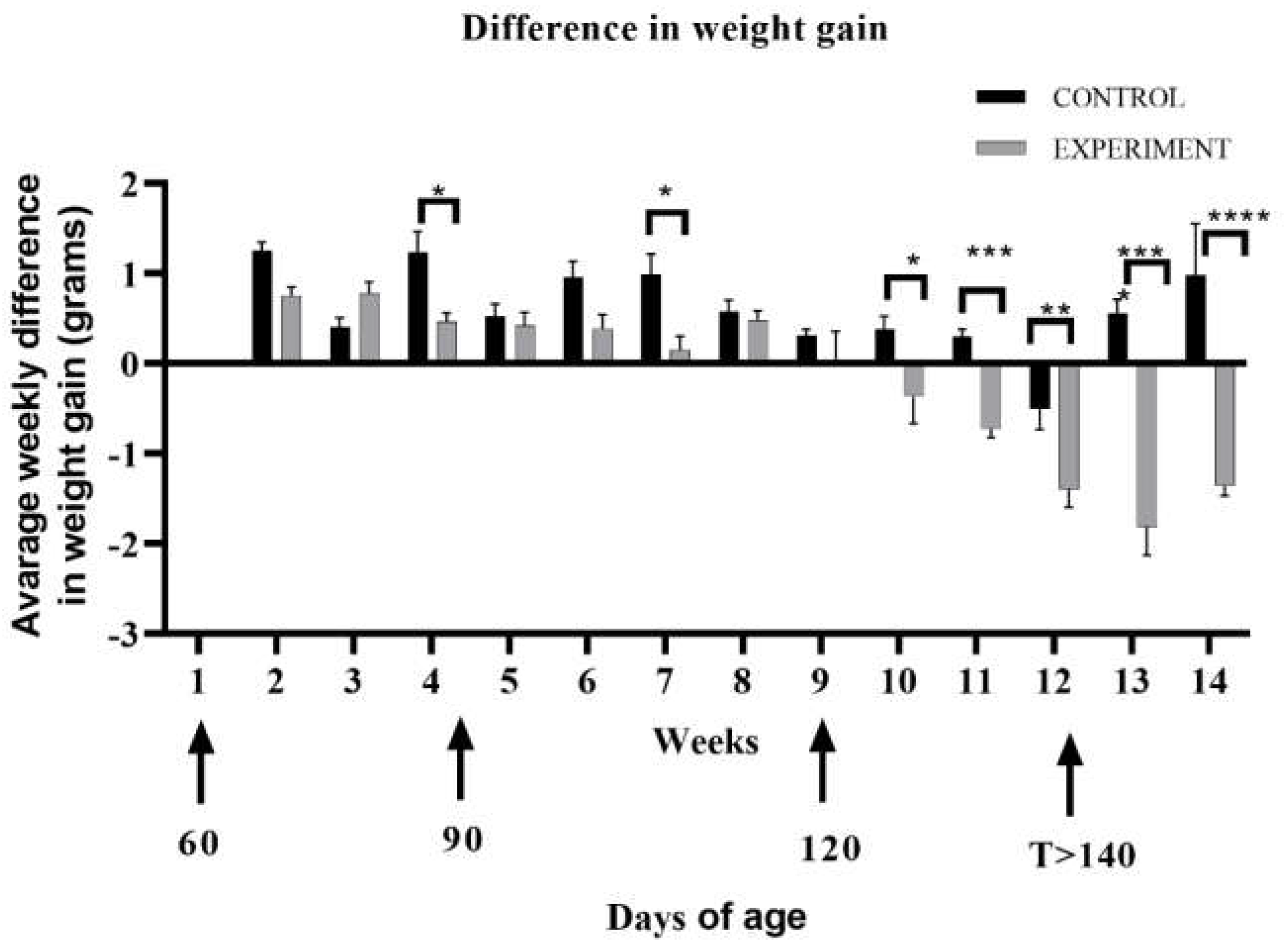
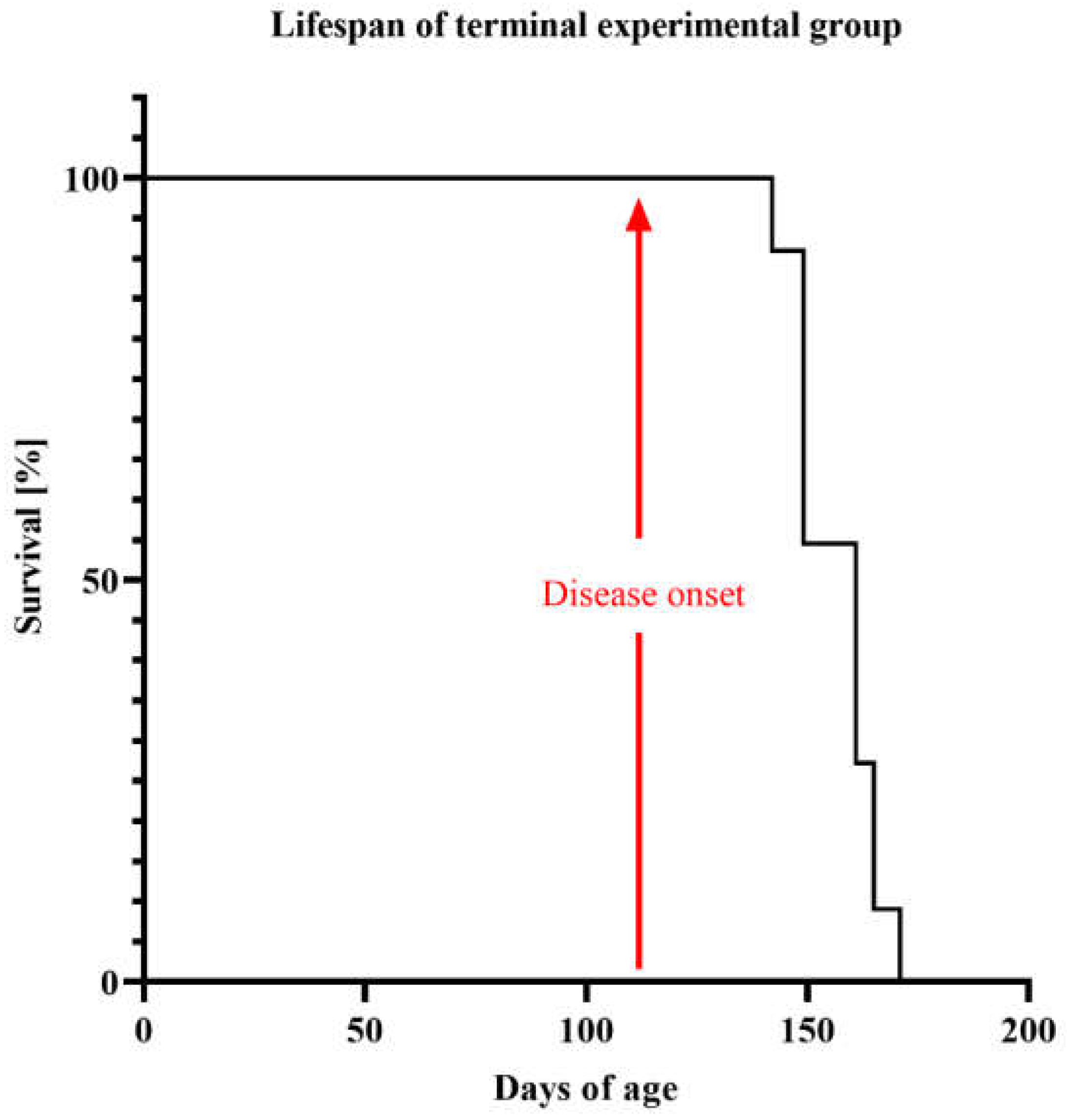
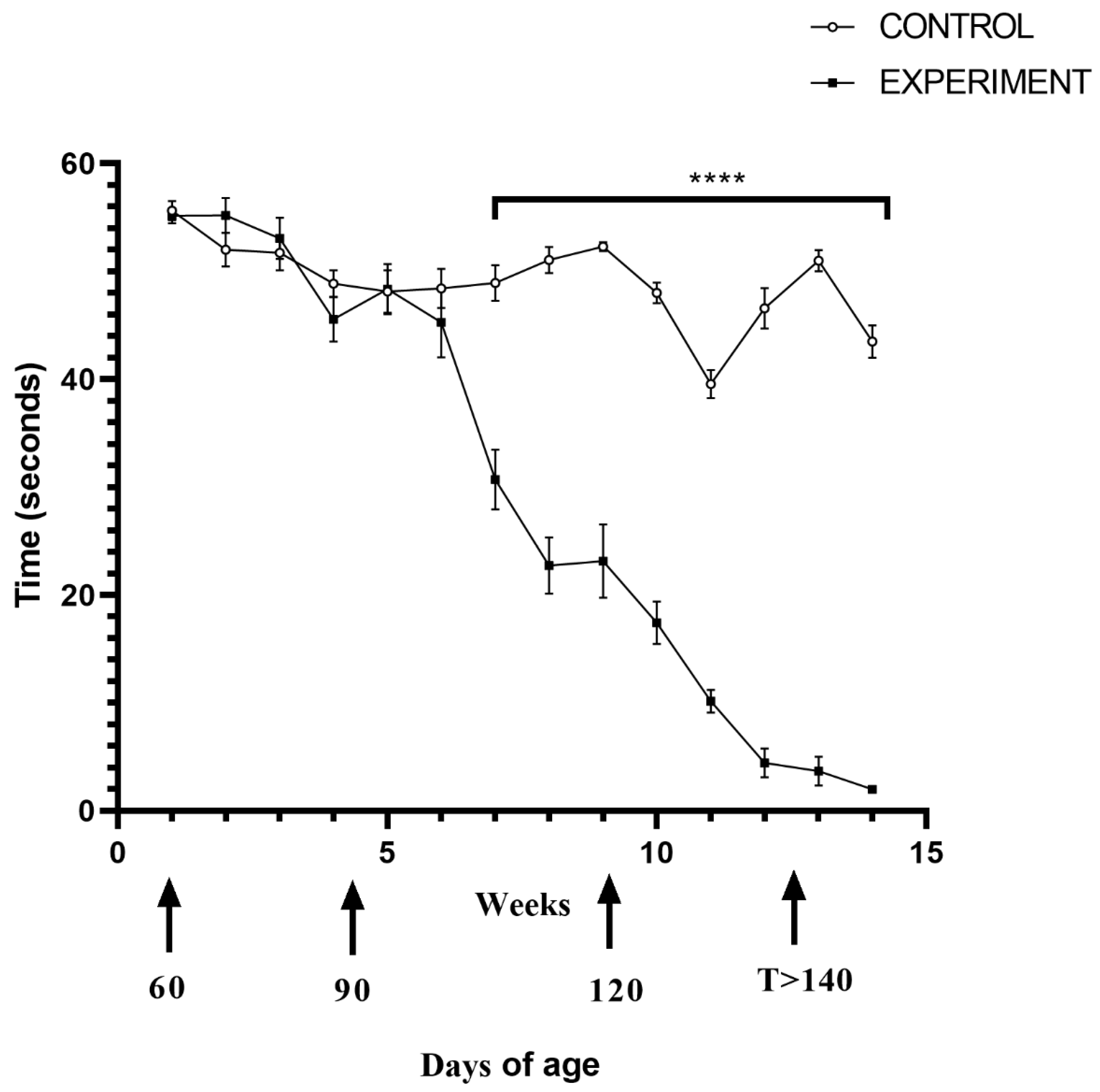
2.1. Life Quality, Mobility and Lifespan of Mice with Congenital ALS
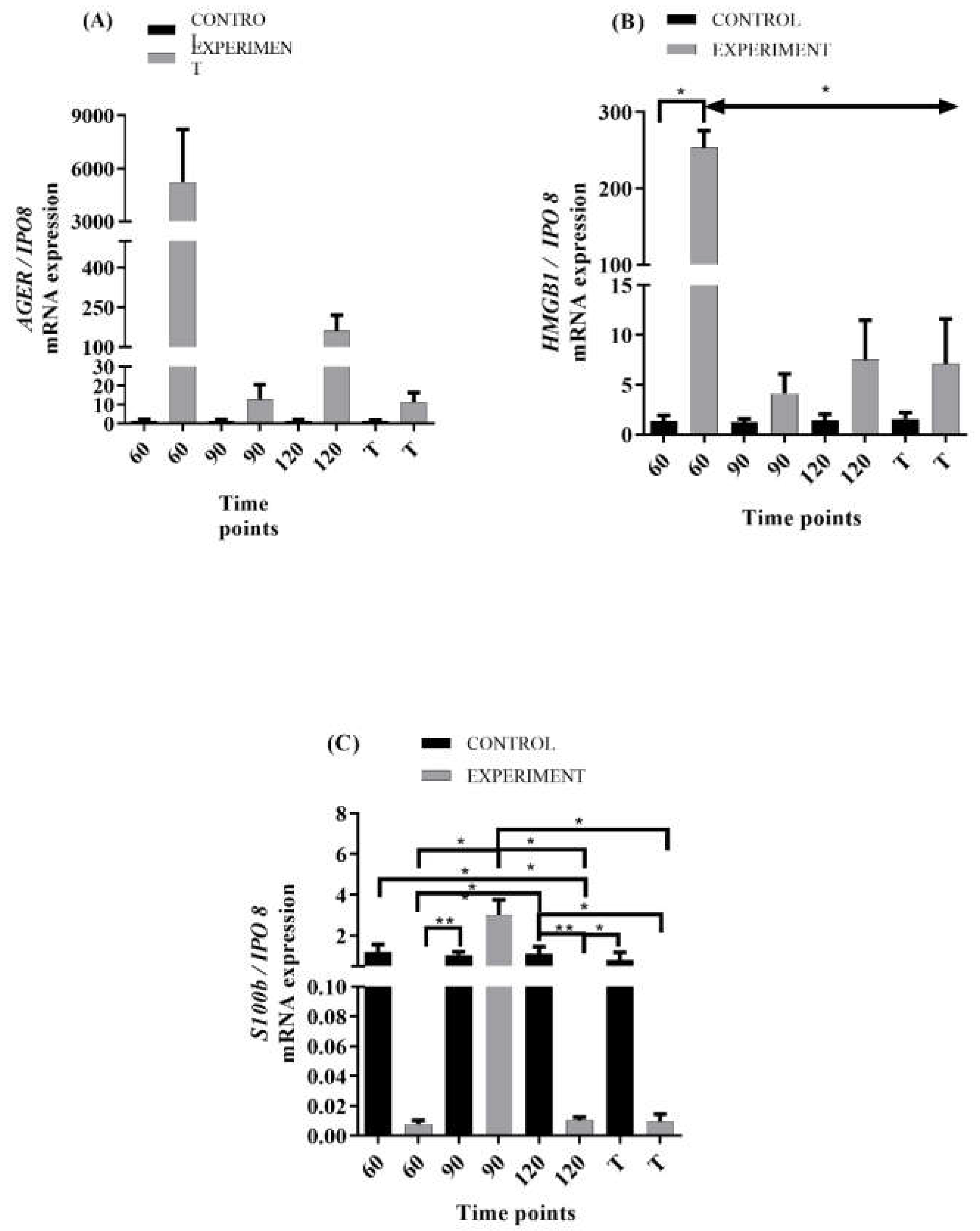
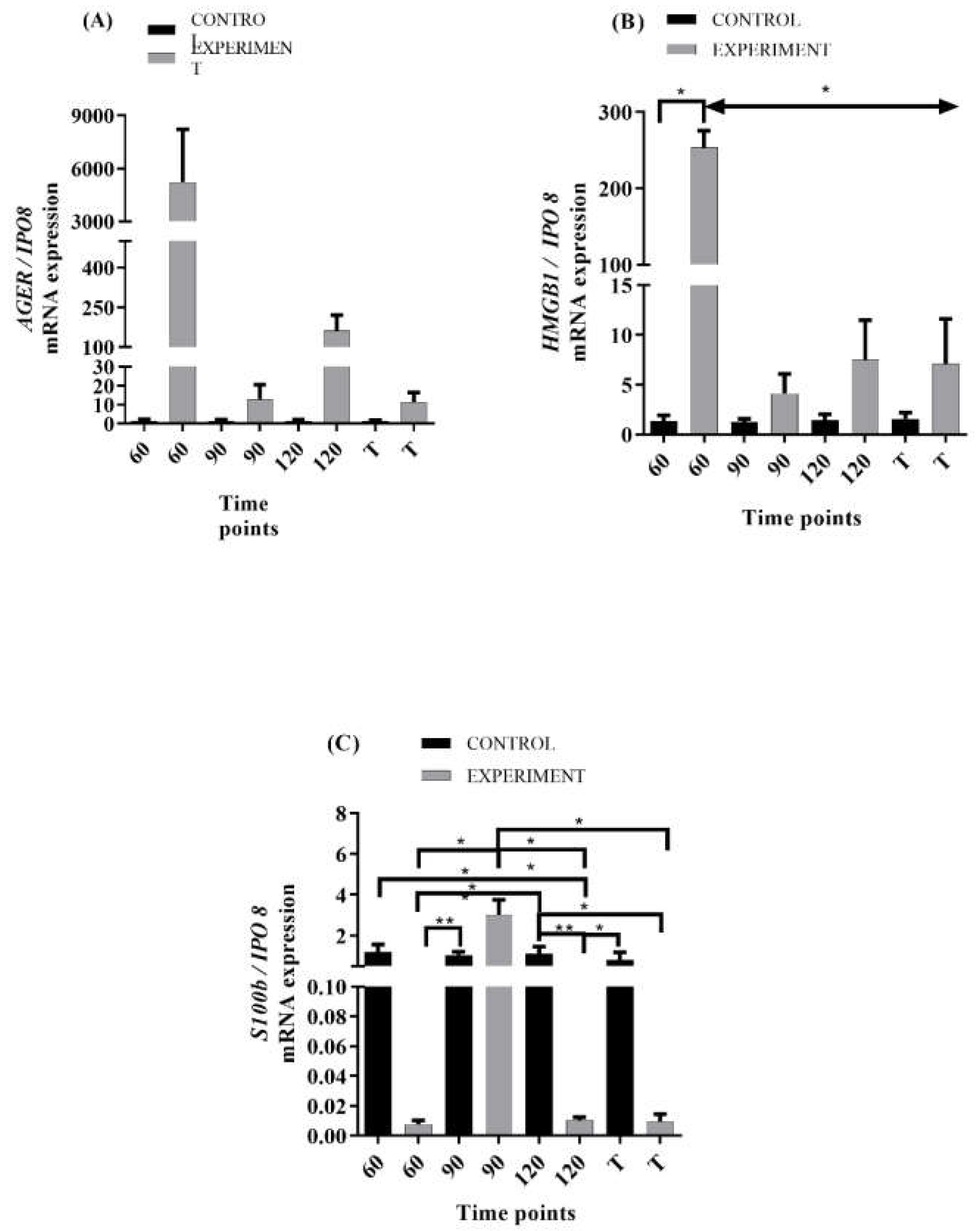
2.2. mRNA Levels of RAGE and Its Ligands Transcripts in ALS Mouse Lumbar Spinal Cord
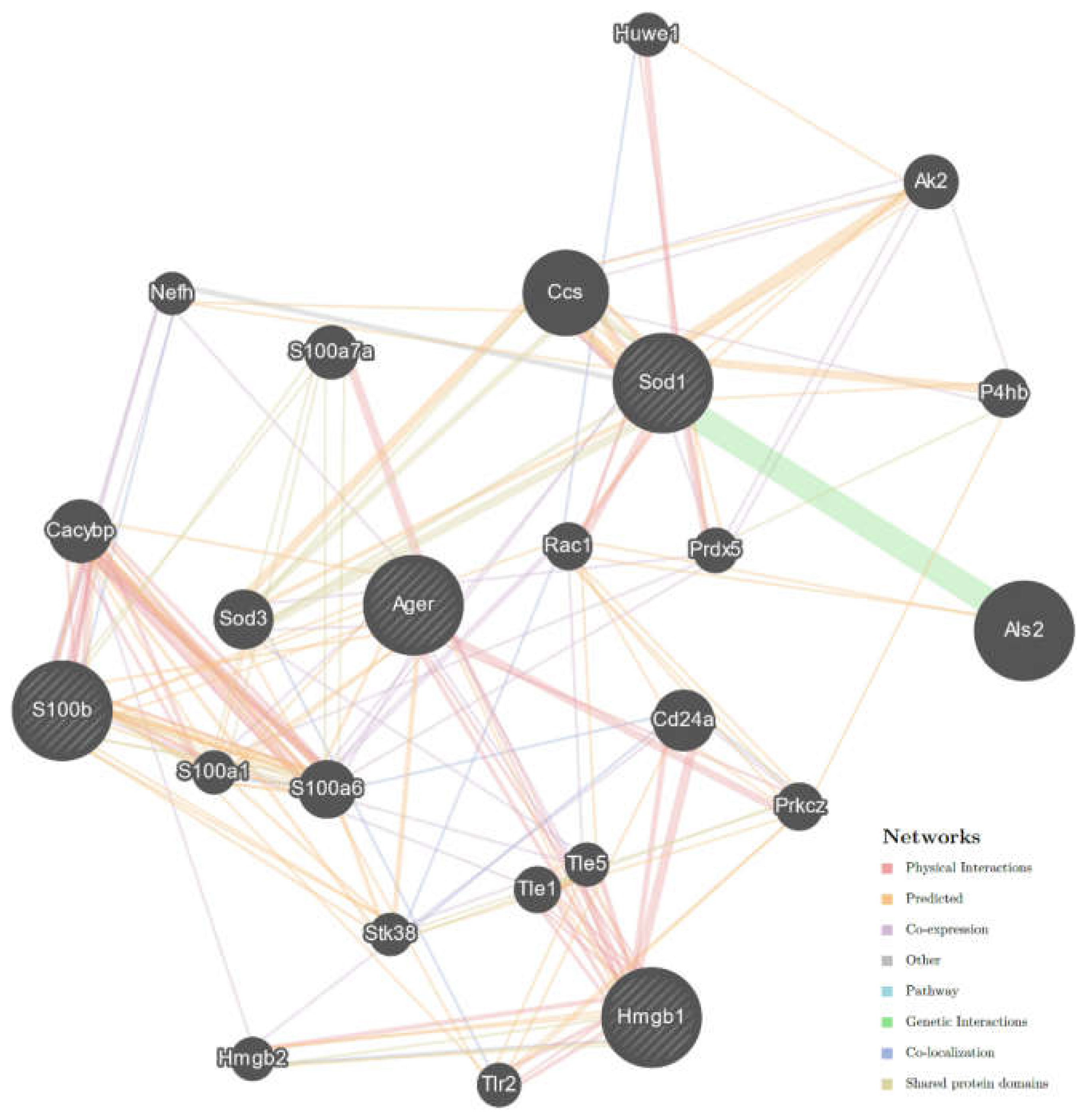
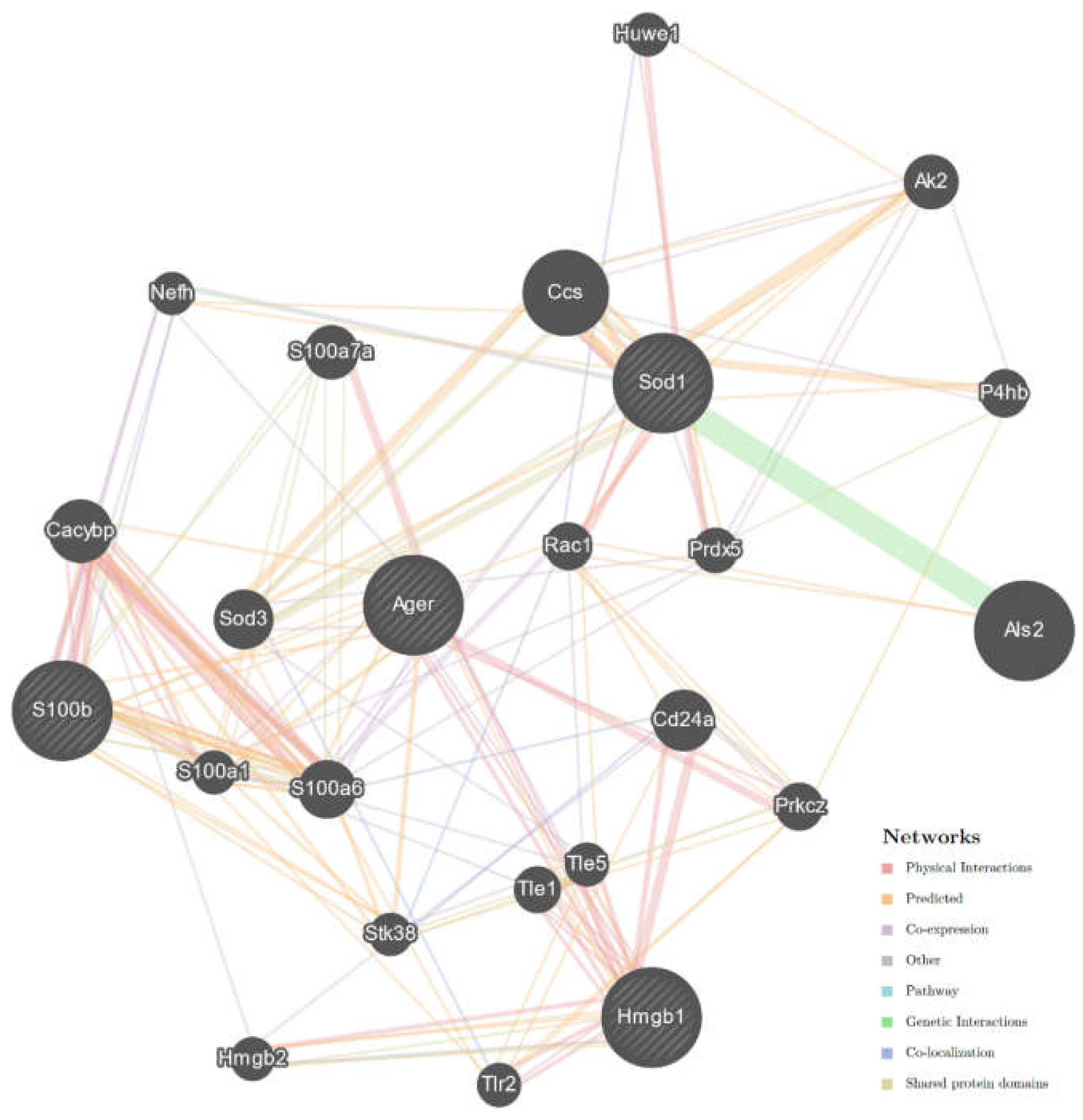
2.3. Interaction Network of Studied Genes in ALS
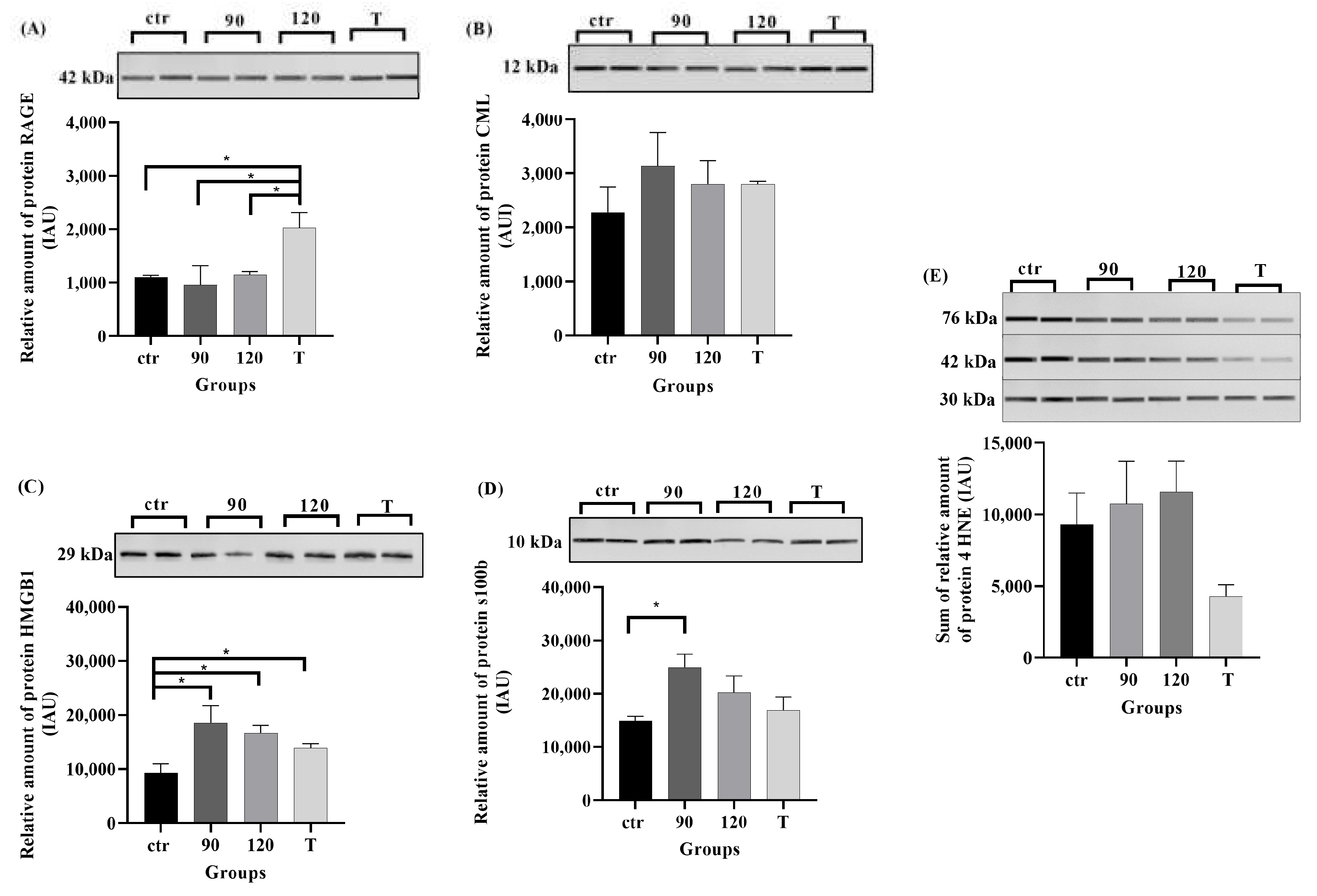
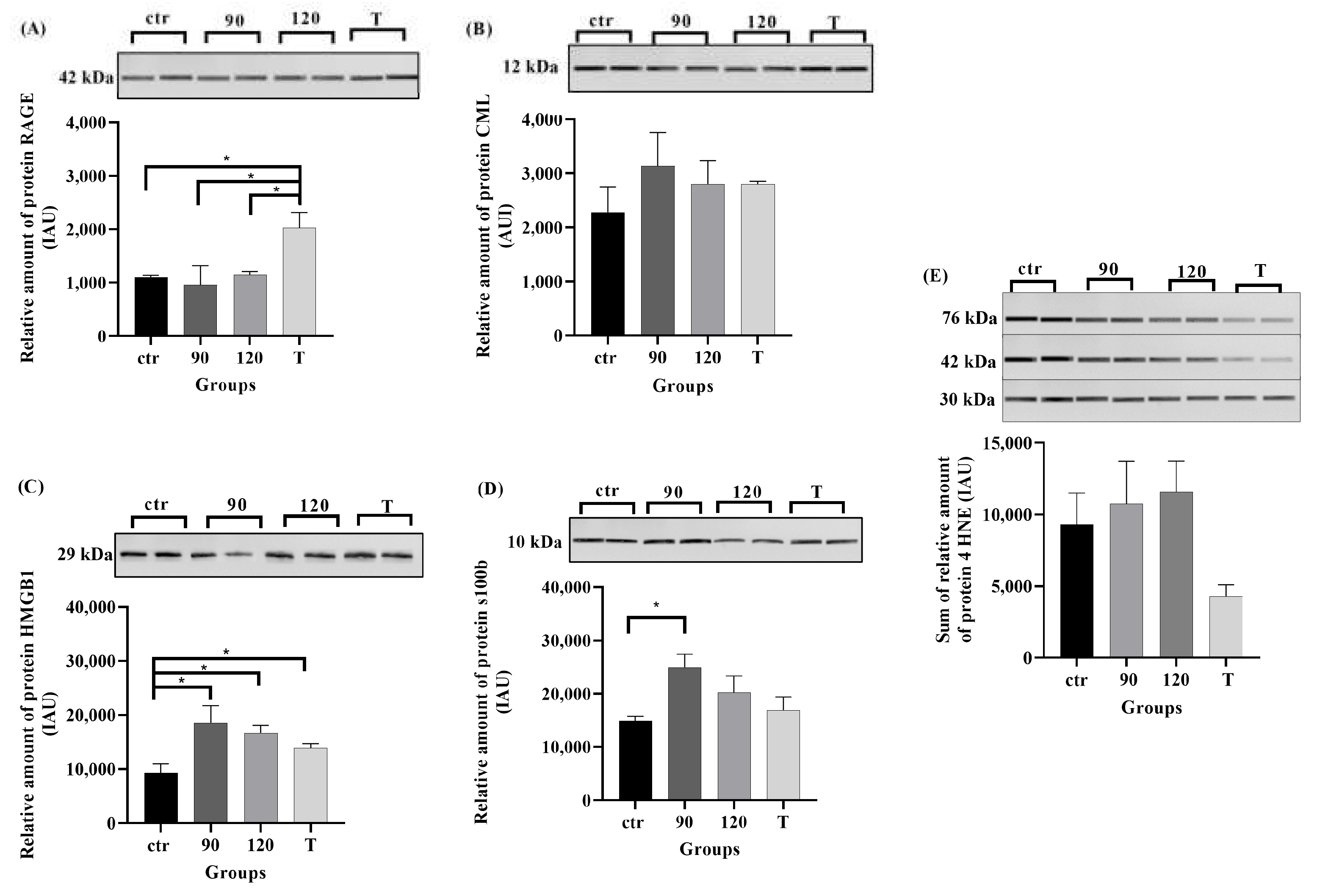
2.4. Protein Levels of RAGE, Its Ligands, and 4HNE in ALS Mouse Lumbar Spinal Cord
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Motor Function Test
4.3. RNA and Protein Extraction
4.4. Reverse Transcription and Quantitative PCR
4.5. Molecular Interactions between Studied Genes in ALS
4.6. Western Blot Analysis
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; Van den Berg, L.H. Amyotrophic lateral sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar]
- Nowicka, N.; Juranek, J.; Juranek, J.K.; Wojtkiewicz, J. Risk Factors and Emerging Therapies in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2019, 20, 2616. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, J.D.; Borasio, G.D. Amyotrophic lateral sclerosis. Lancet 2007, 369, 2031–2041. [Google Scholar] [CrossRef]
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [PubMed]
- Davis, A.A.; Leyns, C.E.G.; Holtzman, D.M. Intercellular Spread of Protein Aggregates in Neurodegenerative Disease. Annu. Rev. Cell Dev. Biol. 2018, 34, 545–568. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.; Cozzolino, M.; Carri, M.T. Old versus New Mechanisms in the Pathogenesis of ALS. Brain Pathol. 2016, 26, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Bentmann, E.; Haass, C.; Dormann, D. Stress granules in neurodegeneration—Lessons learnt from TAR DNA binding protein of 43 kDa and fused in sarcoma. FEBS J. 2013, 280, 4348–4370. [Google Scholar] [PubMed]
- Cai, Z.; Liu, N.; Wang, C.; Qin, B.; Zhou, Y.; Xiao, M.; Zhao, B. Role of RAGE in Alzheimer’s Disease. Cell Mol. Neurobiol. 2016, 36, 483–495. [Google Scholar] [PubMed]
- Chung, C.G.; Lee, H.; Lee, S.B. Mechanisms of protein toxicity in neurodegenerative diseases. Cell Mol. Life Sci. 2018, 75, 3159–3180. [Google Scholar] [PubMed] [Green Version]
- Ganguly, G.; Chakrabarti, S.; Chatterjee, U.; Saso, L. Proteinopathy, oxidative stress and mitochondrial dysfunction: Cross talk in Alzheimer’s disease and Parkinson’s disease. Drug Des. Devel. Ther. 2017, 11, 797–810. [Google Scholar] [CrossRef] [Green Version]
- Neeper, M.; Schmidt, A.M.; Brett, J.; Yan, S.D.; Wang, F.; Pan, Y.C.; Elliston, K.; Stern, D.; Shaw, A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J. Biol. Chem. 1992, 267, 14998–15004. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Mora, R.; Cao, R.; Yan, S.D.; Brett, J.; Ramakrishnan, R.; Tsang, T.C.; Simionescu, M.; Stern, D. The endothelial cell binding site for advanced glycation end products consists of a complex: An integral membrane protein and a lactoferrin-like polypeptide. J. Biol. Chem. 1994, 269, 9882–9888. [Google Scholar] [CrossRef]
- Derk, J.; MacLean, M.; Juranek, J.; Schmidt, A.M. The Receptor for Advanced Glycation Endproducts (RAGE) and Mediation of Inflammatory Neurodegeneration. J. Alzheimers Dis. Park. 2018, 8, 421. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, R.; Yan, S.F.; Schmidt, A.M. Advanced glycation endproducts: From precursors to RAGE: Round and round we go. Amino Acids 2012, 42, 1151–1161. [Google Scholar] [PubMed] [Green Version]
- Rabbani, N.; Thornalley, P.J. Dicarbonyl stress in cell and tissue dysfunction contributing to ageing and disease. Biochem. Biophys Res. Commun. 2015, 458, 221–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorci, G.; Riuzzi, F.; Giambanco, I.; Donato, R. RAGE in tissue homeostasis, repair and regeneration. Biochim. Biophys Acta 2013, 1833, 101–109. [Google Scholar]
- Kim, M.J.; Vargas, M.R.; Harlan, B.A.; Killoy, K.M.; Ball, L.E.; Comte-Walters, S.; Gooz, M.; Yamamoto, Y.; Beckman, J.S.; Barbeito, L.; et al. Nitration and Glycation Turn Mature NGF into a Toxic Factor for Motor Neurons: A Role for p75(NTR) and RAGE Signaling in ALS. Antioxid Redox Signal. 2018, 28, 1587–1602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, R.; Juranek, J.K.; Rai, V. RAGE axis in neuroinflammation, neurodegeneration and its emerging role in the pathogenesis of amyotrophic lateral sclerosis. Neurosci. Biobehav. Rev. 2016, 62, 48–55. [Google Scholar]
- Tu, D.; Zhu, Z.; Zhou, A.; Yun, C.-H.; Lee, K.-E.; Toms, A.V.; Li, Y.; Dunn, G.P.; Chan, E.; Thai, T.; et al. Structure and ubiquitination-dependent activation of TANK-binding kinase 1. Cell Rep. 2013, 3, 747–758. [Google Scholar]
- Paudel, Y.N.; Shaikh, M.F.; Chakraborti, A.; Kumari, Y.; Aledo-Serrano, A.; Aleksovska, K.; Alvim, M.K.M.; Othman, I. HMGB1: A Common Biomarker and Potential Target for TBI, Neuroinflammation, Epilepsy, and Cognitive Dysfunction. Front. Neurosci. 2018, 12, 628. [Google Scholar] [PubMed] [Green Version]
- Yang, H.; Antoine, D.J.; Andersson, U.; Tracey, K.J. The many faces of HMGB1: Molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J. Leukoc. Biol. 2013, 93, 865–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michetti, F.; Corvino, V.; Geloso, M.C.; Lattanzi, W.; Bernardini, C.; Serpero, L.; Gazzolo, D. The S100B protein in biological fluids: More than a lifelong biomarker of brain distress. J. Neurochem. 2012, 120, 644–659. [Google Scholar]
- Serrano, A.; Donno, C.; Giannetti, S.; Perić, M.; Andjus, P.; D’Ambrosi, N.; Michetti, F. The Astrocytic S100B Protein with Its Receptor RAGE Is Aberrantly Expressed in SOD1(G93A) Models, and Its Inhibition Decreases the Expression of Proinflammatory Genes. Mediators Inflamm. 2017, 2017, 1626204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruminis-Kaszkiel, E.; Juranek, J.; Maksymowicz, W.; Wojtkiewicz, J. CRISPR/Cas9 Technology as an Emerging Tool for Targeting Amyotrophic Lateral Sclerosis (ALS). Int. J. Mol. Sci. 2018, 19, 906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruminis-Kaszkiel, E.; Wojtkiewicz, J.; Maksymowicz, W. Glial-restricted precursors as potential candidates for ALS cell-replacement therapy. Acta Neurobiol. Exp. 2014, 74, 233–241. [Google Scholar]
- Czarzasta, J.; Habich, A.; Siwek, T.; Czaplinski, A.; Maksymowicz, W.; Wojtkiewicz, J. Stem cells for ALS: An overview of possible therapeutic approaches. Int. J. Dev. Neurosci. 2017, 57, 46–55. [Google Scholar] [CrossRef]
- Di Domenico, F.; Tramutola, A.; Butterfield, D.A. Role of 4-hydroxy-2-nonenal (HNE) in the pathogenesis of alzheimer disease and other selected age-related neurodegenerative disorders. Free Radic. Biol. Med. 2017, 111, 253–261. [Google Scholar]
- Di Domenico, F.; Pupo, G.; Tramutola, A.; Giorgi, A.; Schininà, M.E.; Coccia, R.; Head, E.; Butterfield, D.A.; Perluigi, M. Redox proteomics analysis of HNE-modified proteins in Down syndrome brain: Clues for understanding the development of Alzheimer disease. Free Radic. Biol. Med. 2014, 71, 270–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poon, H.F.; Hensley, K.; Thongboonkerd, V.; Merchant, M.L.; Lynn, B.C.; Pierce, W.M.; Klein, J.B.; Calabrese, V.; Butterfield, D.A. Redox proteomics analysis of oxidatively modified proteins in G93A-SOD1 transgenic mice--a model of familial amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2005, 39, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Juranek, J.K.; Daffu, G.K.; Geddis, M.S.; Li, H.; Rosario, R.; Kaplan, B.J.; Kelly, L.; Schmidt, A.M. Soluble RAGE Treatment Delays Progression of Amyotrophic Lateral Sclerosis in SOD1 Mice. Front. Cell Neurosci. 2016, 10, 117. [Google Scholar]
- Franz, M.; Rodriguez, H.; Lopes, C.; Zuberi, K.; Montojo, J.; Bader, G.D.; Morris, Q. GeneMANIA update 2018. Nucleic Acids Res. 2018, 46, W60–W64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.D.; Liu, N.; Levin, S.C.; Ottosson, L.; Andersson, U.; Harris, H.E.; Woodruff, T.M. Therapeutic blockade of HMGB1 reduces early motor deficits, but not survival in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. J. Neuroinflamm. 2019, 16, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Bickel, P.J.; Biggin, M.D. System wide analyses have underestimated protein abundances and the importance of transcription in mammals. PeerJ 2014, 2, e270. [Google Scholar]
- Liscic, R.M.; Alberici, A.; Cairns, N.J.; Romano, M.; Buratti, E. From basic research to the clinic: Innovative therapies for ALS and FTD in the pipeline. Mol. Neurodegener. 2020, 15, 31. [Google Scholar] [PubMed]
- Steinle, J.J. Role of HMGB1 signaling in the inflammatory process in diabetic retinopathy. Cell Signal. 2020, 73, 109687. [Google Scholar] [PubMed]
- Lee, J.Y.; Lee, J.D.; Phipps, S.; Noakes, P.G.; Woodruff, T.M. Absence of toll-like receptor 4 (TLR4) extends survival in the hSOD1 G93A mouse model of amyotrophic lateral sclerosis. J. Neuroinflamm. 2015, 12, 90. [Google Scholar] [CrossRef] [Green Version]
- Casula, M.; Iyer, A.; Spliet, W.; Anink, J.; Steentjes, K.; Sta, M.; Troost, D.; Aronica, E. Toll-like receptor signaling in amyotrophic lateral sclerosis spinal cord tissue. Neuroscience 2011, 179, 233–243. [Google Scholar] [CrossRef]
- Sims, G.P.; Rowe, D.C.; Rietdijk, S.T.; Herbst, R.; Coyle, A.J. HMGB1 and RAGE in inflammation and cancer. Annu. Rev. Immunol. 2010, 28, 367–388. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Hyeon, S.J.; Im, H.; Ryu, H.; Kim, Y.; Ryu, H. Astrocytes and Microglia as Non-cell Autonomous Players in the Pathogenesis of ALS. Exp. Neurobiol. 2016, 25, 233–240. [Google Scholar]
- Fu, M.X.; Requena, J.R.; Jenkins, A.J.; Lyons, T.J.; Baynes, J.W.; Thorpe, S.R. The advanced glycation end product, Nepsilon-(carboxymethyl)lysine, is a product of both lipid peroxidation and glycoxidation reactions. J. Biol. Chem. 1996, 271, 9982–9986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikuchi, S.; Shinpo, K.; Ogata, A.; Tsuji, S.; Takeuchi, M.; Makita, Z.; Tashiro, K. Detection of N epsilon-(carboxymethyl)lysine (CML) and non-CML advanced glycation end-products in the anterior horn of amyotrophic lateral sclerosis spinal cord. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2002, 3, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Castro, J.P.; Jung, T.; Grune, T.; Siems, W. 4-Hydroxynonenal (HNE) modified proteins in metabolic diseases. Free Radic. Biol. Med. 2017, 111, 309–315. [Google Scholar] [PubMed] [Green Version]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Mel’nikova, T.I.; Porozov, Y.B.; Terentiev, A.A. Oxidative Stress and Advanced Lipoxidation and Glycation End Products (ALEs and AGEs) in Aging and Age-Related Diseases. Oxid. Med. Cell Longev. 2019, 2019, 3085756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, L.J.; Wong, M. Aberrant regulation of DNA methylation in amyotrophic lateral sclerosis: A new target of disease mechanisms. Neurotherapeutics 2013, 10, 722–733. [Google Scholar] [PubMed] [Green Version]
- Nabais, M.F.; The Australian Imaging Biomarkers and Lifestyle study; Laws, S.M.; Lin, T.; Vallerga, C.L.; Armstrong, N.J.; Blair, I.P.; Kwok, J.B.; Mather, K.A.; Mellick, G.D.; et al. Meta-analysis of genome-wide DNA methylation identifies shared associations across neurodegenerative disorders. Genome Biol. 2021, 22, 90. [Google Scholar] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol (Official) | Primer Sequences | Target Sequence Accession Number | Amplicon Length |
|---|---|---|---|
| AGER | F: 5′-CTTAGCTGGCACTTAGATGG-3′ R:5′-GAAACTGCAGGAGAAGGTAG-3′ | L33412.1 | 168 nt |
| HMGB1 | F: 5′-GAGAAGGATATTGCTGCCTAC-3′ R: 5′-CTTCATCTTCGTCTTCCTCTTC-3′ | U00431.1 | 160 nt |
| S100b | F: 5′-GTCAGAACTGAAGGAGCTTATC-3′ R: 5′-CATGTTCAAAGAACTCATGGC-3′ | NM_009115.3 | 185 nt |
| IPO8 | F: 5′-CTATGCTCTCGTTCAGTATGC-3′ R: 5′-GAGCCCACTTCTTACACTTC-3′ | NM_001081113.1 | 173 nt |
| Primary Antibodies | ||||||
|---|---|---|---|---|---|---|
| Antigen | Code | Species | Working Dilution | Supplier | ||
| RAGE | ab37647 | Rabbit | 1:1000 | Abcam, Cambridge, UK | ||
| HMGB1 | ab18256 | Rabbit | 1:1000 | |||
| S100b | ab52642 | Rabbit | 1:1000 | |||
| CML | ab27684 | Rabbit | 1:5000 | |||
| 4HNE | Ab46545 | Rabbit | 1:5000 | |||
| Secondary Antibody | ||||||
| Reagents | Code | Working Dilution | Supplier | |||
| StarBright Blue 700 Goat Anti-Rabbit IgG, | 12004162 | 1:10,000 | BioRad Hercules, CA, USA | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nowicka, N.; Szymańska, K.; Juranek, J.; Zglejc-Waszak, K.; Korytko, A.; Załęcki, M.; Chmielewska-Krzesińska, M.; Wąsowicz, K.; Wojtkiewicz, J. The Involvement of RAGE and Its Ligands during Progression of ALS in SOD1 G93A Transgenic Mice. Int. J. Mol. Sci. 2022, 23, 2184. https://doi.org/10.3390/ijms23042184
Nowicka N, Szymańska K, Juranek J, Zglejc-Waszak K, Korytko A, Załęcki M, Chmielewska-Krzesińska M, Wąsowicz K, Wojtkiewicz J. The Involvement of RAGE and Its Ligands during Progression of ALS in SOD1 G93A Transgenic Mice. International Journal of Molecular Sciences. 2022; 23(4):2184. https://doi.org/10.3390/ijms23042184
Chicago/Turabian StyleNowicka, Natalia, Kamila Szymańska, Judyta Juranek, Kamila Zglejc-Waszak, Agnieszka Korytko, Michał Załęcki, Małgorzata Chmielewska-Krzesińska, Krzysztof Wąsowicz, and Joanna Wojtkiewicz. 2022. "The Involvement of RAGE and Its Ligands during Progression of ALS in SOD1 G93A Transgenic Mice" International Journal of Molecular Sciences 23, no. 4: 2184. https://doi.org/10.3390/ijms23042184
APA StyleNowicka, N., Szymańska, K., Juranek, J., Zglejc-Waszak, K., Korytko, A., Załęcki, M., Chmielewska-Krzesińska, M., Wąsowicz, K., & Wojtkiewicz, J. (2022). The Involvement of RAGE and Its Ligands during Progression of ALS in SOD1 G93A Transgenic Mice. International Journal of Molecular Sciences, 23(4), 2184. https://doi.org/10.3390/ijms23042184