
Straight to the Point—The Novel Strategies to Cure Pediatric AML


Abstract
1. Introduction
2. Genetic Subtypes and Characterization
3. Genetics Influence the Success of Treatment
3.1. Low-Risk Genetics
3.1.1. RUNX1-RUNX1T1
3.1.2. CBFβ::MYH11
3.1.3. Mutated NPM1 without FLT3/ITD
3.1.4. CEBPA
3.1.5. PML::RARA
3.2. High Risk Genetic
3.2.1. FLT3/ITD Mutation
3.2.2. 11q23/KMT2A Rearrangements
3.2.3. 11p15/NUP98::NSD1 Rearrangements
3.2.4. MNX::ETV6
3.2.5. Aberrations Involving GATA2 and MECOM (EVI1)
3.3. Intermediate Risk
3.3.1. MYST3::CREBBP
3.3.2. Trisomy 21
3.3.3. KIT Mutations
3.3.4. FLT3/TKD
3.3.5. BCR::ABL1
3.3.6. NPM1::MLF1
3.3.7. t(16;21)
3.3.8. RBM15::MKL1
3.3.9. Trisomy 8
3.3.10. Monosomy 7/5 or Del(5q)
3.3.11. Hyperdiploid and Complex Karyotypes
3.4. Mutations That May Significantly Affect Prognosis

{kind=link}
| Molecular Alteration | Most Common Secondary Cytogenetic Factors | Influence on Prognosis | References |
|---|---|---|---|
| NPM1 gene mutations | FLT3/ITD Normal karyotype | Improve the prognosis | [12,16,123] |
| CEBPA gene mutations | FLT3/ITD GATA2 mutations CBFB::MYH11 Normal karyotype | Improve the prognosis | [12,16,110,123] |
| FLT3/ITD | Normal karyotype PML::RARA RUNX1::RUNX1T1 CBFB::MYH11 NUP98::NSD1 DEK::NUP214 trisomy 8 CEBPAdm mutated WT1 | Generally, worsens the prognosis with a few exceptions (e.g., NPM1) | [13,14,54,123] |
| Mutated WT1 | NUP98::NSD1 FLT3/ITD CEBPA NPM1 N-RAS K-RAS | Worsens the prognosis | [12,110,123] |
4. New Therapeutic Achievements
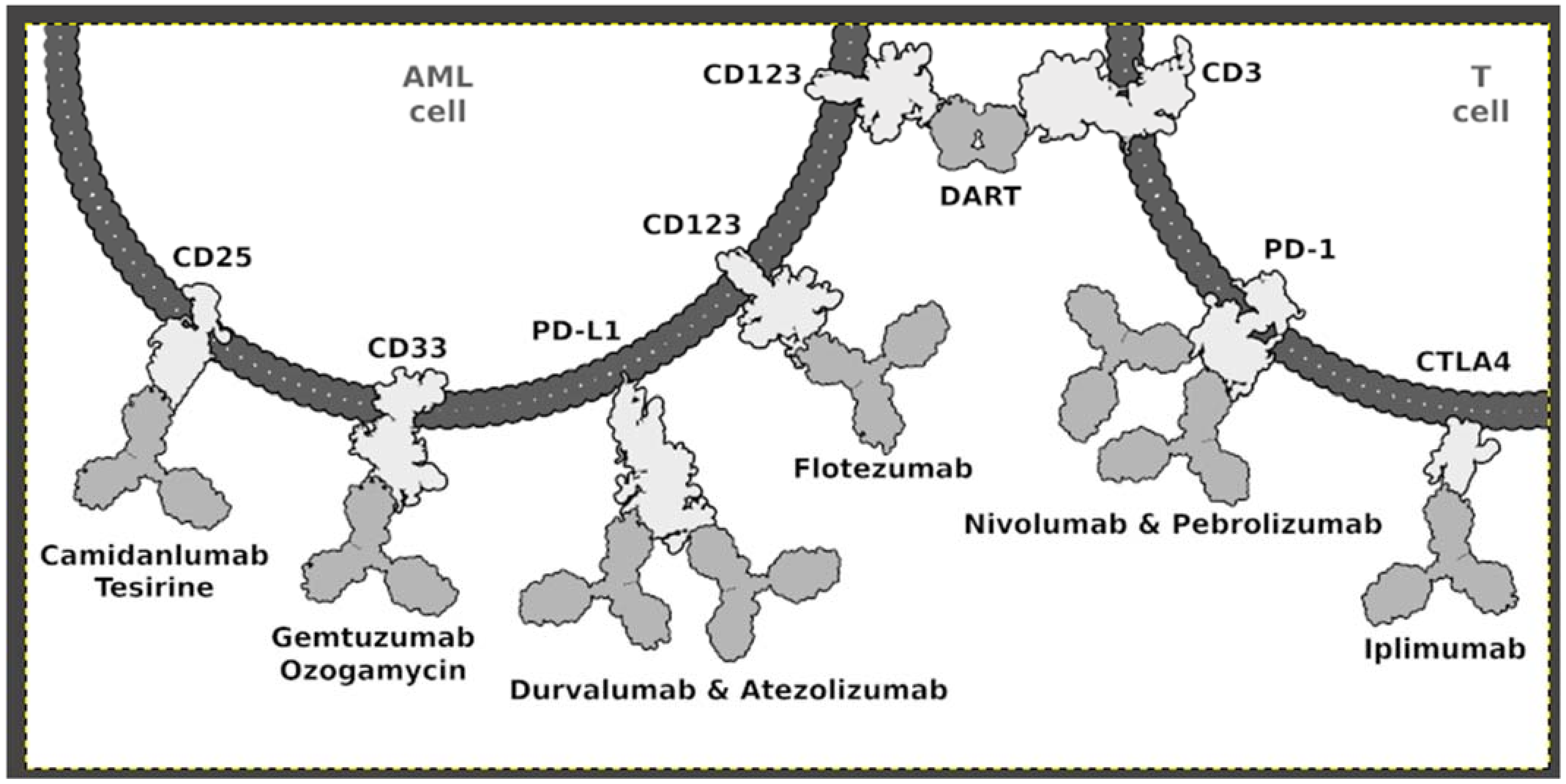
4.1. Immunotherapy
4.2. CART-T
4.3. Other Therapeutical Achievements
4.3.1. CPX-351 (Vyxeos®)
4.3.2. HDAC Inhibitors
5. NGS—Predisposition in Pediatric AML
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ksiazek, T.; Czogala, M.; Kaczowka, P.; Sadowska, B.; Pawinska-Wasikowska, K.; Bik-Multanowski, M.; Sikorska-Fic, B.; Matysiak, M.; Skalska-Sadowska, J.; Wachowiak, J.; et al. High Frequency of Fusion Gene Transcript Resulting From t(10;11)(P12;Q23) Translocation in Pediatric Acute Myeloid Leukemia in Poland. Front. Pediatrics 2020, 8, 278. [Google Scholar] [CrossRef] [PubMed]
- Song, M.K.; Park, B.B.; Uhm, J.E. Targeted Therapeutic Approach Based on Understanding of Aberrant Molecular Pathways Leading to Leukemic Proliferation in Patients with Acute Myeloid Leukemia. Int. J. Mol. Sci. 2021, 22, 5789. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.H.; Carroll, W.L.; Meshinchi, S.; Arceci, R.J. Biology, Risk Stratification, and Therapy of Pediatric Acute Leukemias: An Update. J. Clin. Oncol. 2011, 29, 551–565. [Google Scholar] [CrossRef] [PubMed]
- de Rooij, J.; Zwaan, C.; van den Heuvel-Eibrink, M. Pediatric AML: From Biology to Clinical Management. J. Clin. Med. 2015, 4, 127–149. [Google Scholar] [CrossRef] [PubMed]
- Creutzig, U.; Kutny, M.A.; Barr, R.; Schlenk, R.F.; Ribeiro, R.C. Acute Myelogenous Leukemia in Adolescents and Young Adults. Pediatric Blood Cancer 2018, 65, e27089. [Google Scholar] [CrossRef] [PubMed]
- Creutzig, U.; Büchner, T.; Sauerland, M.C.; Zimmermann, M.; Reinhardt, D.; Döhner, H.; Schlenk, R.F. Significance of Age in Acute Myeloid Leukemia Patients Younger than 30 Years: A Common Analysis of the Pediatric Trials AML-BFM 93/98 and the Adult Trials AMLCG 92/99 and AMLSG HD93/98A. Cancer 2008, 112, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Bonifant, C.L.; Velasquez, M.P.; Gottschalk, S. Advances in Immunotherapy for Pediatric Acute Myeloid Leukemia. Expert Opin. Biol. Ther. 2018, 18, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Lamble, A.J.; Tasian, S.K. Opportunities for Immunotherapy in Childhood Acute Myeloid Leukemia. Blood Adv. 2019, 3, 3750–3758. [Google Scholar] [CrossRef]
- Grimwade, D.; Ivey, A.; Huntly, B.J.P. Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood J. Am. Soc. Hematol. 2016, 127, 29–41. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Creutzig, U.; Zimmermann, M.; Reinhardt, D.; Rasche, M.; von Neuhoff, C.; Alpermann, T.; Dworzak, M.; Perglerová, K.; Zemanova, Z.; Tchinda, J.; et al. Changes in Cytogenetics and Molecular Genetics in Acute Myeloid Leukemia from Childhood to Adult Age Groups. Cancer 2016, 122, 3821–3830. [Google Scholar] [CrossRef] [PubMed]
- Bolouri, H.; Farrar, J.E.; Triche, T.; Ries, R.E.; Lim, E.L.; Alonzo, T.A.; Ma, Y.; Moore, R.; Mungall, A.J.; Marra, M.A.; et al. The Molecular Landscape of Pediatric Acute Myeloid Leukemia Reveals Recurrent Structural Alterations and Age-Specific Mutational Interactions. Nat. Med. 2018, 24, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.J.; Hills, R.K.; Moorman, A.v.; Grimwade, D.J.; Hann, I.; Webb, D.K.H.; Wheatley, K.; de Graaf, S.S.N.; van den Berg, E.; Burnett, A.K.; et al. Cytogenetics of Childhood Acute Myeloid Leukemia: United Kingdom Medical Research Council Treatment Trials AML 10 and 12. J. Clin. Oncol. 2010, 28, 2674–2681. [Google Scholar] [CrossRef] [PubMed]
- Von Neuhoff, C.; Reinhardt, D.; Sander, A.; Zimmermann, M.; Bradtke, J.; Betts, D.R.; Zemanova, Z.; Stary, J.; Bourquin, J.P.; Haas, O.A.; et al. Prognostic Impact of Specific Chromosomal Aberrations in a Large Group of Pediatric Patients with Acute Myeloid Leukemia Treated Uniformly According to Trial AML-BFM 98. J. Clin. Oncol. 2010, 28, 2682–2689. [Google Scholar] [CrossRef]
- Kutny, M.A.; Alonzo, T.A.; Gerbing, R.B.; Wang, Y.C.; Raimondi, S.C.; Hirsch, B.A.; Fu, C.H.; Meshinchi, S.; Gamis, A.S.; Feusner, J.H.; et al. Arsenic Trioxide Consolidation Allows Anthracycline Dose Reduction for Pediatric Patients with Acute Promyelocytic Leukemia: Report from the Children’s Oncology Group Phase III Historically Controlled Trial AAML0631. J. Clin. Oncol. 2017, 35, 3021–3029. [Google Scholar] [CrossRef] [PubMed]
- Hollink, I.H.I.M.; Zwaan, C.M.; Zimmermann, M.; Arentsen-Peters, T.C.J.M.; Pieters, R.; Cloos, J.; Kaspers, G.J.L.; de Graaf, S.S.N.; Harbott, J.; Creutzig, U.; et al. Favorable Prognostic Impact of NPM1 Gene Mutations in Childhood Acute Myeloid Leukemia, with Emphasis on Cytogenetically Normal AML. Leukemia 2009, 23, 262–270. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Goudswaard, C.S.; Van Putten, W.; Bijl, M.A.; Sanders, M.A.; Hugens, W.; Uitterlinden, A.G.; Erpelinck, C.A.J.; Delwel, R.; Löwenberg, B.; et al. Mutations in Nucleophosmin (NPM1) in Acute Myeloid Leukemia (AML): Association with Other Gene Abnormalities and Previously Established Gene Expression Signatures and Their Favorable Prognostic Significance. Blood 2005, 106, 3747–3754. [Google Scholar] [CrossRef]
- Hollink, I.H.I.M.; van den Heuvel-Eibrink, M.M.; Arentsen-Peters, S.T.C.J.M.; Zimmermann, M.; Peeters, J.K.; Valk, P.J.M.; Balgobind, B.v.; Sonneveld, E.; Kaspers, G.J.L.; de Bont, E.S.J.M.; et al. Characterization of CEBPA Mutations and Promoter Hypermethylation in Pediatric Acute Myeloid Leukemia. Haematologica 2011, 96, 384–392. [Google Scholar] [CrossRef]
- Fasan, A.; Haferlach, C.; Alpermann, T.; Jeromin, S.; Grossmann, V.; Eder, C.; Weissmann, S.; Dicker, F.; Kohlmann, A.; Schindela, S.; et al. The Role of Different Genetic Subtypes of CEBPA Mutated AML. Leukemia 2014, 28, 794–803. [Google Scholar] [CrossRef] [PubMed]
- Noort, S.; Zimmermann, M.; Reinhardt, D.; Cuccuini, W.; Pigazzi, M.; Smith, J.; Ries, R.E.; Alonzo, T.A.; Hirsch, B.; Tomizawa, D.; et al. Prognostic Impact of t(16;21)(P11;Q22) and t(16;21)(Q24;Q22) in Pediatric AML: A Retrospective Study by the I-BFM Study Group. Blood 2018, 132, 1584–1592. [Google Scholar] [CrossRef] [PubMed]
- Shiba, N.; Yoshida, K.; Hara, Y.; Yamato, G.; Shiraishi, Y.; Matsuo, H.; Okuno, Y.; Chiba, K.; Tanaka, H.; Kaburagi, T.; et al. Transcriptome Analysis Offers a Comprehensive Illustration of the Genetic Background of Pediatric Acute Myeloid Leukemia. Blood Adv. 2019, 3, 3157–3169. [Google Scholar] [CrossRef] [PubMed]
- Taga, T. Treatment strategy for myeloid leukemia with Down syndrome. Rinsho ketsueki. Jpn. J. Clin. Hematol. 2020, 61, 1382–1387. [Google Scholar] [CrossRef]
- Forestier, E.; Izraeli, S.; Beverloo, B.; Haas, O.; Pession, A.; Michalová, K.; Stark, B.; Harrison, C.J.; Teigler-Schlegel, A.; Johansson, B. Cytogenetic Features of Acute Lymphoblastic and Myeloid Leukemias in Pediatric. Patients with Down Syndrome: An IBFM-SG Study. Blood 2008, 111, 1575–1583. [Google Scholar] [CrossRef] [PubMed]
- Elgarten, C.W.; Aplenc, R. Pediatric Acute Myeloid Leukemia: Updates on Biology, Risk Stratification, and Therapy. Curr. Opin. Pediatrics 2020, 32, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Meshinchi, S.; Alonzo, T.A.; Stirewalt, D.L.; Zwaan, M.; Zimmerman, M.; Reinhardt, D.; Kaspers, G.J.L.; Heerema, N.A.; Gerbing, R.; Lange, B.J.; et al. Clinical Implications of FLT3 Mutations in Pediatric AML. Blood 2006, 108, 3654–3661. [Google Scholar] [CrossRef]
- Coenen, E.; Harbott, J.; Zwaan, C.; Raimondi, S.; van den Heuvel-Eibrink, M. 11Q23 Rearrangements in De Novo Childhood Acute Myeloid Leukemia. Atlas Genet. Cytogenet. Oncol. Haematol. 2012, 16, 574–581. [Google Scholar] [CrossRef]
- Chen, Y.; Kantarjian, H.; Pierce, S.; Faderl, S.; O’Brien, S.; Qiao, W.; Abruzzo, L.; De Lima, M.; Kebriaei, P.; Jabbour, E.; et al. Prognostic Significance of 11q23 Aberrations in Adult Acute Myeloid Leukemia and the Role of Allogeneic Stem Cell Transplantation. Leukemia 2013, 27, 836–842. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Climent, J.A.; Espinosa, R., III; Thirman, M.J.; Le Beau, M.M.; Rowley, J.D. Abnormalities of Chromosome Band 11q23 and the MLL Gene in Pediatric Myelomonocytic and Monoblastic Leukemias. Identification of the t(9;11) as an Indicator of Long Survival. J. Pediatric Hematol./Oncol. 1995, 17, 277–283. [Google Scholar] [CrossRef]
- Meyer, C.; Burmeister, T.; Gröger, D.; Tsaur, G.; Fechina, L.; Renneville, A.; Sutton, R.; Venn, N.C.; Emerenciano, M.; Pombo-De-Oliveira, M.S.; et al. The MLL Recombinome of Acute Leukemias in 2017. Leukemia 2018, 32, 273–284. [Google Scholar] [CrossRef]
- Balgobind, B.V.; Raimondi, S.C.; Harbott, J.; Zimmermann, M.; Alonzo, T.A.; Auvrignon, A.; Beverloo, H.B.; Chang, M.; Creutzig, U.; Dworzak, M.N.; et al. Novel Prognostic Subgroups in Childhood 11q23/MLL-Rearranged Acute Myeloid Leukemia: Results of an International Retrospective Study. Blood 2009, 114, 2489–2496. [Google Scholar] [CrossRef]
- Akiki, S.; Dyer, S.A.; Grimwade, D.; Ivey, A.; Abou-Zeid, N.; Borrow, J.; Jeffries, S.; Caddick, J.; Newell, H.; Begum, S.; et al. NUP98-NSD1 Fusion in Association with FLT3-ITD Mutation Identifies a Prognostically Relevant Subgroup of Pediatric Acute Myeloid Leukemia Patients Suitable for Monitoring by Real Time Quantitative PCR. Genes Chromosomes Cancer 2013, 52, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Balgobind, B.V.; Hollink, I.H.I.M.; Arentsen-Peters, S.T.C.J.M.; Zimmermann, M.; Harbott, J.; Berna Beverloo, H.; von Bergh, A.R.M.; Cloos, J.; Kaspers, G.J.L.; de Haas, V.; et al. Integrative Analysis of Type-I and Type-II Aberrations Underscores the Genetic Heterogeneity of Pediatric Acute Myeloid Leukemia. Haematologica 2011, 96, 1478–1487. [Google Scholar] [CrossRef]
- Radtke, I.; Mullighan, C.G.; Ishii, M.; Su, X.; Cheng, J.; Ma, J.; Ganti, R.; Cai, Z.; Goorha, S.; Pounds, S.B.; et al. Genomic Analysis Reveals Few Genetic Alterations in Pediatric Acute Myeloid Leukemia. Proc. Natl. Acad. Sci. USA 2009, 106, 12944–12949. [Google Scholar] [CrossRef] [PubMed]
- Struski, S.; Lagarde, S.; Bories, P.; Puiseux, C.; Prade, N.; Cuccuini, W.; Pages, M.P.; Bidet, A.; Gervais, C.; Lafage-Pochitaloff, M.; et al. NUP98 Is Rearranged in 3.8% of Pediatric AML Forming a Clinical and Molecular Homogenous Group with a Poor Prognosis. Leukemia 2017, 31, 565–572. [Google Scholar] [CrossRef]
- Hara, Y.; Shiba, N.; Ohki, K.; Tabuchi, K.; Yamato, G.; Park, M.J.; Tomizawa, D.; Kinoshita, A.; Shimada, A.; Arakawa, H.; et al. Prognostic Impact of Specific Molecular Profiles in Pediatric Acute Megakaryoblastic Leukemia in Non-Down Syndrome. Genes Chromosomes Cancer 2017, 56, 394–404. [Google Scholar] [CrossRef]
- Noort, S.; Wander, P.; Alonzo, T.A.; Smith, J.; Ries, R.E.; Gerbing, R.B.; Dolman, E.M.M.; Locatelli, F.; Reinhardt, D.; Baruchel, A.; et al. The Clinical and Biological Characteristics of NUP98-KDM5A Pediatric Acute Myeloid Leukemia. Haematologica 2021, 106, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Espersen, A.D.L.; Noren-Nyström, U.; Abrahamsson, J.; Ha, S.Y.; Pronk, C.J.; Jahnukainen, K.; Jónsson, Ó.G.; Lausen, B.; Palle, J.; Zeller, B.; et al. Acute Myeloid Leukemia (AML) with t(7;12)(Q36;P13) Is Associated with Infancy and Trisomy 19: Data from Nordic Society for Pediatric Hematology and Oncology (NOPHO-AML) and Review of the Literature. Genes Chromosomes Cancer 2018, 57, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Johnston, D.; Alonzo, T.A.; Gerbing, R.B.; Hirsch, B.; Heerema, N.A.; Ravindranath, Y.; Woods, W.G.; Lange, B.J.; Gamis, A.S.; Raimondi, S.C. Outcome of Pediatric Patients with Acute Myeloid Leukemia (AML) and -5/5q- Abnormalities from Five Pediatric AML Treatment Protocols: A Report from the Children’s Oncology Group. Pediatric Blood Cancer 2013, 60, 2073–2078. [Google Scholar] [CrossRef] [PubMed]
- Masetti, R.; Pigazzi, M.; Togni, M.; Astolfi, A.; Indio, V.; Manara, E.; Casadio, R.; Pession, A.; Basso, G.; Locatelli, F. CBFA2T3-GLIS2 Fusion Transcript Is a Novel Common Feature in Pediatric, Cytogenetically Normal AML, Not Restricted to FAB M7 Subtype. Blood 2013, 121, 3469–3472. [Google Scholar] [CrossRef]
- Creutzig, U.; Van Den Heuvel-Eibrink, M.M.; Gibson, B.; Dworzak, M.N.; Adachi, S.; De Bont, E.; Harbott, J.; Hasle, H.; Johnston, D.; Kinoshita, A.; et al. Diagnosis and Management of Acute Myeloid Leukemia in Children and Adolescents: Recommendations from an International Expert Panel. Blood 2012, 120, 3167–3205. [Google Scholar] [CrossRef]
- Lugthart, S.; Gröschel, S.; Beverloo, H.B.; Kayser, S.; Valk, P.J.M.; Van Zelderen-Bhola, S.L.; Ossenkoppele, G.J.; Vellenga, E.; Van Den Berg-De Ruiter, E.; Schanz, U.; et al. Clinical, Molecular, and Prognostic Significance of WHO Type Inv(3)(Q21q26.2)/t(3;3)(Q21;Q26.2) and Various Other 3q Abnormalities in Acute Myeloid Leukemia. J. Clin. Oncol. 2010, 28, 3890–3898. [Google Scholar] [CrossRef] [PubMed]
- Grimwade, D.; Hills, R.K.; Moorman, A.V.; Walker, H.; Chatters, S.; Goldstone, A.H.; Wheatley, K.; Harrison, C.J.; Burnett, A.K. Refinement of Cytogenetic Classification in Acute Myeloid Leukemia: Determination of Prognostic Significance of Rare Recurring Chromosomal Abnormalities among 5876 Younger Adult Patients Treated in the United Kingdom Medical Research Council Trials. Blood 2010, 116, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Neuendorff, N.R.; Hemmati, P.; Arnold, R.; Ihlow, J.; Dörken, B.; Müller-Tidow, C.; Westermann, J. BCR-ABL1 Acute Myeloid Leukemia: Are We Always Dealing with a High-Risk Disease? Blood Adv. 2018, 2, 1409–1411. [Google Scholar] [CrossRef] [PubMed]
- Borel, C.; Dastugue, N.; Cances-Lauwers, V.; Mozziconacci, M.J.; Prebet, T.; Vey, N.; Pigneux, A.; Lippert, E.; Visanica, S.; Legrand, F.; et al. PICALM-MLLT10 Acute Myeloid Leukemia: A French Cohort of 18 Patients. Leuk. Res. 2012, 36, 1365–1369. [Google Scholar] [CrossRef] [PubMed]
- Arbor, A. Spontaneous Remission of Congenital Acute Myeloid Leukemia. Pediatric Blood Cancer 2011, 331–332. [Google Scholar] [CrossRef]
- Wong, K.; Frcp, H.Y.; Siu, L.L.P.; Pang, A.; Kwong, Y. T(8;16)(P11;P13) Predisposes to a Transient but Potentially Recurring Neonatal Leukemia. Hum. Pathol. 2008, 39, 1702–1707. [Google Scholar] [CrossRef]
- Boissel, N.; Leroy, H.; Brethon, B.; Philippe, N.; De Botton, S.; Auvrignon, A.; Raffoux, E.; Leblanc, T.; Thomas, X.; Hermine, O. Incidence and Prognostic Impact of C-Kit, FLT3, and Ras Gene Mutations in Core Binding Factor Acute Myeloid Leukemia (CBF-AML). Leukemia 2006, 965–970. [Google Scholar] [CrossRef]
- Pollard, J.A.; Alonzo, T.A.; Gerbing, R.B.; Ho, P.A.; Zeng, R.; Ravindranath, Y.; Dahl, G.; Lacayo, N.J.; Becton, D.; Chang, M.; et al. Prevalence and Prognostic Significance of KIT Mutations in Pediatric Patients with Core Binding Factor AML Enrolled on Serial Pediatric Cooperative Trials for de Novo AML. Blood 2016, 115, 2372–2380. [Google Scholar] [CrossRef]
- Duployez, N.; Marceau-Renaut, A.; Boissel, N.; Petit, A.; Bucci, M.; Geffroy, S.; Lapillonne, H.; Renneville, A.; Ragu, C.; Figeac, M.; et al. Comprehensive Mutational Profiling of Core Binding Factor Acute Myeloid Leukemia. Blood 2016, 127, 2451–2459. [Google Scholar] [CrossRef]
- Inaba, H.; Zhou, Y.; Abla, O.; Adachi, S.; Auvrignon, A.; Beverloo, H.B.; De Bont, E.; Chang, T.T.; Creutzig, U.; Dworzak, M.; et al. Heterogeneous Cytogenetic Subgroups and Outcomes in Childhood Acute Megakaryoblastic Leukemia: A Retrospective International Study. Blood 2015, 126, 1575–1584. [Google Scholar] [CrossRef]
- De Rooij, J.D.E.; Masetti, R.; Van Den Heuvel-Eibrink, M.M.; Cayuela, J.M.; Trka, J.; Reinhardt, D.; Rasche, M.; Sonneveld, E.; Alonzo, T.A.; Fornerod, M.; et al. Recurrent Abnormalities Can Be Used for Risk Group Stratification in Pediatric AMKL: A Retrospective Intergroup Study. Blood 2016, 127, 3424–3430. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Nicoletti, I.; Bolli, N.; Paola Martelli, M.; Liso, A.; Gorello, P.; Mandelli, F.; Mecucci, C.; Fabrizio Martelli, M.; Sapienza, L.; et al. Translocations and Mutations Involving the Nucleophosmin (NPM1) Gene in Lymphomas and Leukemias. Haematology 2007, 92, 519–532. [Google Scholar] [CrossRef]
- Lim, G.; Choi, J.R.; Kim, M.J.; Kim, S.Y.; Lee, H.J.; Suh, J.T.; Yoon, H.J.; Lee, J.; Lee, S.; Lee, W.I.; et al. Detection of t(3;5) and NPM1/MLF1 Rearrangement in an Elderly Patient with Acute Myeloid Leukemia: Clinical and Laboratory Study with Review of the Literature. Cancer Genet. Cytogenet. 2010, 199, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Laursen, A.C.L.; Sandahl, J.D.; Kjeldsen, E.; Abrahamsson, J.; Asdahl, P.; Ha, S.Y.; Heldrup, J.; Jahnukainen, K. Trisomy 8 in Pediatric Acute Myeloid Leukemia: A NOPHO-AML Study. Genes Chromosomes Cancer 2016, 55, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Rasche, M.; von Neuhoff, C.; Dworzak, M.; Bourquin, J.P.; Bradtke, J.; Göhring, G.; Escherich, G.; Fleischhack, G.; Graf, N.; Gruhn, B.; et al. Genotype-Outcome Correlations in Pediatric AML: The Impact of a Monosomal Karyotype in Trial AML-BFM 2004. Leukemia 2017, 31, 2807–2814. [Google Scholar] [CrossRef] [PubMed]
- Sandahl, J.D.; Kjeldsen, E.; Abrahamsson, J.; Ha, S.Y.; Heldrup, J.; Jahnukainen, K.; Jonsson, O.G.; Lausen, B.; Palle, J.; Zeller, B.; et al. Ploidy and Clinical Characteristics of Childhood Acute Myeloid Leukemia: A NOPHO-AML Study Julie. Genes Chromosomes Cancer 2014, 53, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Chilton, L.; Hills, R.K.; Harrison, C.J.; Burnett, A.K.; Grimwade, D.; Moorman, A.V. Hyperdiploidy with 49–65 Chromosomes Represents a Heterogeneous Cytogenetic Subgroup of Acute Myeloid Leukemia with Differential Outcome. Leukemia 2014, 28, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Nicoletti, I.; Martelli, M.F.; Mecucci, C. Acute Myeloid Leukemia Carrying Cytoplasmic/Mutated Nucleophosmin (NPMc+ AML): Biologic and Clinical Features. Blood 2007, 109, 874–885. [Google Scholar] [CrossRef] [PubMed]
- Dufour, A.; Schneider, F.; Metzeler, K.H.; Hoster, E.; Schneider, S.; Zellmeier, E.; Benthaus, T.; Sauerland, M.-C.; Berdel, W.E.; Büchner, T.; et al. Acute Myeloid Leukemia with Biallelic CEBPA Gene Mutations and Normal Karyotype Represents a Distinct Genetic Entity Associated with a Favorable Clinical Outcome. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 570–577. [Google Scholar] [CrossRef]
- Wouters, B.J.; Löwenberg, B.; Erpelinck-Verschueren, C.A.J.; van Putten, W.L.J.; Valk, P.J.M.; Delwel, R. Double CEBPA Mutations, but Not Single CEBPA Mutations, Define a Subgroup of Acute Myeloid Leukemia with a Distinctive Gene Expression Profile That Is Uniquely Associated with a Favorable Outcome. Blood 2009, 113, 3088–3091. [Google Scholar] [CrossRef]
- Kottaridis, P.D.; Gale, R.E.; Frew, M.E.; Harrison, G.; Langabeer, S.E.; Belton, A.A.; Walker, H.; Wheatley, K.; Bowen, D.T.; Burnett, A.K.; et al. The Presence of a FLT3 Internal Tandem Duplication in Patients with Acute Myeloid Leukemia (AML) Adds Important Prognostic Information to Cytogenetic Risk Group and Response to the First Cycle of Chemotherapy: Analysis of 854 Patients from the United King. Blood 2001, 98, 1752–1759. [Google Scholar] [CrossRef] [PubMed]
- Bill, M.; Mrózek, K.; Kohlschmidt, J.; Eisfeld, A.K.; Walker, C.J.; Nicolet, D.; Papaioannou, D.; Blachly, J.S.; Orwick, S.; Carroll, A.J.; et al. Mutational Landscape and Clinical Outcome of Patients with de Novo Acute Myeloid Leukemia and Rearrangements Involving 11q23/KMT2A. Proc. Natl. Acad. Sci. USA 2020, 117, 26340–26346. [Google Scholar] [CrossRef] [PubMed]
- Smol, T.; Collonge-Rame, M.-A. t(8;16)(P11;P13) KAT6A/CREBBP. Atlas Genet. Cytogenet. Oncol. Haematol. 2017, 11–16. [Google Scholar] [CrossRef]
- Ayatollahi, H.; Shajiei, A.; Sadeghian, M.H.; Sheikhi, M.; Yazdandoust, E.; Ghazanfarpour, M.; Shams, S.F.; Shakeri, S. Prognostic Importance of C-KIT Mutations in Core Binding Factor Acute Myeloid Leukemia: A Systematic Review. Hematol./Oncol. Stem Cell Ther. 2017, 10, 1–7. [Google Scholar] [CrossRef]
- Allan, J.M. Genetic Susceptibility to Breast Cancer in Lymphoma Survivors. Blood 2019, 133, 1004–1006. [Google Scholar] [CrossRef]
- Al-Harbi, S.; Aljurf, M.; Mohty, M.; Almohareb, F.; Ahmed, S.O.A. An Update on the Molecular Pathogenesis and Potential Therapeutic Targeting of AML with t(8;21)(Q22;Q22.1);RUNX1-RUNX1T1. Blood Adv. 2020, 4, 229–238. [Google Scholar] [CrossRef]
- Sakamoto, K.; Shiba, N.; Deguchi, T.; Kiyokawa, N.; Hashii, Y.; Moriya-Saito, A.; Tomizawa, D.; Taga, T.; Adachi, S.; Horibe, K.; et al. Negative CD19 Expression Is Associated with Inferior Relapse-Free Survival in Children with RUNX1-RUNX1T1–Positive Acute Myeloid Leukaemia: Results from the Japanese Paediatric Leukaemia/Lymphoma Study Group AML-05 Study. Br. J. Haematol. 2019, 187, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Kundu, M.; Liu, P.P. Function of the Inv(16) Fusion Gene CBFB-MYH11. Curr. Opin. Hematol. 2001, 8, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Huret, J. Inv(16)(P13Q22)-T(16;16)(P13;Q22)-Del(16)(Q22). Atlas Genet. Cytogenet. Oncol. Haematol. 2011, 3, 147–149. [Google Scholar] [CrossRef][Green Version]
- Schoch, C.; Kern, W.; Schnittger, S.; Büchner, T.; Hiddemann, W.; Haferlach, T. The Influence of Age on Prognosis of de Novo Acute Myeloid Leukemia Differs According to Cytogenetic Subgroups. Haematologica 2004, 89, 1082–1090. [Google Scholar]
- Hann, I.M.; Webb, D.K.W.; Gibson, B.E.S.; Harrison, C.J. MRC Trials in Childhood Acute Myeloid Leukaemia. Ann. Hematol. 2004, 83 (Suppl. 1), S108–S112. [Google Scholar] [CrossRef] [PubMed]
- Paschka, P.; Marcucci, G.; Ruppert, A.S.; Mrózek, K.; Chen, H.; Kittles, R.A.; Vukosavljevic, T.; Perrotti, D.; Vardiman, J.W.; Carroll, A.J.; et al. Adverse Prognostic Significance of KIT Mutations in Adult Acute Myeloid Leukemia with Inv(16) and t(8;21): A Cancer and Leukemia Group B Study. J. Clin. Oncol. 2006, 24, 3904–3911. [Google Scholar] [CrossRef] [PubMed]
- Hollink, I.H.I.M.; Van Den Heuvel-Eibrink, M.M.; Zimmermann, M.; Balgobind, B.V.; Arentsen-Peters, S.T.C.J.M.; Alders, M.; Willasch, A.; Kaspers, G.J.L.; Trka, J.; Baruchel, A.; et al. Clinical Relevance of Wilms Tumor 1 Gene Mutations in Childhood Acute Myeloid Leukemia. Blood 2009, 113, 5951–5960. [Google Scholar] [CrossRef] [PubMed]
- Colombo, E.; Marine, J.C.; Danovi, D.; Falini, B.; Pelicci, P.G. Nucleophosmin Regulates the Stability and Transcriptional Activity of P53. Nat. Cell Biol. 2002, 4, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Brady, S.N.; Yu, Y.; Maggi, L.B.; Weber, J.D. ARF Impedes NPM/B23 Shuttling in an Mdm2-Sensitive Tumor Suppressor Pathway. Mol. Cell. Biol. 2004, 24, 9327–9338. [Google Scholar] [CrossRef]
- Brown, P.; McIntyre, E.; Rau, R.; Meshinchi, S.; Lacayo, N.; Dahl, G.; Alonzo, T.A.; Chang, M.; Arceci, R.J.; Small, D. The Incidence and Clinical Significance of Nucleophosmin Mutations in Childhood AML. Blood 2007, 110, 979–985. [Google Scholar] [CrossRef]
- Cazzaniga, G.; Dell’Oro, M.G.; Mecucci, C.; Giarin, E.; Masetti, R.; Rossi, V.; Locatelli, F.; Martelli, M.F.; Basso, G.; Pession, A.; et al. Nucleophosmin Mutations in Childhood Acute Myelogenous Leukemia with Normal Karyotype. Blood 2005, 106, 1419–1422. [Google Scholar] [CrossRef]
- Falini, B.; Mecucci, C.; Tiacci, E.; Alcalay, M.; Rosati, R.; Pasqualucci, L.; La Starza, R.; Diverio, D.; Colombo, E.; Santucci, A.; et al. Cytoplasmic Nucleophosmin in Acute Myelogenous Leukemia with a Normal Karyotype. N. Engl. J. Med. 2005, 352, 254–266. [Google Scholar] [CrossRef]
- Schnittger, S.; Schoch, C.; Kern, W.; Mecucci, C.; Tschulik, C.; Martelli, M.F.; Haferlach, T.; Hiddemann, W.; Falini, B. Nucleophosmin Gene Mutations Are Predictors of Favorable Prognosis in Acute Myelogenous Leukemia with a Normal Karyotype. Blood 2005, 106, 3733–3739. [Google Scholar] [CrossRef]
- Thiede, C.; Koch, S.; Creutzig, E.; Steudel, C.; Illmer, T.; Schaich, M.; Ehninger, G. Prevalence and Prognostic Impact of NPM1 Mutations in 1485 Adult Patients with Acute Myeloid Leukemia (AML). Blood 2006, 107, 4011–4020. [Google Scholar] [CrossRef]
- Falini, B.; Brunetti, L.; Sportoletti, P.; Paola Martelli, M. NPM1-Mutated Acute Myeloid Leukemia: From Bench to Bedside. Blood 2020, 136, 1707–1721. [Google Scholar] [CrossRef]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 Mutations in AML: Review of Current Knowledge and Evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Sciabolacci, S.; Falini, L.; Brunetti, L.; Martelli, M.P. Diagnostic and Therapeutic Pitfalls in NPM1-Mutated AML: Notes from the Field. Leukemia 2021, 35, 3113–3126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Iwasaki-Arai, J.; Iwasaki, H.; Fenyus, M.L.; Dayaram, T.; Owens, B.M.; Shigematsu, H.; Levantini, E.; Huettner, C.S.; Lekstrom-Himes, J.A.; et al. Enhancement of Hematopoietic Stem Cell Repopulating Capacity and Self-Renewal in the Absence of the Transcription Factor C/EBPα. Immunity 2004, 21, 853–863. [Google Scholar] [CrossRef]
- Wouters, B.J.; Jordà, M.A.; Keeshan, K.; Louwers, I.; Erpelinck-Verschueren, C.A.J.; Tielemans, D.; Langerak, A.W.; He, Y.; Yashiro-Ohtani, Y.; Zhang, P.; et al. Distinct Gene Expression Profiles of Acute Myeloid/T-Lymphoid Leukemia with Silenced CEBPA and Mutations in NOTCH1. Blood 2007, 110, 3706–3714. [Google Scholar] [CrossRef]
- Radomska, H.S.; Bassères, D.S.; Zheng, R.; Zhang, P.; Dayaram, T.; Yamamoto, Y.; Sternberg, D.W.; Lokker, N.; Giese, N.A.; Bohlander, S.K.; et al. Block of C/EBPα Function by Phosphorylation in Acute Myeloid Leukemia with FLT3 Activating Mutations. J. Exp. Med. 2006, 203, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Helbling, D.; Mueller, B.U.; Timchenko, N.A.; Schardt, J.; Eyer, M.; Betts, D.R.; Jotterand, M.; Meyer-Monard, S.; Fey, M.F.; Pabst, T. CBFB-SMMHC Is Correlated with Increased Calreticulin Expression and Suppresses the Granulocytic Differentiation Factor CEBPA in AML with Inv(16). Blood 2005, 106, 1369–1375. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schmidt, L.; Heyes, E.; Scheiblecker, L.; Eder, T.; Volpe, G.; Frampton, J.; Nerlov, C.; Valent, P.; Grembecka, J.; Grebien, F. CEBPA-Mutated Leukemia Is Sensitive to Genetic and Pharmacological Targeting of the MLL1 Complex. Leukemia 2019, 33, 1608–1619. [Google Scholar] [CrossRef]
- Mannelli, F.; Ponziani, V.; Bencini, S.; Bonetti, M.I.; Benelli, M.; Cutini, I.; Gianfaldoni, G.; Scappini, B.; Pancani, F.; Piccini, M.; et al. CEBPA–Double-Mutated Acute Myeloid Leukemia Displays a Unique Phenotypic Profile: A Reliable Screening Method and Insight into Biological Features. Haematologica 2017, 102, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Conneely, S.E.; Stevens, A.M. Advances in Pediatric Acute Promyelocytic Leukemia. Children 2020, 7, 11. [Google Scholar] [CrossRef]
- De Braekeleer, E.; Douet-Guilbert, N.; De Braekeleer, M. RARA Fusion Genes in Acute Promyelocytic Leukemia: A Review. Expert Rev. Hematol. 2014, 7, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Taga, T.; Tomizawa, D.; Takahashi, H.; Adachi, S. Acute Myeloid Leukemia in Children: Current Status and Future Directions. Pediatrics Int. 2016, 58, 71–80. [Google Scholar] [CrossRef]
- Iland, H.J.; Collins, M.; Bradstock, K.; Supple, S.G.; Catalano, A.; Hertzberg, M.; Browett, P.; Grigg, A.; Firkin, F.; Campbell, L.J.; et al. Use of Arsenic Trioxide in Remission Induction and Consolidation Therapy for Acute Promyelocytic Leukaemia in the Australasian Leukaemia and Lymphoma Group (ALLG) APML4 Study: A Non-Randomised Phase 2 Trial. Lancet Haematol. 2015, 2, e357–e366. [Google Scholar] [CrossRef]
- Jabbar, N.; Khayyam, N.; Arshad, U.; Maqsood, S.; Hamid, S.A.; Mansoor, N. An Outcome Analysis of Childhood Acute Promyelocytic Leukemia Treated with Atra and Arsenic Trioxide, and Limited Dose Anthracycline. Indian J. Hematol. Blood Transfus. 2021, 37, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, A.; Asou, N.; Atsuta, Y.; Sakura, T.; Ueda, Y.; Sawa, M.; Dobashi, N.; Taniguchi, Y.; Suzuki, R.; Nakagawa, M.; et al. Tamibarotene Maintenance Improved Relapse-Free Survival of Acute Promyelocytic Leukemia: A Final Result of Prospective, Randomized, JALSG-APL204 Study. Leukemia 2019, 33, 358–370. [Google Scholar] [CrossRef]
- Lagunas-Rangel, F.A.; Chávez-Valencia, V. FLT3–ITD and Its Current Role in Acute Myeloid Leukaemia. Med. Oncol. 2017, 34. [Google Scholar] [CrossRef]
- Janke, H.; Pastore, F.; Schumacher, D.; Herold, T.; Hopfner, K.P.; Schneider, S.; Berdel, W.E.; Büchner, T.; Woermann, B.J.; Subklewe, M.; et al. Activating FLT3 Mutants Show Distinct Gain-of-Function Phenotypes in Vitro and a Characteristic Signaling Pathway Profile Associated with Prognosis in Acute Myeloid Leukemia. PLoS ONE 2014, 9, e89560. [Google Scholar] [CrossRef]
- Haghi, A.; Salami, M.; Kian, M.M.; Nikbakht, M.; Mohammadi, S.; Chahardouli, B.; Rostami, S.; Malekzadeh, K. Effects of Sorafenib and Arsenic Trioxide on U937 and KG-1 Cell Lines: Apoptosis or Autophagy? Cell J. 2020, 22, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Inaba, H.; Rubnitz, J.E.; Coustan-Smith, E.; Li, L.; Furmanski, B.D.; Mascara, G.P.; Heym, K.M.; Christensen, R.; Onciu, M.; Shurtleff, S.A.; et al. Phase I Pharmacokinetic and Pharmacodynamic Study of the Multikinase Inhibitor Sorafenib in Combination with Clofarabine and Cytarabine in Pediatric Relapsed/Refractory Leukemia. J. Clin. Oncol. 2011, 29, 3293–3300. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.A.; Alonzo, T.A.; Brown, P.A.; Gerbing, R.B.; Fox, E.; Choi, J.K.; Fisher, B.T.; Hirsch, B.A.; Kahwash, S.; Levine, J.E.; et al. Sorafenib in Combination with Standard Chemotherapy for Children with High Allelic Ratio FLT3/ITD+ AML Improves Event-Free Survival and Reduces Relapse Risk: A Report from the Children’s Oncology Group Protocol AAML1031. Blood 2019, 134, 292. [Google Scholar] [CrossRef]
- Burchert, A.; Bug, G.; Fritz, L.V.; Finke, J.; Stelljes, M.; Röllig, C.; Wollmer, E.; Wäsch, R.; Bornhäuser, M.; Berg, T.; et al. Sorafenib Maintenance after Allogeneic Hematopoietic Stem Cell Transplantation for Acute Myeloid Leukemia with FLT3-Internal Tandem Duplication Mutation (SORMAIN). J. Clin. Oncol. 2020, 38, 2993–3002. [Google Scholar] [CrossRef] [PubMed]
- Coenen, E.A.; Raimondi, S.C.; Harbott, J.; Zimmermann, M.; Alonzo, T.A.; Auvrignon, A.; Beverloo, H.B.; Chang, M.; Creutzig, U.; Dworzak, M.N.; et al. Prognostic Significance of Additional Cytogenetic Aberrations in 733 de Novo Pediatric 11q23/MLL-Rearranged AML Patients: Results of an International Study. Blood 2011, 117, 7102–7111. [Google Scholar] [CrossRef] [PubMed]
- Mercher, T.; Schwaller, J. Pediatric Acute Myeloid Leukemia (AML): From Genes to Models Toward Targeted Therapeutic Intervention. Front. Pediatrics 2019, 7, 401. [Google Scholar] [CrossRef] [PubMed]
- Lonetti, A.; Indio, V.; Laginestra, M.A.; Tarantino, G.; Chiarini, F.; Astolfi, A.; Bertuccio, S.N.; Martelli, A.M.; Locatelli, F.; Pession, A.; et al. Inhibition of Methyltransferase Dot1l Sensitizes to Sorafenib Treatment Aml Cells Irrespective of Mll-Rearrangements: A Novel Therapeutic Strategy for Pediatric Aml. Cancers 2020, 12, 1972. [Google Scholar] [CrossRef] [PubMed]
- Shiba, N.; Ichikawa, H.; Taki, T.; Park, M.-J.; Jo, A.; Mitani, S.; Kobayashi, T.; Shimada, A.; Sotomatsu, M.; Arakawa, H.; et al. NUP98-NSD1 Gene Fusion and Its Related Gene Expression Signature Are Strongly Associated with a Poor Prognosis in Pediatric Acute Myeloid Leukemia. Genes Chromosomes Cancer 2013, 52, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, S.; Jyotsana, N.; Sharma, A.; Othman, B.; Kloos, A.; Mandhania, M.; Schottmann, R.; Ramsay, E.; Vornlocher, H.-P.; Ganser, A.; et al. Targeted Inhibition of the NUP98-NSD1 Fusion Oncogene in AML. Blood 2019, 134, 2545. [Google Scholar] [CrossRef]
- Schmoellerl, J.; Barbosa, I.A.M.; Eder, T.; Brandstoetter, T.; Schmidt, L.; Maurer, B.; Troester, S.; Pham, H.T.T.; Sagarajit, M.; Ebner, J.; et al. CDK6 Is an Essential Direct Target of NUP98 Fusion Proteins in Acute Myeloid Leukemia. Blood 2020, 136, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Naiel, A.; Vetter, M.; Plekhanova, O.; Fleischman, E.; Sokova, O.; Tsaur, G.; Harbott, J.; Tosi, S. A Novel Three-Colour Fluorescence in Situ Hybridization Approach for the Detection of t(7;12)(Q36;P13) in Acute Myeloid Leukaemia Reveals New Cryptic Three Way Translocation t(7;12;16). Cancers 2013, 5, 281–295. [Google Scholar] [CrossRef]
- Tosi, S.; Mostafa Kamel, Y.; Owoka, T.; Federico, C.; Truong, T.H.; Saccone, S. Paediatric Acute Myeloid Leukaemia with the t(7;12)(Q36;P13) Rearrangement: A Review of the Biological and Clinical Management Aspects. Biomark. Res. 2015, 3, 1–11. [Google Scholar] [CrossRef]
- Ballabio, E.; Cantarella, C.D.; Federico, C.; Di Mare, P.; Hall, G.; Harbott, J.; Hughes, J.; Saccone, S.; Tosi, S. Ectopic Expression of the HLXB9 Gene Is Associated with an Altered Nuclear Position in t(7;12) Leukaemias. Leukemia 2009, 23, 1179–1182. [Google Scholar] [CrossRef]
- von Bergh, A.R.M.; van Drunen, E.; van Wering, E.R.; van Zutven, L.J.C.M.; Hainmann, I.; Lo¨nnerholm, G.; Meijerink, J.P.; Pieters, R.; Beverloo, H.B. High Incidence of t(7;12)(Q36;P13) in Infant AML but Not in Infant ALL, with a Dismal Outcome and Ectopic Expression of HLXB9. Genes Chromosomes Cancer 2006, 45, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Gröschel, S.; Sanders, M.A.; Hoogenboezem, R.; De Wit, E.; Bouwman, B.A.M.; Erpelinck, C.; Van Der Velden, V.H.J.; Havermans, M.; Avellino, R.; Van Lom, K.; et al. A Single Oncogenic Enhancer Rearrangement Causes Concomitant EVI1 and GATA2 Deregulation in Leukemia. Cell 2014, 157, 369–381. [Google Scholar] [CrossRef]
- Gervais, C.; Murati, A.; Helias, C.; Struski, S.; Eischen, A.; Lippert, E.; Tigaud, I.; Penther, D.; Bastard, C.; Mugneret, F.; et al. Acute Myeloid Leukaemia with 8p11 (MYST3) Rearrangement: An Integrated Cytologic, Cytogenetic and Molecular Study by the Groupe Francophone de Cytogénétique Hématologique. Leukemia 2008, 11, 1567–1575. [Google Scholar] [CrossRef] [PubMed]
- Coenen, E.A.; Zwaan, C.M.; Reinhardt, D.; Harrison, C.J.; Haas, O.A.; De Haas, V.; Mih, V.; De Moerloose, B.; Jeison, M.; Rubnitz, J.E.; et al. Pediatric Acute Myeloid Leukemia with t(8;16)(P11;P13), a Distinct Clinical and Biological Entity: A Collaborative Study by the AML-Study Group. Blood J. Am. Soc. Hematol. 2016, 122, 2704–2713. [Google Scholar] [CrossRef]
- Gamis, A.S.; Woods, W.G.; Alonzo, T.A.; Buxton, A.; Lange, B.; Barnard, D.R.; Gold, S.; Smith, F.O. Increased Age at Diagnosis Has a Significantly Negative Effect on Outcome in Children with Down Syndrome and Acute Myeloid Leukemia: A Report from the Children’s Cancer Group Study 2891. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2003, 21, 3415–3422. [Google Scholar] [CrossRef] [PubMed]
- Quessada, J.; Cuccuini, W.; Saultier, P.; Loosveld, M.; Harrison, C.J.; Lafage-Pochitaloff, M. Cytogenetics of Pediatric Acute Myeloid Leukemia: A Review of the Current Knowledge. Genes 2021, 12, 924. [Google Scholar] [CrossRef] [PubMed]
- Cairoli, R.; Beghini, A.; Grillo, G.; Nadali, G.; Elice, F.; Ripamonti, C.B.; Colapietro, P.; Nichelatti, M.; Pezzetti, L.; Lunghi, M.; et al. Prognostic Impact of C-KIT Mutations in Core Binding Factor Leukemias: An Italian Retrospective Study. Blood 2006, 107, 3463–3468. [Google Scholar] [CrossRef]
- Renneville, A.; Roumier, C.; Biggio, V.; Nibourel, O.; Boissel, N.; Fenaux, P.; Preudhomme, C. Cooperating Gene Mutations in Acute Myeloid Leukemia: A Review of the Literature. Leukemia 2008, 22, 915–931. [Google Scholar] [CrossRef]
- Jabbour, E.; Kantarjian, H. Chronic Myeloid Leukemia: 2020 Update on Diagnosis, Therapy and Monitoring. Am. J. Hematol. 2020, 95, 691–709. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. Clinical Practice Guidelines in Oncology on Acute Myeloid Leukemia. Version 3. 2017. Available online: https://www.nccn.org/professionals/physician_gls/pdf/aml.pdf (accessed on 15 December 2021).
- Middeke, J.M.; Beelen, D.; Stadler, M.; Göhring, G.; Schlegelberger, B.; Baurmann, H.; Bug, G.; Bellos, F.; Mohr, B.; Buchholz, S.; et al. Outcome of High-Risk Acute Myeloid Leukemia after Allogeneic Hematopoietic Cell Transplantation: Negative Impact of Abnl(17p) and -5/5q-. Blood 2012, 120, 2521–2528. [Google Scholar] [CrossRef]
- Falini, B.; Mason, D.Y. Proteins Encoded by Genes Involved in Chromosomal Alterations in Lymphoma and Leukemia: Clinical Value of Their Detection by Immunocytochemistry. Blood 2002, 99, 409–426. [Google Scholar] [CrossRef] [PubMed]
- Arceci, R.J.; Sande, J.; Lange, B.; Shannon, K.; Franklin, J.; Hutchinson, R.; Vik, T.A.; Flowers, D.; Aplenc, R.; Berger, M.S.; et al. Safety and Efficacy of Gemtuzumab Ozogamicin in Pediatric Patients with Advanced CD33+ Acute Myeloid Leukemia. Blood 2005, 106, 1183–1188. [Google Scholar] [CrossRef] [PubMed]
- Ehninger, A.; Kramer, M.; Röllig, C.; Thiede, C.; Bornhäuser, M.; Von Bonin, M.; Wermke, M.; Feldmann, A.; Bachmann, M.; Ehninger, G.; et al. Distribution and Levels of Cell Surface Expression of CD33 and CD123 in Acute Myeloid Leukemia. Blood Cancer J. 2014, 4, e218. [Google Scholar] [CrossRef] [PubMed]
- Gamis, A.S.; Alonzo, T.A.; Meshinchi, S.; Sung, L.; Gerbing, R.B.; Raimondi, S.C.; Hirsch, B.A.; Kahwash, S.B.; Heerema-McKenney, A.; Winter, L.; et al. Gemtuzumab Ozogamicin in Children and Adolescents with de Novo Acute Myeloid Leukemia Improves Event-Free Survival by Reducing Relapse Risk: Results from the Randomized Phase III Children’s Oncology Group Trial AAML0531. J. Clin. Oncol. 2014, 32, 3021–3032. [Google Scholar] [CrossRef]
- Park, S.H.; You, E.; Park, C.J.; Jang, S.; Cho, Y.U.; Yoon, C.H.; Koh, K.N.; Im, H.J.; Seo, J.J. The Incidence and Immunophenotypic and Genetic Features of JL1 Expressing Cells and the Therapeutic Potential of an Anti-JL1 Antibody in de Novo Pediatric Acute Leukemias. Ann. Lab. Med. 2019, 39, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Uy, G.L.; Aldoss, I.; Foster, M.C.; Sayre, P.H.; Wieduwilt, M.J.; Advani, A.S.; Godwin, J.E.; Arellano, M.L.; Sweet, K.L.; Emadi, A.; et al. Flotetuzumab as Salvage Immunotherapy for Refractory Acute Myeloid Leukemia. Blood 2021, 137, 751–762. [Google Scholar] [CrossRef]
- Malesa, A.; Nowak, J.; Skorka, K.; Karp, M.; Giannopoulos, K. Monoclonal Antibodies against PD-1/PD-L1 Pathway. Acta Haematol. Pol. 2018, 49, 207–227. [Google Scholar] [CrossRef]
- Labanieh, L.; Majzner, R.G.; Mackall, C.L. Programming CAR-T Cells to Kill Cancer. Nat. Biomed. Eng. 2018, 2, 377–391. [Google Scholar] [CrossRef]
- Gill, S.I. How Close Are We to CAR T-Cell Therapy for AML? Best Pract. Res. Clin. Haematol. 2019, 32, 101104. [Google Scholar] [CrossRef]
- Schneider, D.; Xiong, Y.; Hu, P.; Wu, D.; Chen, W.; Ying, T.; Zhu, Z.; Dimitrov, D.S.; Dropulic, B.; Orentas, R.J. A Unique Human Immunoglobulin Heavy Chain Variable Domain-Only CD33 CAR for the Treatment of Acute Myeloid Leukemia. Front. Oncol. 2018, 8, 1–16. [Google Scholar] [CrossRef]
- Qin, H.; Yang, L.; Chukinas, J.A.; Shah, N.; Tarun, S.; Pouzolles, M.; Chien, C.D.; Niswander, L.M.; Welch, A.R.; Taylor, N.; et al. Systematic Preclinical Evaluation of CD33-Directed Chimeric Antigen Receptor T Cell Immunotherapy for Acute Myeloid Leukemia Defines Optimized Construct Design. J. Immunother. Cancer 2021, 9, e003149. [Google Scholar] [CrossRef] [PubMed]
- O’Hear, C.; Heiber, J.F.; Schubert, I.; Fey, G.; Geiger, T.L. Anti-CD33 Chimeric Antigen Receptor Targeting of Acute Myeloid Leukemia. Haematologica 2015, 100, 336. [Google Scholar] [CrossRef]
- Kim, M.Y.; Yu, K.-R.; Kenderian, S.S.; Ruella, M.; Chen, S.; Shin, T.-H.; Aljanahi, A.A.; Schreeder, D.; Klichinsky, M.; Shestova, O.; et al. Genetic Inactivation of CD33 in Hematopoietic Stem Cells to Enable CAR T Cell Immunotherapy for Acute Myeloid Leukemia. Cell 2018, 173, 1439–1453.e19. [Google Scholar] [CrossRef]
- Testa, U.; Pelosi, E.; Castelli, G. CD123 as a Therapeutic Target in the Treatment of Hematological Malignancies. Cancers 2019, 11, 1358. [Google Scholar] [CrossRef]
- Gill, S.; Tasian, S.K.; Ruella, M.; Shestova, O.; Li, Y.; Porter, D.L.; Carroll, M.; Danet-Desnoyers, G.; Scholler, J.; Grupp, S.A.; et al. Preclinical Targeting of Human Acute Myeloid Leukemia and Myeloablation Using Chimeric Antigen Receptor-Modified T Cells. Blood 2014, 123, 2343–2354. [Google Scholar] [CrossRef] [PubMed]
- Ruella, M.; Barrett, D.M.; Kenderian, S.S.; Shestova, O.; Hofmann, T.J.; Perazzelli, J.; Klichinsky, M.; Aikawa, V.; Nazimuddin, F.; Kozlowski, M.; et al. Dual CD19 and CD123 Targeting Prevents Antigen-Loss Relapses after CD19-Directed Immunotherapies. J. Clin. Investig. 2016, 126, 3814–3826. [Google Scholar] [CrossRef]
- Mardiros, A.; Dos Santos, C.; McDonald, T.; Brown, C.E.; Wang, X.; Budde, L.E.; Hoffman, L.; Aguilar, B.; Chang, W.-C.; Bretzlaff, W.; et al. T Cells Expressing CD123-Specific Chimeric Antigen Receptors Exhibit Specific Cytolytic Effector Functions and Antitumor Effects against Human Acute Myeloid Leukemia. Blood 2013, 122, 3138–3148. [Google Scholar] [CrossRef]
- Loff, S.; Dietrich, J.; Meyer, J.E.; Riewaldt, J.; Spehr, J.; von Bonin, M.; Gründer, C.; Swayampakula, M.; Franke, K.; Feldmann, A.; et al. Rapidly Switchable Universal CAR-T Cells for Treatment of CD123-Positive Leukemia. Mol. Ther.-Oncolyt. 2020, 17, 408–420. [Google Scholar] [CrossRef]
- Available online: https://Clinicaltrials.Gov (accessed on 15 December 2021).
- Chichili, G.R.; Huang, L.; Li, H.; Burke, S.; He, L.; Tang, Q.; Jin, L.; Gorlatov, S.; Ciccarone, V.; Chen, F.; et al. A CD3xCD123 Bispecific DART for Redirecting Host T Cells to Myelogenous Leukemia: Preclinical Activity and Safety in Nonhuman Primates. Sci. Transl. Med. 2015, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mayer, L.D.; Tardi, P.; Louie, A.C. CPX-351: A Nanoscale Liposomal Co-Formulation of Daunorubicin and Cytarabine with Unique Biodistribution and Tumor Cell Uptake Properties. Int. J. Nanomed. 2019, 14, 3819–3830. [Google Scholar] [CrossRef]
- José-Enériz, E.S.; Gimenez-Camino, N.; Agirre, X.; Prosper, F. HDAC Inhibitors in Acute Myeloid Leukemia. Cancers 2019, 11, 1794. [Google Scholar] [CrossRef]
- Huang, Y.; Dong, G.; Li, H.; Liu, N.; Zhang, W.; Sheng, C. Discovery of Janus Kinase 2 (JAK2) and Histone Deacetylase (HDAC) Dual Inhibitors as a Novel Strategy for the Combinational Treatment of Leukemia and Invasive Fungal Infections. J. Med. Chem. 2018, 61, 6056–6074. [Google Scholar] [CrossRef]
- Morganti, S.; Tarantino, P.; Ferraro, E.; D’Amico, P.; Duso, B.A.; Curigliano, G. Next Generation Sequencing (NGS): A Revolutionary Technology in Pharmacogenomics and Personalized Medicine in Cancer. Adv. Exp. Med. Biol. 2019, 1168, 9–30. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.K.; Miller, D.W.; Lynch, J.A.; Lemoff, A.S.; Cai, Z.; Pounds, S.B.; Radtke, I.; Yan, B.; Schuetz, J.D.; Rubnitz, J.E.; et al. IDH1 and IDH2 mutations in pediatric acute leukemia. Leukemia 2011, 25, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Mondesir, J.; Willekens, C.; Touat, M.; de Botton, S. IDH1 and IDH2 mutations as novel theraputic targets: Current perspectives. J. Blood Med. 2016, 7, 171–180. [Google Scholar]
- Drazer, M.W.; Kadri, S.; Sukhanova, M.; Patil, S.A.; West, A.H.; Feurstein, S.; Calderon, D.A.; Jones, M.F.; Weipert, C.M.; Daugherty, C.K.; et al. Prognostic tumor sequencing panels frequently identify germ line variants associated with hereditary hematopoietic malignancies. Blood Adv. 2018, 92, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Obrochta, E.; Godley, L.A. Identifying Patients with Genetic Predisposition to Acute Myeloid Leukemia. Best Pract. Res. Clin. Haematol. 2018, 31, 373–378. [Google Scholar] [CrossRef]

| Molecular Alteration | Frequency in Pediatric AML | Prognosis | References |
|---|---|---|---|
| t(8;21)(q22;q22) RUNX1::RUNX1T1 | 10–12% | Very good 5-year OS 80–90% | [13,14] |
| inv(16)(p13.1q22) or t(16;16)(p13.1;q22); CBFB::MYH11 | 10% | Very good 5-year OS 80–90% | [13,14] |
| t(15;17)(q24.1;q21.2) PML::RARA | 5–10% 2% in infants | Very good due to advanced therapy 5-year OS 95% 5-year EFS 90% | [15] |
| NPM1 gene mutations | 10% (50–60% in adult AML) | Very good 5-year OS 85% Better prognosis without FLT3/ITD | [16,17] |
| CEBPA gene double mutations | 5.6% Approximately 70% of CEBPAmt cases | Very good 5-year OS 80% | [18,19] |
| CEBPA gene single mutations | 2.4% | Poor 5-year OS 25% | [18,19] |
| t(16;21)(q24;q22) RUNX1::CBFA2T3 | 0.2% | Good/intermediate 4-year EFS 77% | [20,21] |
| Trisomy 21 | 15% | Good Higher sensitivity to chemotherapeutic agents | [22,23] |
| FLT3/ITD mutation | Frequency increases with age 1.5% in infants 7% in children aged 1–5 17% in adolescents and young adults | Poor 5-year OS 30–40% for patients with high allelic ratios | [24,25] |
| 11q23 (KMT2A) rearrangements | 20% Most frequently in infants | Varies depending on KMT2A fusion partner | [12,26,27,28,29,30] |
| t(9;11)(p22;q23) KMT2A::AF9(MLLT3) | 6–9% | Intermediate | A/M |
| t(11;19)(q23;p13.1) KMT2A::ELL | 1–2% | Intermediate | A/M |
| t(11;19)(q23;p13.3) KMT2A::ENL(MLLT1) | 1–2% | Intermediate | A/M |
| t(10;11)(p12;q23) or ins(10;11)(p12;q23q13) KMT2A::AF10(MLLT10) | 2–3% | Poor | A/M |
| t(6;11)(q27;q23) KMT2A::AF6(MLLT4) | 1–2% | Poor | A/M |
| t(5;11)(q35;p15) NUP98::NSD1 | 3–4% Strong association with FLT3-ITD | Poor 4-year EFS below 10% | [31,32,33] |
| t(11;12)(p15;p13) NUP98::KMD5A | 1–2% | Poor 5-year OS 33% | [34,35,36] |
| t(7;12)(q36;p13) MNX1::ETV6 | Below 30% Only in infants (<2 y.) | Poor 3-year EFS below 24% | [24,37] |
| Monosomy 7 | 4% | Poor 10-year OS 32% 10-year EFS 29% | [13] |
| Monosomy 5/del(5q) | 1.2% | Poor 5-yer OS 27% 5-year EFS 23% | [38] |
| t(6;9)(p22;q34) DEK::NUP214 | 1.2–4% | Poor 5–10 year OS 30–40% 10-year EFS 30% | [13,38] |
| inv(16)(p13.3;q24.3) CBFA2T3::GLIS2 | 2% | Poor 5-year EFS 25–30% | [39] |
| inv(3)(q21q26.2) or t(3;3)(q21;q26.2) EVI1 (former MECOM) | 1–2% | Poor Long-term OS < 10% | [11,40,41] |
| t(16;21)(p11;q22) FUS::ERG | 0.4% | Poor 4-year EFS 7% | [20,21] |
| t(9;22)(q34;q11) BCR::ABL1 | 1% in adults | Uncertain significance Mainly depends on coexisting aberrations | [42,43] |
| t(10;11)(p12;q14) PICALM::MLLT10 | <1% | Uncertain significance | [21,44] |
| t(8;16)(p11;p13) MYST3::CREBBP | 10% | Spontaneous remission has been observed | [24,45,46] |
| KIT gene mutations | 5% overall 25% in CBF-AML population | Uncertain significance May negatively impact response to therapy | [47,48] |
| FLT3/TKD mutations | 7% inv(16) AML | Uncertain significance | [49] |
| t(1;22)(p13;q13) RBM15::MKL1 | 0.3% | Uncertain significance 5-year EFS 54.5%, 5-year OS 58.2% | [35,50,51] |
| t(3;5)(q25;q35 NPM1::MLF1 | <0.5% | Uncertain significance | [42,52,53] |
| Trisomy 8 | 3% | Uncertain/discussed significance 5-year EFS 25% | [13,54,55] |
| Hyperdiploid karyotype | 11% | Uncertain significance | [56,57] |
| Complex karyotype | 8–15% | Uncertain/discussed significance | [13,14] |
| Molecular Alteration | Pediatric AML | Adult AML | ||||
|---|---|---|---|---|---|---|
| Frequency | Prognosis | References | Frequency | Prognosis | References | |
| t(8;21)(q22;q22) RUNX1::RUNX1T1 | 10–12% | Very good | [13,14] | 3.5% | Good | [11] |
| inv(16)(p13.1q22) or t(16;16)(p13.1;q22) CBFB::MYH11 | 10% | Very good | [13,14] | 3% | Good | [11] |
| t(15;17)(q24.1;q21.2) PML::RARA | 5–10% 2% in infants | Very good due to advanced therapy | [15] | 6% | Good | [11] |
| NPM1 gene mutations | 10% | Very good Better prognosis without FLT3/ITD | [16,17] | 50–60% | Very good | [58] |
| CEBPA gene double mutations | 5.6% | Very good | [18,19] | 5% | Very good | [59,60] |
| CEBPA gene single mutations | 2.4% | Poor | [18,19] | 2.3% | Poor | [59,60] |
| FLT3/ITD mutation | Frequency increases with age 1.5–17% | Poor | [24,25] | 20–35% | Good/intermediate | [25,61] |
| 11q23 (KMT2A) rearrangements | 20% | Mostly intermediate and poor | [12,26,27,28,29,30] | 15–20% | Mostly intermediate and poor | [62] |
| t(6;9)(p22;q34) DEK::NUP214 | 1.2–4% | Poor | [13,38] | 0.5% | Poor | [11] |
| t(9;22)(q34;q11) BCR::ABL1 | The vast majority of adult cases | Uncertain | [42,43] | 1% | Uncertain | [42,43] |
| t(8;16)(p11;p13) MYST3::CREBBP | 10% | Uncertain | [24,45,46] | 0.2–0.4% | Uncertain | [63] |
| KIT gene mutations | 5% | Uncertain | [47,48] | 12.8–46.1% in CBF leukemia | Uncertain | [64] |
| FLT3/TKD mutations | 7% in inv(16) AML | Uncertain | [49] | 28% in inv(16) AML | Uncertain | [49] |
| Trisomy 8 | 11% | Uncertain | [13,54,55] | 5% | Uncertain | [11] |
| Complex karyotype | 8–15% | Uncertain | [13,14] | 14% | Uncertain | [11] |
| t(11;12)(p15;p13) NUP98::KMD5A | 1–2% | Poor | [34,35,36] | Lesions typical of pediatric AML | ||
| inv(16)(p13.3;q24.3) CBFA2T3::GLIS2 | 2% | Poor | [39] | |||
| t(7;12)(q36;p13) MNX1::ETV6 | Below 30% | Poor | [24,37] | |||
| inv(3)(q21q26.2) or t(3;3)(q21;q26.2) EVI1 (former MECOM) | 1–2% | Poor | [11,40,41] | |||
| t(1;22)(p13;q13) RBM15::MKL1 | 0.3% | Uncertain | [35,50,51] | |||
| t(3;5)(q25;q35 NPM1::MLF1 | <0.5% | Uncertain | [42,52,53] | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lejman, M.; Dziatkiewicz, I.; Jurek, M. Straight to the Point—The Novel Strategies to Cure Pediatric AML. Int. J. Mol. Sci. 2022, 23, 1968. https://doi.org/10.3390/ijms23041968
Lejman M, Dziatkiewicz I, Jurek M. Straight to the Point—The Novel Strategies to Cure Pediatric AML. International Journal of Molecular Sciences. 2022; 23(4):1968. https://doi.org/10.3390/ijms23041968
Chicago/Turabian StyleLejman, Monika, Izabela Dziatkiewicz, and Mateusz Jurek. 2022. "Straight to the Point—The Novel Strategies to Cure Pediatric AML" International Journal of Molecular Sciences 23, no. 4: 1968. https://doi.org/10.3390/ijms23041968
APA StyleLejman, M., Dziatkiewicz, I., & Jurek, M. (2022). Straight to the Point—The Novel Strategies to Cure Pediatric AML. International Journal of Molecular Sciences, 23(4), 1968. https://doi.org/10.3390/ijms23041968