
Vancomycin-Associated Acute Kidney Injury: A Narrative Review from Pathophysiology to Clinical Application



Abstract
:1. Introduction
2. Pharmacokinetics and Pharmacodynamics of Vancomycin
3. Pathological Manifestations of VA-AKI
4. Pathophysiological Mechanisms of VA-AKI
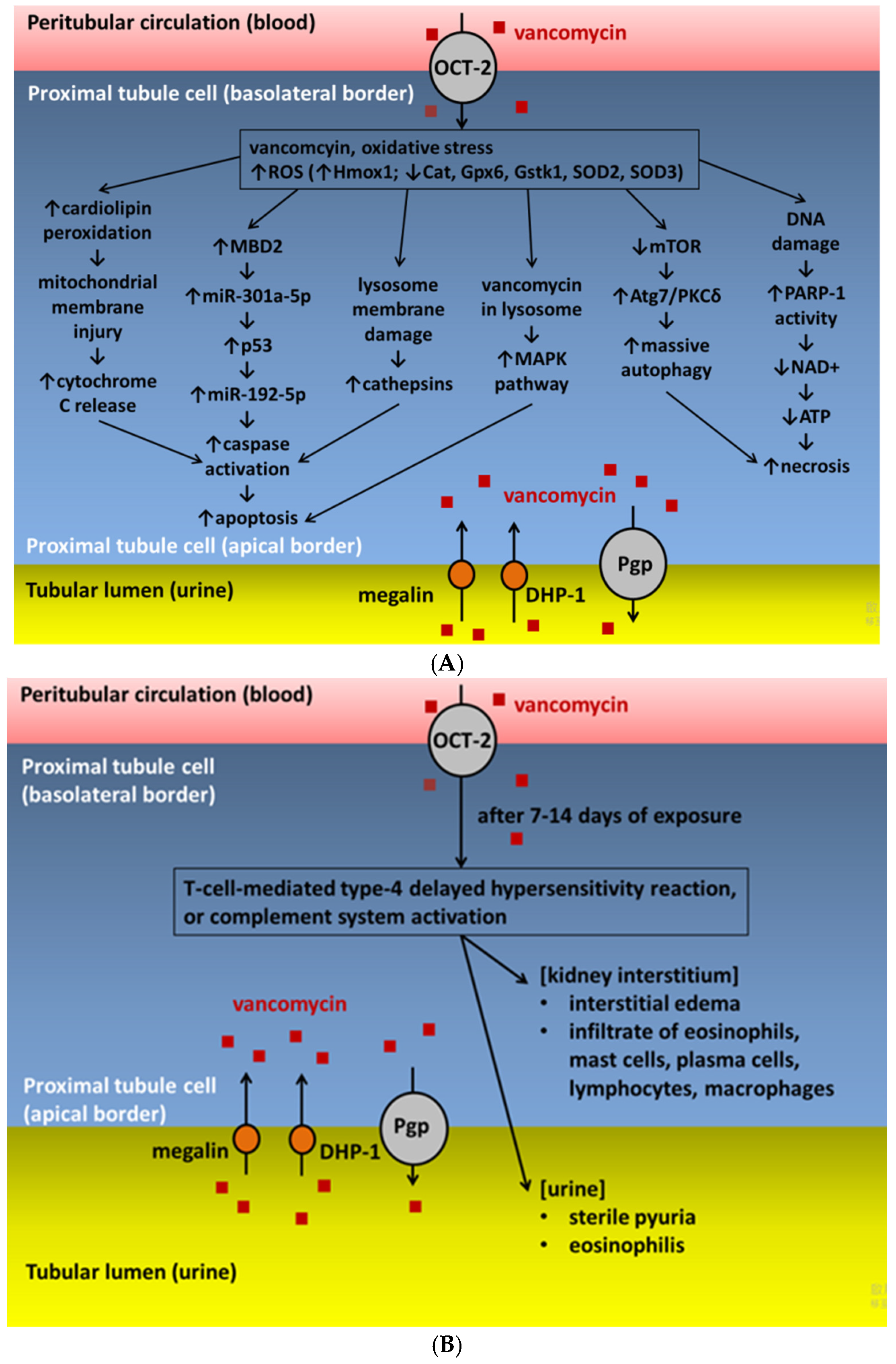
4.1. Oxidative Stress
4.2. Allergic Reaction
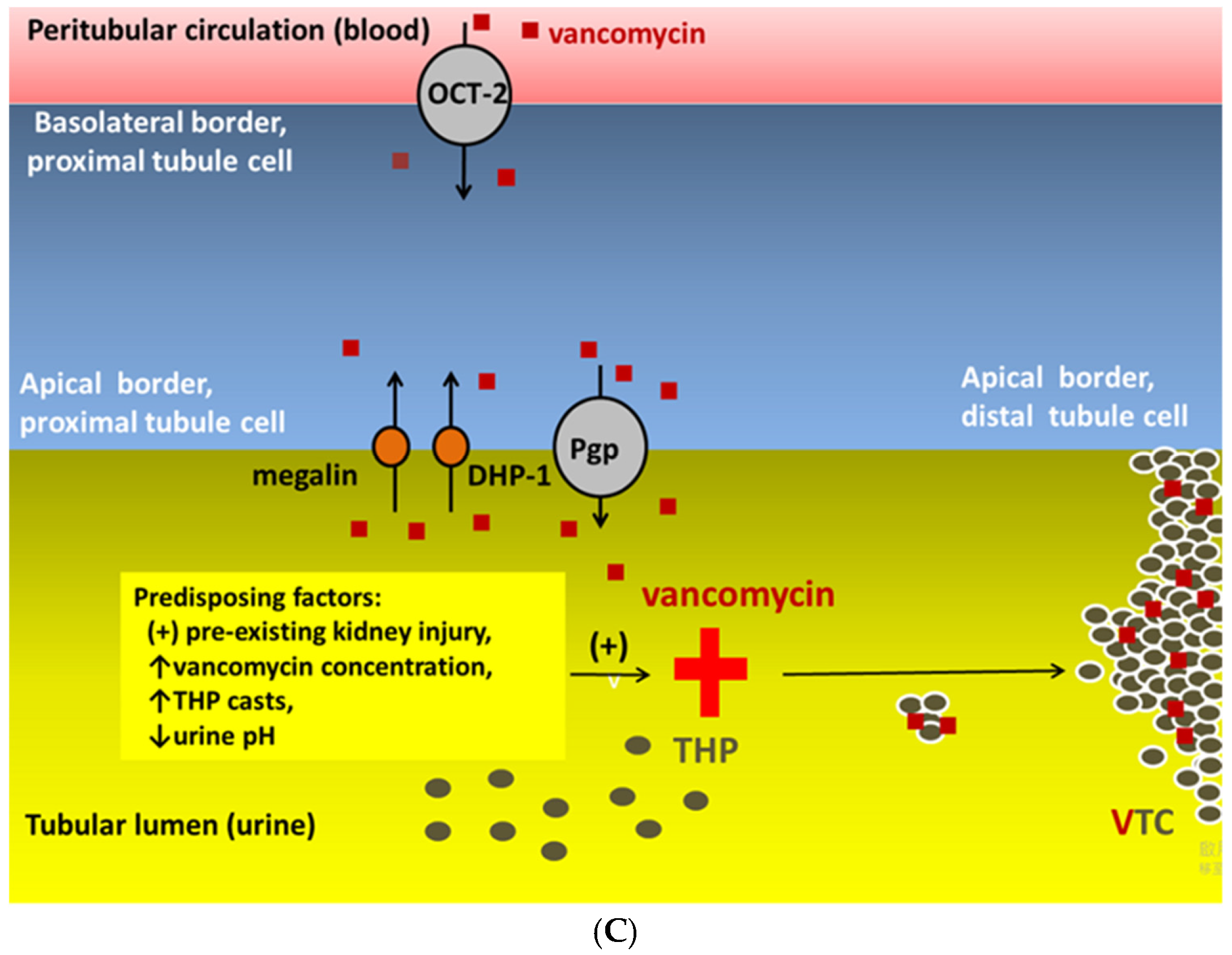
4.3. Vancomycin-Associated Tubular Cast
5. Some Crucial Issues Regarding VA-AKI
5.1. Concomitant Administration with Piperacillin–Tazobactam
5.2. Administration Dosage and VA-AKI
5.3. Administration Patterns and VA-AKI
6. Biomarkers for Detecting VA-AKI
7. Potential Risk Factors Associated with VA-AKI
8. Treatment of VA-AKI
9. Preventive Strategies for VA-AKI
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mas-Font, S.; Ros-Martinez, J.; Perez-Calvo, C.; Villa-Diaz, P.; Aldunate-Calvo, S.; Moreno-Clari, E. Prevention of acute kidney injury in Intensive Care Units. Med. Intensiva 2017, 41, 116–126. [Google Scholar] [CrossRef]
- Hoste, E.A.; Bagshaw, S.M.; Bellomo, R.; Cely, C.M.; Colman, R.; Cruz, D.N.; Edipidis, K.; Forni, L.G.; Gomersall, C.D.; Govil, D.; et al. Epidemiology of acute kidney injury in critically ill patients: The multinational AKI-EPI study. Intensive Care Med. 2015, 41, 1411–1423. [Google Scholar] [CrossRef]
- Wu, V.C.; Huang, T.M.; Lai, C.F.; Shiao, C.C.; Lin, Y.F.; Chu, T.S.; Wu, P.C.; Chao, C.T.; Wang, J.Y.; Kao, T.W.; et al. Acute-on-chronic kidney injury at hospital discharge is associated with long-term dialysis and mortality. Kidney Int. 2011, 80, 1222–1230. [Google Scholar] [CrossRef] [Green Version]
- Druml, W. Systemic consequences of acute kidney injury. Curr. Opin. Crit. Care 2014, 20, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Petejova, N.; Martinek, A.; Zadrazil, J.; Kanova, M.; Klementa, V.; Sigutova, R.; Kacirova, I.; Hrabovsky, V.; Svagera, Z.; Stejskal, D. Acute kidney injury in septic patients treated by selected nephrotoxic antibiotic agents-pathophysiology and biomarkers-a review. Int. J. Mol. Sci 2020, 21, 7115. [Google Scholar] [CrossRef] [PubMed]
- Morales-Alvarez, M.C. Nephrotoxicity of antimicrobials and antibiotics. Adv. Chronic. Kidney Dis. 2020, 27, 31–37. [Google Scholar] [CrossRef]
- Kim, S.J.; Matsuoka, S.; Patti, G.J.; Schaefer, J. Vancomycin derivative with damaged D-Ala-D-Ala binding cleft binds to cross-linked peptidoglycan in the cell wall of Staphylococcus aureus. Biochemistry 2008, 47, 3822–3831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitevska, E.; Wong, B.; Surewaard, B.G.J.; Jenne, C.N. The prevalence, risk, and management of methicillin-resistant staphylococcus aureus infection in diverse populations across Canada: A systematic review. Pathogens 2021, 10, 393. [Google Scholar] [CrossRef] [PubMed]
- Diallo, O.O.; Baron, S.A.; Abat, C.; Colson, P.; Chaudet, H.; Rolain, J.M. Antibiotic resistance surveillance systems: A review. J. Glob. Antimicrob. Resist. 2020, 23, 430–438. [Google Scholar] [CrossRef]
- Geraci, J.E.; Heilman, F.R.; Nichols, D.R.; Wellman, W.E. Antibiotic therapy of bacterial endocarditis. VII. Vancomycin for acute micrococcal endocarditis; preliminary report. Proc. Staff Meet. Mayo Clin. 1958, 33, 172–181. [Google Scholar]
- Sinha Ray, A.; Haikal, A.; Hammoud, K.A.; Yu, A.S. Vancomycin and the risk of AKI: A systematic review and meta-analysis. Clin. J. Am. Soc. Nephrol. 2016, 11, 2132–2140. [Google Scholar] [CrossRef] [Green Version]
- Awdishu, L.; Le, A.; Amato, J.; Jani, V.; Bal, S.; Mills, R.H.; Carrillo-Terrazas, M.; Gonzalez, D.J.; Tolwani, A.; Acharya, A.; et al. Urinary exosomes identify inflammatory pathways in vancomycin associated acute kidney injury. Int. J. Mol. Sci. 2021, 22, 2784. [Google Scholar] [CrossRef]
- Jorgensen, S.C.J.; Murray, K.P.; Lagnf, A.M.; Melvin, S.; Bhatia, S.; Shamim, M.D.; Smith, J.R.; Brade, K.D.; Simon, S.P.; Nagel, J.; et al. A multicenter evaluation of vancomycin-associated acute kidney injury in hospitalized patients with acute bacterial skin and skin structure infections. Infect. Dis. Ther. 2020, 9, 89–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tantranont, N.; Luque, Y.; Hsiao, M.; Haute, C.; Gaber, L.; Barrios, R.; Adrogue, H.E.; Niasse, A.; Truong, L.D. Vancomycin-associated tubular casts and vancomycin nephrotoxicity. Kidney Int. Rep. 2021, 6, 1912–1922. [Google Scholar] [CrossRef]
- Tantranont, N.; Obi, C.; Luque, Y.; Truong, L.D. Vancomycin nephrotoxicity: Vancomycin tubular casts with characteristic electron microscopic findings. Clin. Nephrol. Case Stud. 2019, 7, 66–72. [Google Scholar] [CrossRef]
- Kunming, P.; Can, C.; Zhangzhang, C.; Wei, W.; Qing, X.; Xiaoqiang, D.; Xiaoyu, L.; Qianzhou, L. Vancomycin associated acute kidney injury: A longitudinal study in China. Front. Pharmacol. 2021, 12, 632107. [Google Scholar] [CrossRef]
- Zamoner, W.; Prado, I.R.S.; Balbi, A.L.; Ponce, D. Vancomycin dosing, monitoring and toxicity: Critical review of the clinical practice. Clin. Exp. Pharmacol. Physiol. 2019, 46, 292–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiMondi, V.P.; Rafferty, K. Review of continuous-infusion vancomycin. Ann. Pharmacother. 2013, 47, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, J.M.; Patel, N.; Pai, M.P.; Rosano, T.G.; Drusano, G.L.; Lodise, T.P. Refining vancomycin protein binding estimates: Identification of clinical factors that influence protein binding. Antimicrob. Agents Chemother. 2011, 55, 4277–4282. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Yang, Q.; Gui, M.; Ding, L.; Yang, L.; Sun, H.; Li, Z. Changes of renal transporters in the kinetic process of VCM-induced nephrotoxicity in mice. Toxicol. Res. 2021, 10, 687–695. [Google Scholar] [CrossRef]
- Filippone, E.J.; Kraft, W.K.; Farber, J.L. The nephrotoxicity of vancomycin. Clin. Pharmacol. Ther. 2017, 102, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, Y.; Yano, T.; Hanada, Y.; Takeshita, A.; Inagaki, F.; Masuda, S.; Matsunaga, N.; Koyanagi, S.; Ohdo, S. Vancomycin induces reactive oxygen species-dependent apoptosis via mitochondrial cardiolipin peroxidation in renal tubular epithelial cells. Eur. J. Pharmacol. 2017, 800, 48–56. [Google Scholar] [CrossRef]
- Nishino, Y.; Takemura, S.; Minamiyama, Y.; Hirohashi, K.; Ogino, T.; Inoue, M.; Okada, S.; Kinoshita, H. Targeting superoxide dismutase to renal proximal tubule cells attenuates vancomycin-induced nephrotoxicity in rats. Free Radic. Res. 2003, 37, 373–379. [Google Scholar] [CrossRef]
- Fujiwara, K.; Yoshizaki, Y.; Shin, M.; Miyazaki, T.; Saita, T.; Nagata, S. Immunocytochemistry for vancomycin using a monoclonal antibody that reveals accumulation of the drug in rat kidney and liver. Antimicrob. Agents Chemother. 2012, 56, 5883–5891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perazella, M.A. Drug-induced acute kidney injury: Diverse mechanisms of tubular injury. Curr. Opin. Crit. Care 2019, 25, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowska, E.; Domanski, L.; Dziedziejko, V.; Kajdy, A.; Stefanska, K.; Kwiatkowski, S. The mechanism of drug nephrotoxicity and the methods for preventing kidney damage. Int. J. Mol. Sci. 2021, 22, 6109. [Google Scholar] [CrossRef] [PubMed]
- Bellos, I.; Pergialiotis, V.; Perrea, D.N. Kidney biopsy findings in vancomycin-induced acute kidney injury: A pooled analysis. Int. Urol. Nephrol. 2021, 54, 137–148. [Google Scholar] [CrossRef]
- Pais, G.M.; Liu, J.; Zepcan, S.; Avedissian, S.N.; Rhodes, N.J.; Downes, K.J.; Moorthy, G.S.; Scheetz, M.H. Vancomycin-induced kidney injury: Animal models of toxicodynamics, mechanisms of injury, human translation, and potential strategies for prevention. Pharmacotherapy 2020, 40, 438–454. [Google Scholar] [CrossRef]
- Marre, R.; Schulz, E.; Anders, T.; Sack, K. Renal tolerance and pharmacokinetics of vancomycin in rats. J. Antimicrob. Chemother. 1984, 14, 253–260. [Google Scholar] [CrossRef]
- Young, I.S.; Woodside, J.V. Antioxidants in health and disease. J. Clin. Pathol. 2001, 54, 176–186. [Google Scholar] [CrossRef] [Green Version]
- Oktem, F.; Arslan, M.K.; Ozguner, F.; Candir, O.; Yilmaz, H.R.; Ciris, M.; Uz, E. In vivo evidences suggesting the role of oxidative stress in pathogenesis of vancomycin-induced nephrotoxicity: Protection by erdosteine. Toxicology 2005, 215, 227–233. [Google Scholar] [CrossRef]
- Lee, H.S.; Kim, S.M.; Jang, J.H.; Park, H.D.; Lee, S.Y. Serum 5-Hydroxyindoleacetic acid and ratio of 5-hydroxyindoleacetic acid to serotonin as metabolomics indicators for acute oxidative stress and inflammation in vancomycin-associated acute kidney injury. Antioxidants 2021, 10, 895. [Google Scholar] [CrossRef] [PubMed]
- Humanes, B.; Jado, J.C.; Camano, S.; Lopez-Parra, V.; Torres, A.M.; Alvarez-Sala, L.A.; Cercenado, E.; Tejedor, A.; Lazaro, A. Protective effects of cilastatin against vancomycin-induced nephrotoxicity. Biomed Res. Int. 2015, 2015, 704382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, D.W.; Smith, M.A. Proliferative responses observed following vancomycin treatment in renal proximal tubule epithelial cells. Toxicol. Vitr. 2004, 18, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Heller, B.; Wang, Z.Q.; Wagner, E.F.; Radons, J.; Burkle, A.; Fehsel, K.; Burkart, V.; Kolb, H. Inactivation of the poly(ADP-ribose) polymerase gene affects oxygen radical and nitric oxide toxicity in islet cells. J. Biol. Chem. 1995, 270, 11176–11180. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, H.; Qiu, S.; Dong, Z.; Xiang, X.; Zhang, D. MBD2 upregulates miR-301a-5p to induce kidney cell apoptosis during vancomycin-induced A.K.I. Cell Death Dis. 2017, 8, e3120. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Pan, J.; Li, H.; Li, X.; Fang, F.; Wu, D.; Zhou, Y.; Zheng, P.; Xiong, L.; Zhang, D. Atg7 mediates renal tubular cell apoptosis in vancomycin nephrotoxicity through activation of PKC-delta. FASEB J. 2019, 33, 4513–4524. [Google Scholar] [CrossRef]
- Jiang, M.; Wei, Q.; Dong, G.; Komatsu, M.; Su, Y.; Dong, Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012, 82, 1271–1283. [Google Scholar] [CrossRef] [Green Version]
- Boya, P.; Kroemer, G. Lysosomal membrane permeabilization in cell death. Oncogene 2008, 27, 6434–6451. [Google Scholar] [CrossRef] [Green Version]
- Pannu, N.; Nadim, M.K. An overview of drug-induced acute kidney injury. Crit. Care Med. 2008, 36 (Suppl. 4), S216–S223. [Google Scholar] [CrossRef]
- Hosohata, K. Role of oxidative stress in drug-induced kidney injury. Int. J. Mol. Sci. 2016, 17, 1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelfand, M.S.; Cleveland, K.O.; Mazumder, S.A. Vancomycin-induced interstitial nephritis superimposed on coexisting renal disease: The importance of renal biopsy. Am. J. Med. Sci. 2014, 347, 338–340. [Google Scholar] [CrossRef] [PubMed]
- Azar, R.; Bakhache, E.; Boldron, A. Acute interstitial nephropathy induced by vancomycin. Nephrologie 1996, 17, 327–328. [Google Scholar] [PubMed]
- Perazella, M.A.; Markowitz, G.S. Drug-induced acute interstitial nephritis. Nat. Rev. Nephrol. 2010, 6, 461–470. [Google Scholar] [CrossRef]
- Htike, N.L.; Santoro, J.; Gilbert, B.; Elfenbein, I.B.; Teehan, G. Biopsy-proven vancomycin-associated interstitial nephritis and acute tubular necrosis. Clin. Exp. Nephrol. 2012, 16, 320–324. [Google Scholar] [CrossRef]
- Dieterich, C.; Puey, A.; Lin, S.; Swezey, R.; Furimsky, A.; Fairchild, D.; Mirsalis, J.C.; Ng, H.H. Gene expression analysis reveals new possible mechanisms of vancomycin-induced nephrotoxicity and identifies gene markers candidates. Toxicol. Sci. 2009, 107, 258–269. [Google Scholar] [CrossRef] [Green Version]
- Davies, S.W.; Efird, J.T.; Guidry, C.A.; Dietch, Z.C.; Willis, R.N.; Shah, P.M.; Sawyer, R.G. Top guns: The “Maverick” and “Goose” of empiric therapy. Surg. Infect. 2016, 17, 38–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreier, D.J.; Kashani, K.B.; Sakhuja, A.; Mara, K.C.; Tootooni, M.S.; Personett, H.A.; Nelson, S.; Rule, A.D.; Steckelberg, J.M.; Tande, A.J.; et al. Incidence of acute kidney injury among critically ill patients with brief empiric use of antipseudomonal beta-lactams with vancomycin. Clin. Infect. Dis. 2019, 68, 1456–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, Y.H.; Wang, J.L.; Yin, W.J.; Xu, W.H. Vancomycin or daptomycin plus a beta-lactam versus vancomycin or daptomycin alone for methicillin-resistant staphylococcus aureus bloodstream infections: A systematic review and meta-analysis. Microb. Drug. Resist. 2021, 27, 1044–1056. [Google Scholar] [CrossRef]
- Luther, M.K.; Timbrook, T.T.; Caffrey, A.R.; Dosa, D.; Lodise, T.P.; LaPlante, K.L. Vancomycin plus piperacillin-tazobactam and acute kidney injury in adults: A systematic review and meta-analysis. Crit. Care Med. 2018, 46, 12–20. [Google Scholar] [CrossRef]
- Bellos, I.; Karageorgiou, V.; Pergialiotis, V.; Perrea, D.N. Acute kidney injury following the concurrent administration of antipseudomonal β-lactams and vancomycin: A network meta-analysis. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2020, 26, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Kandiah, S.; Patel, M.; Rab, S.; Wong, J.; Xue, W.; Easley, K.; Anderson, A.M. Risk factors for kidney injury during vancomycin and piperacillin/tazobactam administration, including increased odds of injury with combination therapy. BMC Res. Notes 2015, 8, 579. [Google Scholar] [CrossRef] [Green Version]
- Jensen, J.U.; Hein, L.; Lundgren, B.; Bestle, M.H.; Mohr, T.; Andersen, M.H.; Thornberg, K.J.; Loken, J.; Steensen, M.; Fox, Z.; et al. Kidney failure related to broad-spectrum antibiotics in critically ill patients: Secondary end point results from a 1200 patient randomised trial. BMJ Open 2012, 2, e000635. [Google Scholar] [CrossRef]
- Elyasi, S.; Khalili, H.; Dashti-Khavidaki, S.; Mohammadpour, A. Vancomycin-induced nephrotoxicity: Mechanism, incidence, risk factors and special populations. A literature review. Eur. J. Clin. Pharmacol. 2012, 68, 1243–1255. [Google Scholar] [CrossRef]
- Burgess, L.D.; Drew, R.H. Comparison of the incidence of vancomycin-induced nephrotoxicity in hospitalized patients with and without concomitant piperacillin-tazobactam. Pharmacotherapy 2014, 34, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Avedissian, S.N.; Pais, G.M.; Liu, J.; Rhodes, N.J.; Scheetz, M.H. Piperacillin-tazobactam added to vancomycin increases risk for acute kidney injury: Fact or fiction? Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2020, 71, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, V.S.; Ozer, J.S.; Dieterle, F.; Collings, F.B.; Ramirez, V.; Troth, S.; Muniappa, N.; Thudium, D.; Gerhold, D.; Holder, D.J.; et al. Kidney injury molecule-1 outperforms traditional biomarkers of kidney injury in preclinical biomarker qualification studies. Nat. Biotechnol. 2010, 28, 478–485. [Google Scholar] [CrossRef] [Green Version]
- Aljefri, D.M.; Avedissian, S.N.; Rhodes, N.J.; Postelnick, M.J.; Nguyen, K.; Scheetz, M.H. Vancomycin area under the curve and acute kidney injury: A meta-analysis. Clin. Infect. Dis. 2019, 69, 1881–1887. [Google Scholar] [CrossRef]
- Flannery, A.H.; Bissell, B.D.; Bastin, M.T.; Morris, P.E.; Neyra, J.A. Continuous versus intermittent infusion of vancomycin and the risk of acute kidney injury in critically ill adults: A systematic review and meta-analysis. Crit. Care Med. 2020, 48, 912–918. [Google Scholar] [CrossRef]
- O’Donnell, J.N.; Rhodes, N.J.; Lodise, T.P.; Prozialeck, W.C.; Miglis, C.M.; Joshi, M.D.; Venkatesan, N.; Pais, G.; Cluff, C.; Lamar, P.C.; et al. 24-hour pharmacokinetic relationships for vancomycin and novel urinary biomarkers of acute kidney injury. Antimicrob. Agents Chemother. 2017, 61, e00416–e00417. [Google Scholar] [CrossRef] [Green Version]
- Drouet, M.; Chai, F.; Barthelemy, C.; Lebuffe, G.; Debaene, B.; Decaudin, B.; Odou, P. Influence of vancomycin infusion methods on endothelial cell toxicity. Antimicrob. Agents Chemother. 2015, 59, 930–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pais, G.M.; Avedissian, S.N.; O’Donnell, J.N.; Rhodes, N.J.; Lodise, T.P.; Prozialeck, W.C.; Lamar, P.C.; Cluff, C.; Gulati, A.; Fitzgerald, J.C.; et al. Comparative performance of urinary biomarkers for vancomycin-induced kidney injury according to timeline of injury. Antimicrob. Agents Chemother. 2019, 63, e00079-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, H.M.; Qin, X.L.; Liu, T.T.; Wei, W.X.; Cheng, D.H.; Lu, H.; Guo, Q.; Jing, L. Urinary kidney injury molecule-1 and neutrophil gelatinase-associated lipocalin as early biomarkers for predicting vancomycin-associated acute kidney injury: A prospective study. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 4203–4213. [Google Scholar]
- Sampaio de Souza Garms, D.; Cardoso Eid, K.Z.; Burdmann, E.A.; Marcal, L.J.; Antonangelo, L.; Dos Santos, A.; Ponce, D. The role of urinary biomarkers as diagnostic and prognostic predictors of acute kidney injury associated with vancomycin. Front. Pharmacol. 2021, 12, 705636. [Google Scholar] [CrossRef]
- Kane-Gill, S.L.; Ostermann, M.; Shi, J.; Joyce, E.L.; Kellum, J.A. Evaluating renal stress using pharmacokinetic urinary biomarker data in critically ill patients receiving vancomycin and/or piperacillin-tazobactam: A secondary analysis of the multicenter sapphire study. Drug Saf. 2019, 42, 1149–1155. [Google Scholar] [CrossRef]
- Kim, S.M.; Lee, H.S.; Kim, M.J.; Park, H.D.; Lee, S.Y. Diagnostic value of multiple serum biomarkers for vancomycin-induced kidney injury. J. Clin. Med. 2021, 10, 5005. [Google Scholar] [CrossRef]
- Yang, X.; Zhong, H.; Xu, C.; Xu, G. Spotlights on antibiotic-induced acute kidney injury: The evidence to date. Iran. J. Kidney Dis. 2019, 13, 10–20. [Google Scholar] [PubMed]
- Zonozi, R.; Wu, A.; Shin, J.I.; Secora, A.; Coresh, J.; Inker, L.A.; Chang, A.R.; Grams, M.E. Elevated vancomycin trough levels in a tertiary health system: Frequency, risk factors, and prognosis. Mayo Clin. Proc. 2019, 94, 17–26. [Google Scholar] [CrossRef]
- Watkins, R.R.; Deresinski, S. Increasing evidence of the nephrotoxicity of piperacillin/tazobactam and vancomycin combination therapy-what is the clinician to do? Clin. Infect. Dis. 2017, 65, 2137–2143. [Google Scholar] [CrossRef]
- Rybak, M.J.; Le, J.; Lodise, T.P.; Levine, D.P.; Bradley, J.S.; Liu, C.; Mueller, B.A.; Pai, M.P.; Wong-Beringer, A.; Rotschafer, J.C.; et al. Therapeutic monitoring of vancomycin for serious methicillin-resistant Staphylococcus aureus infections: A revised consensus guideline and review by the American Society of Health-System Pharmacists, the Infectious Diseases Society of America, the Pediatric Infectious Diseases Society, and the Society of Infectious Diseases Pharmacists. Am. J. Health Syst. Pharm. 2020, 77, 835–864. [Google Scholar]
- Rossert, J. Drug-induced acute interstitial nephritis. Kidney Int. 2001, 60, 804–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wicklow, B.A.; Ogborn, M.R.; Gibson, I.W.; Blydt-Hansen, T.D. Biopsy-proven acute tubular necrosis in a child attributed to vancomycin intoxication. Pediatr. Nephrol. 2006, 21, 1194–1196. [Google Scholar] [CrossRef] [PubMed]
- DeSoi, C.A.; Sahm, D.F.; Umans, J.G. Vancomycin elimination during high-flux hemodialysis: Kinetic model and comparison of four membranes. Am. J. Kidney Dis. 1992, 20, 354–360. [Google Scholar] [CrossRef]
- Bamgbola, O. Review of vancomycin-induced renal toxicity: An update. Ther. Adv. Endocrinol. Metab. 2016, 7, 136–147. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Biomarkers (Specimen Source) | Subjects | AUC (95%CI) | Cut-off Value/ Sensitivity (%)/ Specificity (%) | References |
|---|---|---|---|---|
| Animal Studies | ||||
| KIM-1 (urine) | 125 rats | 0.82 (0.70–0.95) p = 0.037 * | 6.11 ng/mL 83.8%/89.8% | Pais, 2019 [62] |
| Clusterin (urine) | 125 rats | 0.80 (0.67–0.93) p = 0.060 * | ---- | Pais, 2019 [62] |
| Osteopontin (urine) | 125 rats | 0.70 (0.53–0.86) * | ---- | Pais, 2019 [62] |
| Human studies | ||||
| KIM-1 (urine) | 87 patients | 0.85 (0.75–0.95) p < 0.001 ** | 1.72 ng/mL 81.8%/85.5% | Pang, 2017 [63] |
| NGAL (urine) | 87 patients | 0.82 (0.73–0.92) p = 0.001 ** | 9.07 ng/mL 100.0%/63.2% | Pang, 2017 [63] |
| KIM-1 +NGAL (urine) | 87 patients | 0.85 (0.75–0.95) p < 0.001 ** | 1.72 ng/mL (KIM-1) and 9.07 ng/mL (NGAL); 90.9%/75.0% | Pang, 2017 [63] |
| NGAL (urine) at 96–144 hr | 94 patients | 0.82 (0.61–0.96), p = 0.020 (for predicting VA-AKI by day 5) | 618.8 ng/mL 73.0%/68.0% | Sampaio, 2021 [64] |
| [TIMP-2] × [IGFBP-7] (urine) | 333 patients | ---- | ---- | Kane-Gill, 2019 [65] |
| [TIMP-2] × [IGFBP-7]/Cr at 144–192 h (urine) | 94 patients | 0.71 (0.62–0.98), p = 0.009 (for predicting non-recovery of VA-AKI at discharge) | 2.15 (ng/mL)2/1000 88.0%/64.0% | Sampaio, 2021 [64] |
| 5-HIAA/5-HT ratio (serum) | 97 patients | 0.88 (0.88–0.96) | ---- | Lee, 2021 [32] |
| Cystatin C (serum) | 73 patients | 0.92 | ---- | Kim, 2021 [66] |
| Osteopontin (serum) | 73 patients | 0.79 | ---- | Kim, 2021 [66] |
| TFF3 (serum) | 73 patients | 0.93 | ---- | Kim, 2021 [66] |
| TNF-R1 (serum) | 73 patients | 0.87 | ---- | Kim, 2021 [66] |
| Potential Risk Factors | |
|---|---|
| Modifiable |
|
| Non-modifiable |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kan, W.-C.; Chen, Y.-C.; Wu, V.-C.; Shiao, C.-C. Vancomycin-Associated Acute Kidney Injury: A Narrative Review from Pathophysiology to Clinical Application. Int. J. Mol. Sci. 2022, 23, 2052. https://doi.org/10.3390/ijms23042052
Kan W-C, Chen Y-C, Wu V-C, Shiao C-C. Vancomycin-Associated Acute Kidney Injury: A Narrative Review from Pathophysiology to Clinical Application. International Journal of Molecular Sciences. 2022; 23(4):2052. https://doi.org/10.3390/ijms23042052
Chicago/Turabian StyleKan, Wei-Chih, Yi-Chih Chen, Vin-Cent Wu, and Chih-Chung Shiao. 2022. "Vancomycin-Associated Acute Kidney Injury: A Narrative Review from Pathophysiology to Clinical Application" International Journal of Molecular Sciences 23, no. 4: 2052. https://doi.org/10.3390/ijms23042052
APA StyleKan, W.-C., Chen, Y.-C., Wu, V.-C., & Shiao, C.-C. (2022). Vancomycin-Associated Acute Kidney Injury: A Narrative Review from Pathophysiology to Clinical Application. International Journal of Molecular Sciences, 23(4), 2052. https://doi.org/10.3390/ijms23042052