
Ion Channel Partnerships: Odd and Not-So-Odd Couples Controlling Neuronal Ion Channel Function


Abstract
:1. Introduction
2. Canonical Ion Channel Partnerships
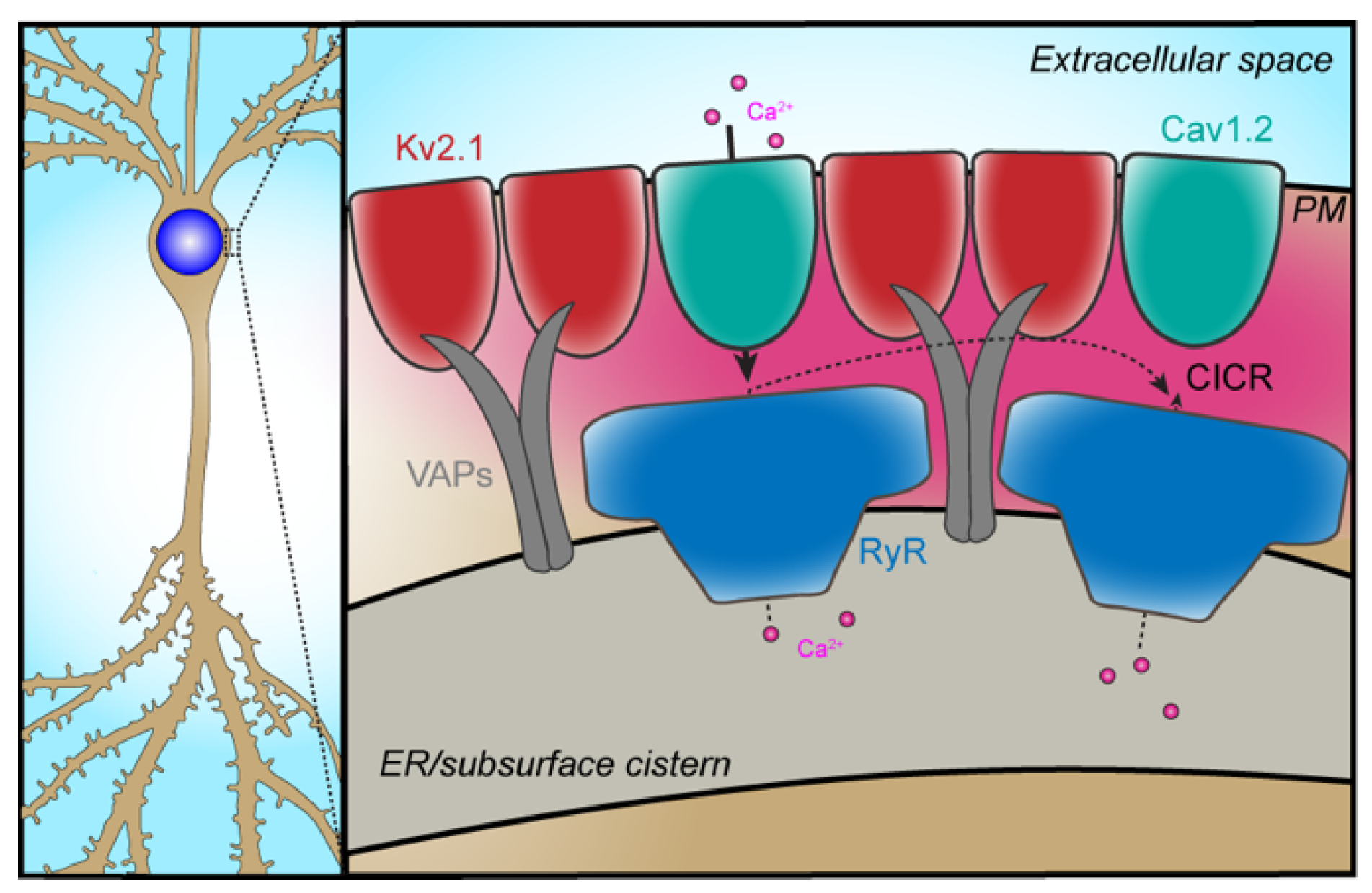
2.1. PM-ER Ion Channel Partnerships
2.2. Cav1-RyR
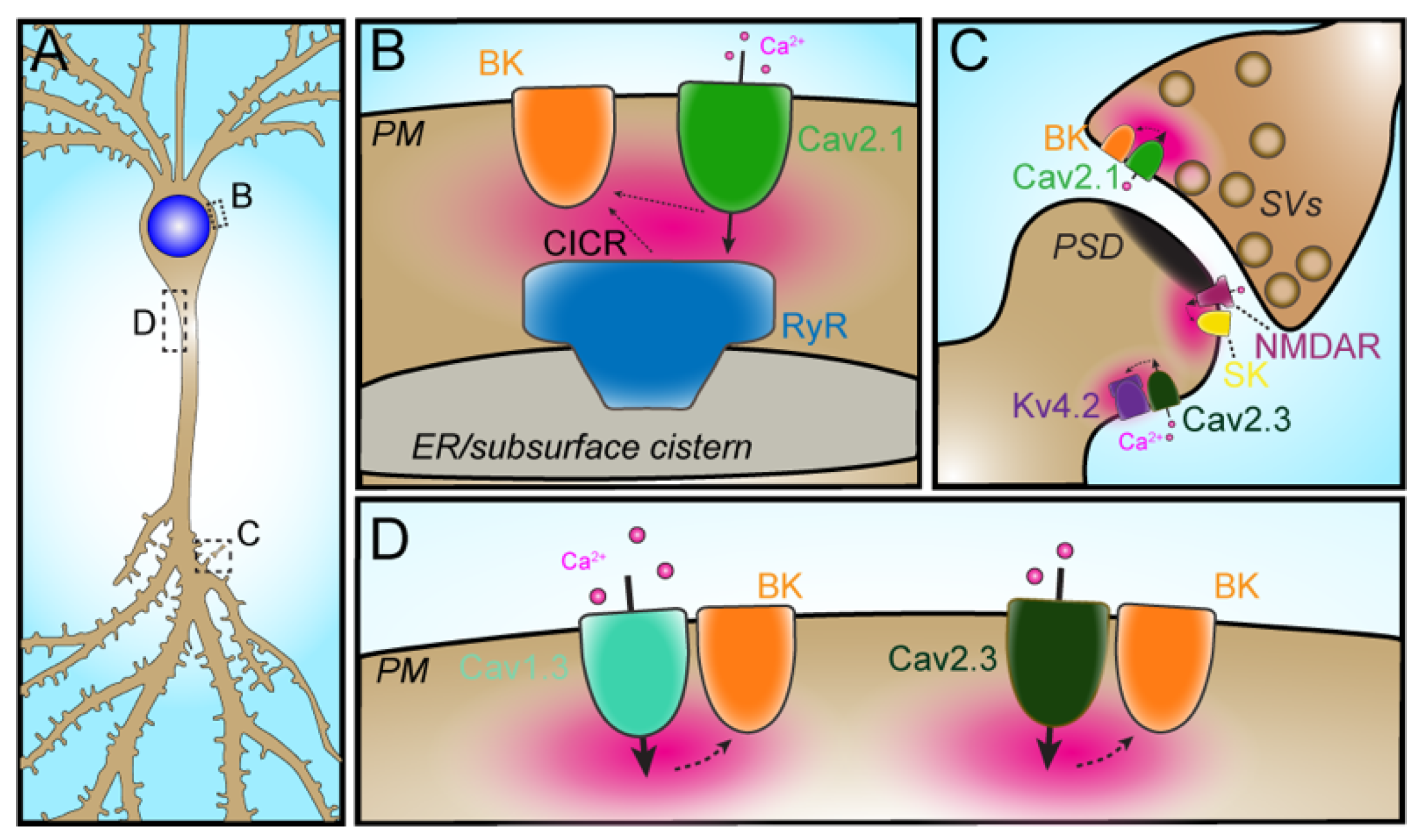
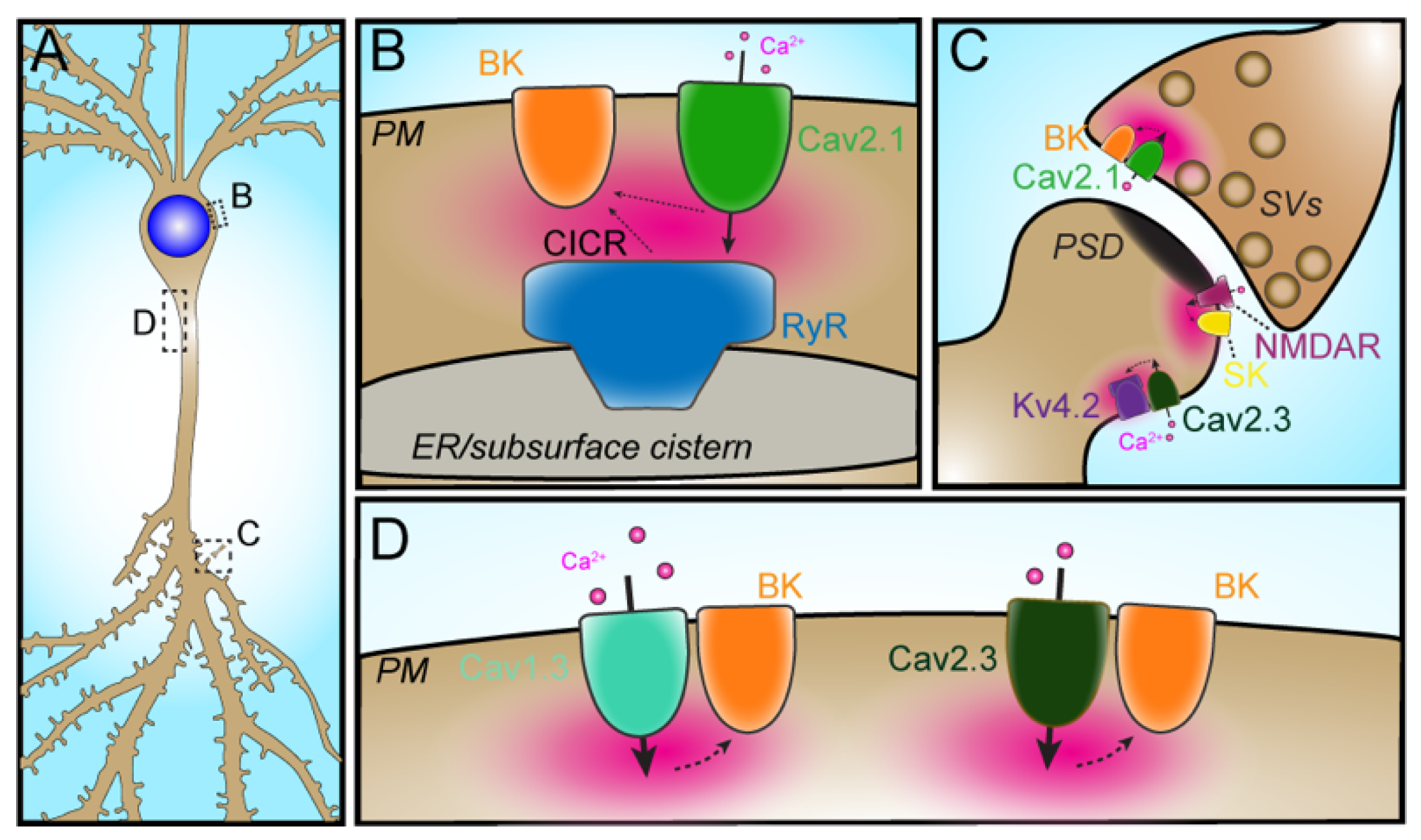
2.3. VGCC–RyR–KCa
2.4. VGCC–BK
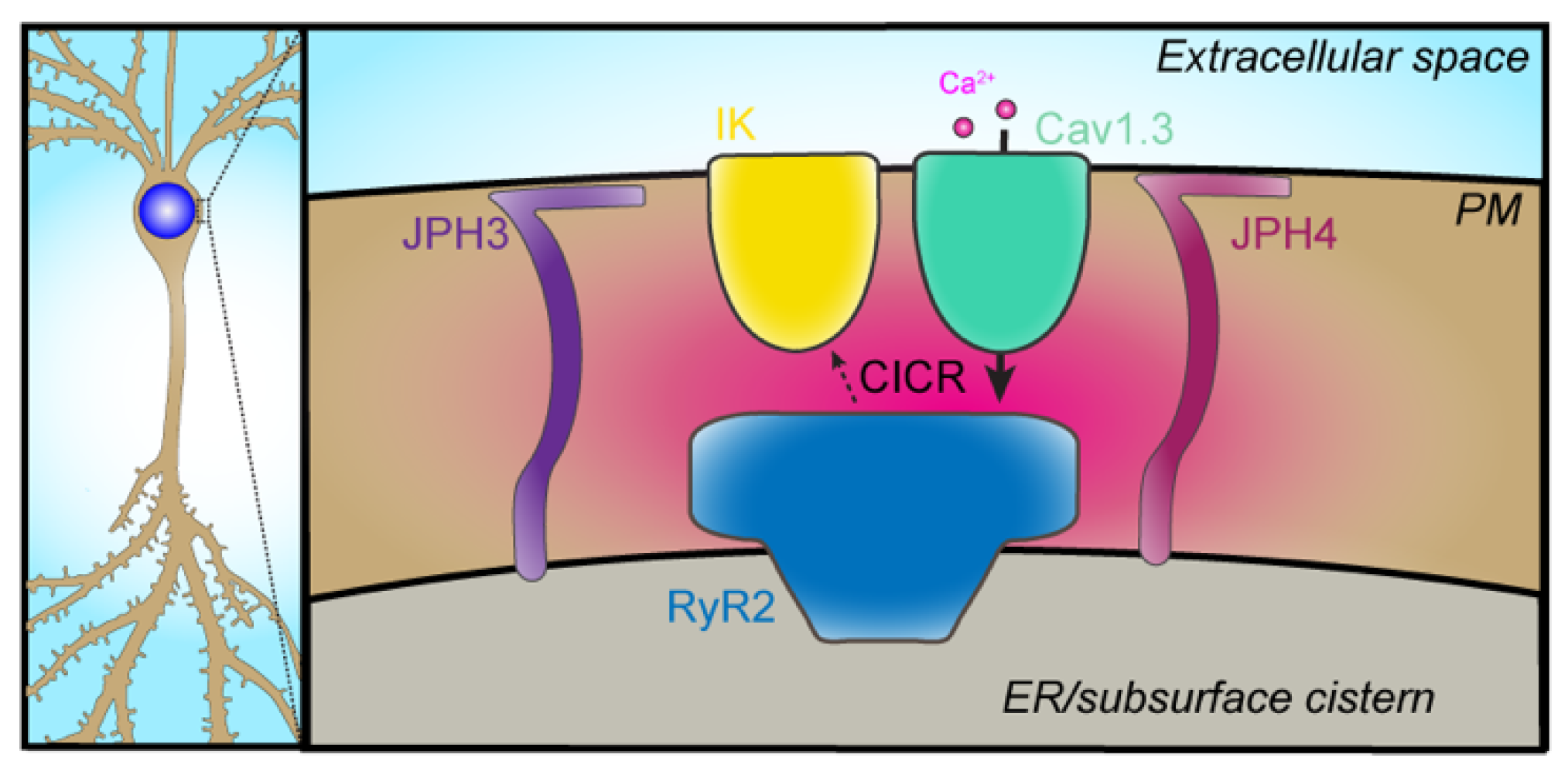
2.5. VGCC–IK
3. Non-Canonical Ion Channel Partnerships
3.1. VGCC–Kv Channel Partnerships
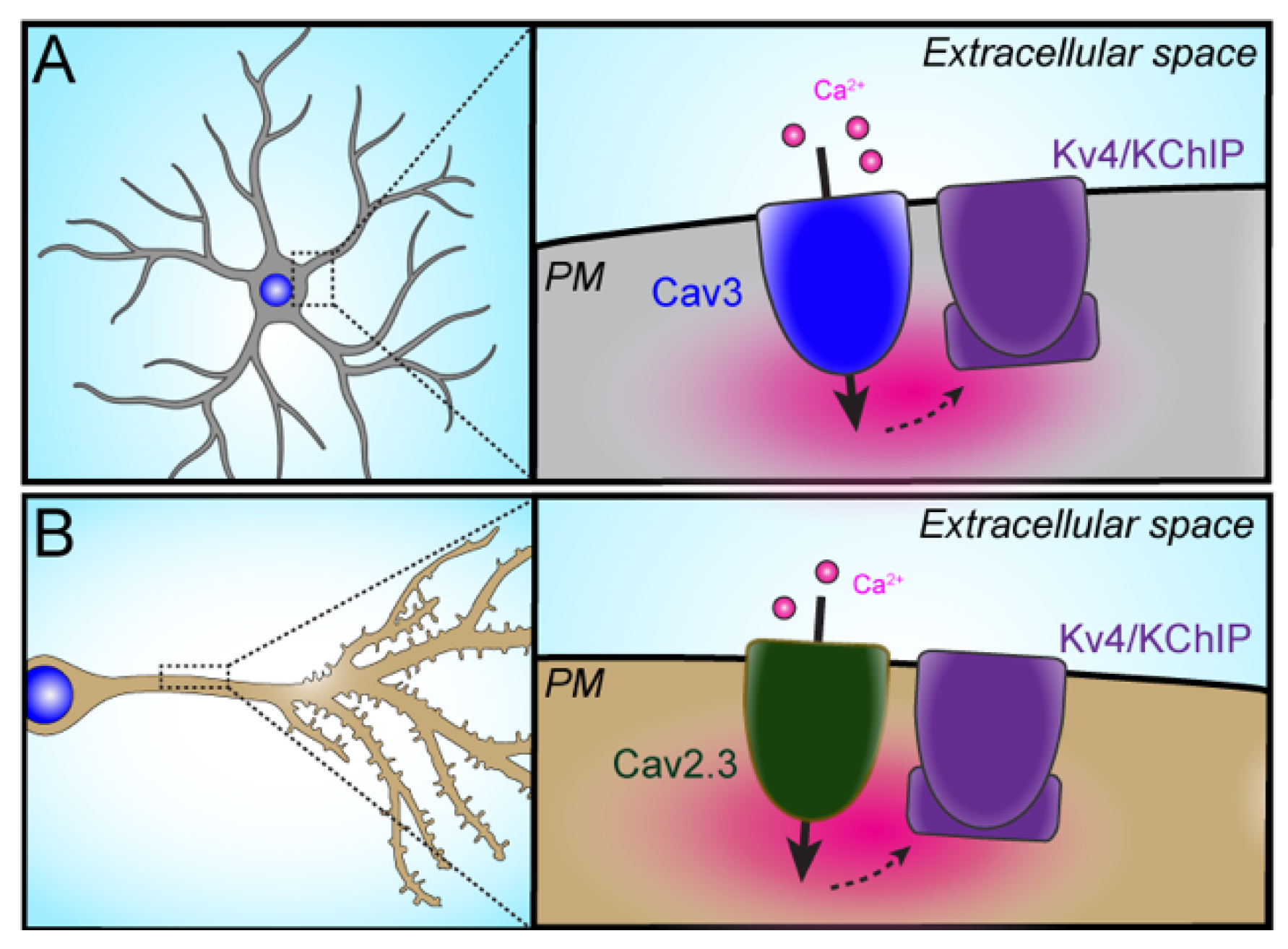
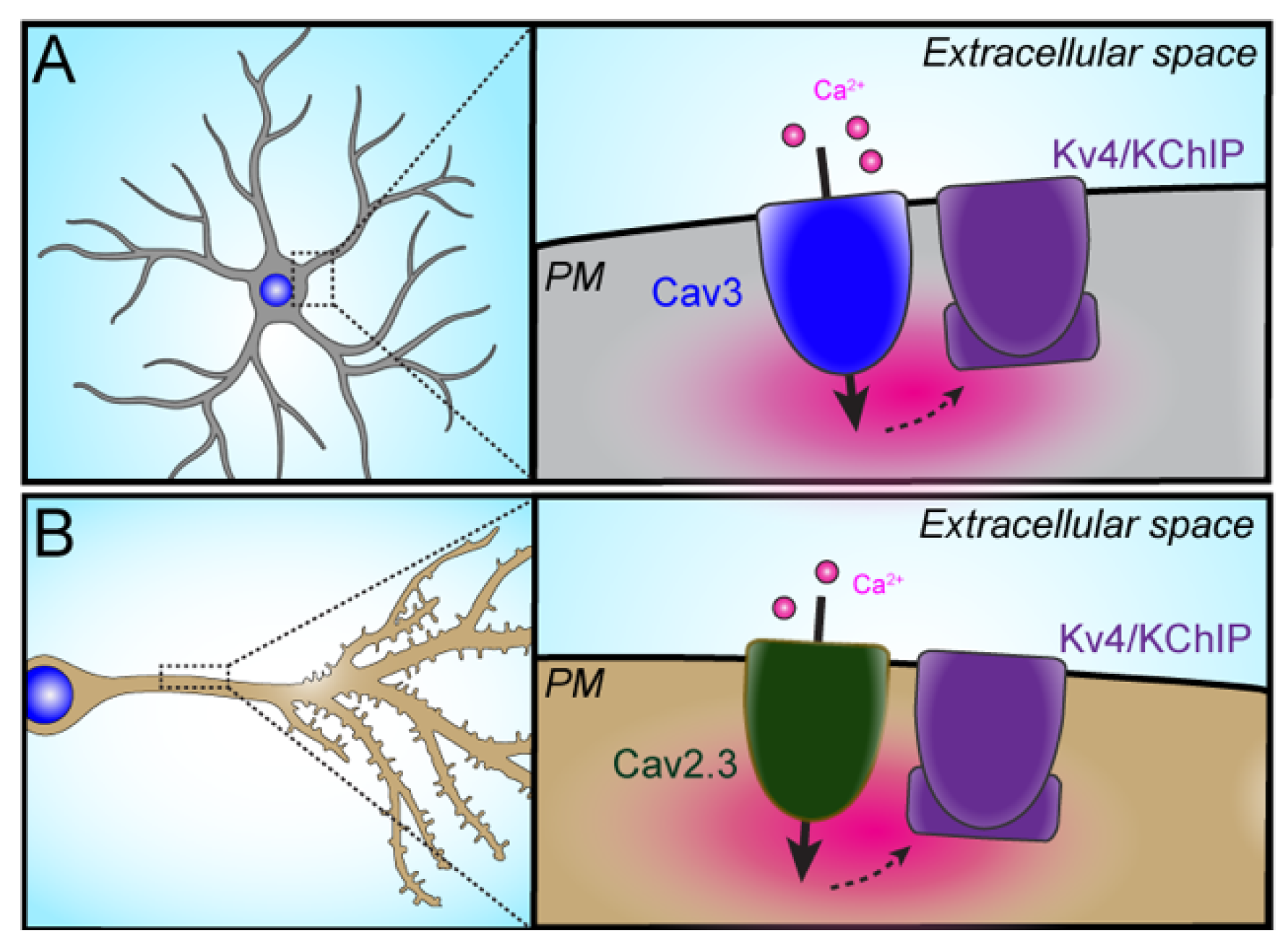
3.1.1. Cav3–Kv4
3.1.2. Cav2.3–Kv4.2
3.2. Ion Channel Partnerships Involving Non-Conducting Functions of Ion Channels
Cav1.2–Kv2.1
4. Restricted Subcellular Compartmentalization of Ion Channel Partnerships
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Dai, S.; Hall, D.D.; Hell, J.W. Supramolecular assemblies and localized regulation of voltage-gated ion channels. Physiol. Rev. 2009, 89, 411–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berridge, M.J. Neuronal calcium signaling. Neuron 1998, 21, 13–26. [Google Scholar] [CrossRef] [Green Version]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augustine, G.J.; Santamaria, F.; Tanaka, K. Local calcium signaling in neurons. Neuron 2003, 40, 331–346. [Google Scholar] [CrossRef] [Green Version]
- Neher, E. Vesicle pools and Ca2+ microdomains: New tools for understanding their roles in neurotransmitter release. Neuron 1998, 20, 389–399. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; Pozzan, T. Microdomains of intracellular Ca2+: Molecular determinants and functional consequences. Physiol. Rev. 2006, 86, 369–408. [Google Scholar] [CrossRef]
- Grienberger, C.; Konnerth, A. Imaging calcium in neurons. Neuron 2012, 73, 862–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catterall, W.A. Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947. [Google Scholar] [CrossRef]
- Simms, B.A.; Zamponi, G.W. Neuronal voltage-gated calcium channels: Structure, function, and dysfunction. Neuron 2014, 82, 24–45. [Google Scholar] [CrossRef] [Green Version]
- Vacher, H.; Mohapatra, D.P.; Trimmer, J.S. Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiol. Rev. 2008, 88, 1407–1447. [Google Scholar] [CrossRef] [Green Version]
- Hell, J.W.; Westenbroek, R.E.; Warner, C.; Ahlijanian, M.K.; Prystay, W.; Gilbert, M.M.; Snutch, T.P.; Catterall, W.A. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. J. Cell Biol. 1993, 123, 949–962. [Google Scholar] [CrossRef]
- Obermair, G.J.; Szabo, Z.; Bourinet, E.; Flucher, B.E. Differential targeting of the L-type Ca2+ channel alpha 1C (CaV1.2) to synaptic and extrasynaptic compartments in hippocampal neurons. Eur. J. Neurosci. 2004, 19, 2109–2122. [Google Scholar] [CrossRef] [PubMed]
- Leitch, B.; Szostek, A.; Lin, R.; Shevtsova, O. Subcellular distribution of L-type calcium channel subtypes in rat hippocampal neurons. Neuroscience 2009, 164, 641–657. [Google Scholar] [CrossRef]
- Stanika, R.I.; Flucher, B.E.; Obermair, G.J. Regulation of postsynaptic stability by the L-type calcium channel Cav1.3 and its interaction with PDZ proteins. Curr. Mol. Pharmacol 2015, 8, 95–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davare, M.A.; Avdonin, V.; Hall, D.D.; Peden, E.M.; Burette, A.; Weinberg, R.J.; Horne, M.C.; Hoshi, T.; Hell, J.W. A beta2 adrenergic receptor signaling complex assembled with the Ca2+ channel Cav1.2. Science 2001, 293, 98–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maximov, A.; Bezprozvanny, I. Synaptic targeting of N-type calcium channels in hippocampal neurons. J. Neurosci. 2002, 22, 6939–6952. [Google Scholar] [CrossRef] [Green Version]
- Mochida, S.; Westenbroek, R.E.; Yokoyama, C.T.; Itoh, K.; Catterall, W.A. Subtype-selective reconstitution of synaptic transmission in sympathetic ganglion neurons by expression of exogenous calcium channels. Proc. Natl. Acad. Sci. USA 2003, 100, 2813–2818. [Google Scholar] [CrossRef] [Green Version]
- Lory, P.; Nicole, S.; Monteil, A. Neuronal Cav3 channelopathies: Recent progress and perspectives. Pflugers Arch. 2020, 472, 831–844. [Google Scholar] [CrossRef]
- Striessnig, J. Voltage-gated Ca2+-channel alpha1-subunit de novo missense mutations: Gain or loss of function—implications for potential therapies. Front. Synaptic Neurosci. 2021, 13, 634760. [Google Scholar] [CrossRef]
- Eggermann, E.; Bucurenciu, I.; Goswami, S.P.; Jonas, P. Nanodomain coupling between Ca2+ channels and sensors of exocytosis at fast mammalian synapses. Nat. Rev. Neurosci. 2011, 13, 7–21. [Google Scholar] [CrossRef]
- Dixon, R.E.; Navedo, M.F.; Binder, M.D.; Santana, L.F. Mechanisms and physiological implications of cooperative gating of ion channels clusters. Physiol. Rev. 2021; online ahead of print. [Google Scholar] [CrossRef]
- Dixon, R.E.; Yuan, C.; Cheng, E.P.; Navedo, M.F.; Santana, L.F. Ca2+ signaling amplification by oligomerization of L-type Cav1.2 channels. Proc. Natl. Acad. Sci. USA 2012, 109, 1749–1754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, R.E.; Moreno, C.M.; Yuan, C.; Opitz-Araya, X.; Binder, M.D.; Navedo, M.F.; Santana, L.F. Graded Ca2+/calmodulin-dependent coupling of voltage-gated Cav1.2 channels. eLife 2015, 4, e05608. [Google Scholar] [CrossRef] [PubMed]
- Rosenbluth, J. Subsurface cisterns and their relationship to the neuronal plasma membrane. J. Cell Biol. 1962, 13, 405–421. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Whiteus, C.; Xu, C.S.; Hayworth, K.J.; Weinberg, R.J.; Hess, H.F.; De Camilli, P. Contacts between the endoplasmic reticulum and other membranes in neurons. Proc. Natl. Acad. Sci. USA 2017, 114, E4859–E4867. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, W.A.; Ferraguti, F.; Fukazawa, Y.; Kasugai, Y.; Shigemoto, R.; Laake, P.; Sexton, J.A.; Ruth, P.; Wietzorrek, G.; Knaus, H.G.; et al. Large-conductance calcium-activated potassium channels in purkinje cell plasma membranes are clustered at sites of hypolemmal microdomains. J. Comp. Neurol. 2009, 515, 215–230. [Google Scholar] [CrossRef]
- Mandikian, D.; Bocksteins, E.; Parajuli, L.K.; Bishop, H.I.; Cerda, O.; Shigemoto, R.; Trimmer, J.S. Cell type-specific spatial and functional coupling between mammalian brain Kv2.1 K+ channels and ryanodine receptors. J. Comp. Neurol. 2014, 522, 3555–3574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arias-Cavieres, A.; Barrientos, G.C.; Sanchez, G.; Elgueta, C.; Munoz, P.; Hidalgo, C. Ryanodine receptor-mediated calcium release has a key role in hippocampal LTD induction. Front. Cell Neurosci. 2018, 12, 403. [Google Scholar] [CrossRef] [Green Version]
- Lobos, P.; Cordova, A.; Vega-Vasquez, I.; Ramirez, O.A.; Adasme, T.; Toledo, J.; Cerda, M.; Hartel, S.; Paula-Lima, A.; Hidalgo, C. RyR-mediated Ca2+ release elicited by neuronal activity induces nuclear Ca2+ signals, CREB phosphorylation, and Npas4/RyR2 expression. Proc. Natl. Acad. Sci. USA 2021, 118, e2102265118. [Google Scholar] [CrossRef]
- Bertan, F.; Wischhof, L.; Sosulina, L.; Mittag, M.; Dalugge, D.; Fornarelli, A.; Gardoni, F.; Marcello, E.; Di Luca, M.; Fuhrmann, M.; et al. Loss of Ryanodine Receptor 2 impairs neuronal activity-dependent remodeling of dendritic spines and triggers compensatory neuronal hyperexcitability. Cell Death Differ. 2020, 27, 3354–3373. [Google Scholar] [CrossRef]
- Sah, P.; McLachlan, E.M. Ca2+-activated K+ currents underlying the afterhyperpolarization in guinea pig vagal neurons: A role for Ca2+-activated Ca2+ release. Neuron 1991, 7, 257–264. [Google Scholar] [CrossRef]
- Wang, B.; Bugay, V.; Ling, L.; Chuang, H.H.; Jaffe, D.B.; Brenner, R. Knockout of the BK beta4-subunit promotes a functional coupling of BK channels and ryanodine receptors that mediate a fAHP-induced increase in excitability. J. Neurophysiol. 2016, 116, 456–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irie, T.; Trussell, L.O. Double-nanodomain coupling of calcium channels, ryanodine receptors, and BK channels controls the generation of burst firing. Neuron 2017, 96, 856–870.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, G.; Okamoto, T.; Morikawa, H. Spontaneous opening of T-type Ca2+ channels contributes to the irregular firing of dopamine neurons in neonatal rats. J. Neurosci. 2004, 24, 11079–11087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipkin, A.M.; Cunniff, M.M.; Spratt, P.W.E.; Lemke, S.M.; Bender, K.J. Functional microstructure of Cav-mediated calcium signaling in the axon initial segment. J. Neurosci. 2021, 41, 3764–3776. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Q.; Song, L.S.; Lakatta, E.G.; Cheng, H. Ca2+ signalling between single L-type Ca2+ channels and ryanodine receptors in heart cells. Nature 2001, 410, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Lederer, W.J. Calcium sparks. Physiol. Rev. 2008, 88, 1491–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manita, S.; Ross, W.N. Synaptic activation and membrane potential changes modulate the frequency of spontaneous elementary Ca2+ release events in the dendrites of pyramidal neurons. J. Neurosci. 2009, 29, 7833–7845. [Google Scholar] [CrossRef] [Green Version]
- Berrout, J.; Isokawa, M. Homeostatic and stimulus-induced coupling of the L-type Ca2+ channel to the ryanodine receptor in the hippocampal neuron in slices. Cell Calcium 2009, 46, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Vierra, N.C.; Kirmiz, M.; van der List, D.; Santana, L.F.; Trimmer, J.S. Kv2.1 mediates spatial and functional coupling of L-type calcium channels and ryanodine receptors in mammalian neurons. eLife 2019, 8, e49953. [Google Scholar] [CrossRef]
- Stern, M.D.; Song, L.S.; Cheng, H.; Sham, J.S.; Yang, H.T.; Boheler, K.R.; Rios, E. Local control models of cardiac excitation-contraction coupling. A possible role for allosteric interactions between ryanodine receptors. J. Gen. Physiol. 1999, 113, 469–489. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, J.M.; Meyer, T. Control of action potential-induced Ca2+ signaling in the soma of hippocampal neurons by Ca2+ release from intracellular stores. J. Neurosci. 1997, 17, 4129–4135. [Google Scholar] [CrossRef] [Green Version]
- Dittmer, P.J.; Wild, A.R.; Dell’Acqua, M.L.; Sather, W.A. STIM1 Ca2+ sensor control of L-type Ca2+-channel-dependent dendritic spine structural plasticity and nuclear signaling. Cell Rep. 2017, 19, 321–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dittmer, P.J.; Dell’Acqua, M.L.; Sather, W.A. Synaptic crosstalk conferred by a zone of differentially regulated Ca2+ signaling in the dendritic shaft adjoining a potentiated spine. Proc. Natl. Acad Sci. USA 2019, 116, 13611–13620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sather, W.A.; Dittmer, P.J. Regulation of voltage-gated calcium channels by the ER calcium sensor STIM1. Curr. Opin. Neurobiol. 2019, 57, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Contet, C.; Goulding, S.P.; Kuljis, D.A.; Barth, A.L. BK channels in the central nervous system. Int. Rev. Neurobiol. 2016, 128, 281–342. [Google Scholar]
- Adelman, J.P.; Maylie, J.; Sah, P. Small-conductance Ca2+-activated K+ channels: Form and function. Annu. Rev. Physiol. 2012, 74, 245–269. [Google Scholar] [CrossRef]
- Kakizawa, S.; Kishimoto, Y.; Hashimoto, K.; Miyazaki, T.; Furutani, K.; Shimizu, H.; Fukaya, M.; Nishi, M.; Sakagami, H.; Ikeda, A.; et al. Junctophilin-mediated channel crosstalk essential for cerebellar synaptic plasticity. EMBO J. 2007, 26, 1924–1933. [Google Scholar] [CrossRef] [Green Version]
- Fakler, B.; Adelman, J.P. Control of K(Ca) channels by calcium nano/microdomains. Neuron 2008, 59, 873–881. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, W.A.; Kasugai, Y.; Ferraguti, F.; Storm, J.F. Two distinct pools of large-conductance calcium-activated potassium channels in the somatic plasma membrane of central principal neurons. Neuroscience 2010, 169, 974–986. [Google Scholar] [CrossRef] [Green Version]
- Indriati, D.W.; Kamasawa, N.; Matsui, K.; Meredith, A.L.; Watanabe, M.; Shigemoto, R. Quantitative localization of Cav2.1 (P/Q-type) voltage-dependent calcium channels in Purkinje cells: Somatodendritic gradient and distinct somatic coclustering with calcium-activated potassium channels. J. Neurosci. 2013, 33, 3668–3678. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, B.; Nicoll, R.A. Properties of two calcium-activated hyperpolarizations in rat hippocampal neurones. J. Physiol. 1987, 389, 187–203. [Google Scholar] [CrossRef] [PubMed]
- Marrion, N.V.; Tavalin, S.J. Selective activation of Ca2+-activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nature 1998, 395, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Berkefeld, H.; Fakler, B.; Schulte, U. Ca2+-activated K+ channels: From protein complexes to function. Physiol. Rev. 2010, 90, 1437–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berkefeld, H.; Sailer, C.A.; Bildl, W.; Rohde, V.; Thumfart, J.O.; Eble, S.; Klugbauer, N.; Reisinger, E.; Bischofberger, J.; Oliver, D.; et al. BKCa-Cav channel complexes mediate rapid and localized Ca2+-activated K+ signaling. Science 2006, 314, 615–620. [Google Scholar] [CrossRef] [Green Version]
- Muller, C.S.; Haupt, A.; Bildl, W.; Schindler, J.; Knaus, H.G.; Meissner, M.; Rammner, B.; Striessnig, J.; Flockerzi, V.; Fakler, B.; et al. Quantitative proteomics of the Cav2 channel nano-environments in the mammalian brain. Proc. Natl. Acad. Sci. USA 2010, 107, 14950–14957. [Google Scholar] [CrossRef] [Green Version]
- Yazejian, B.; DiGregorio, D.A.; Vergara, J.L.; Poage, R.E.; Meriney, S.D.; Grinnell, A.D. Direct measurements of presynaptic calcium and calcium-activated potassium currents regulating neurotransmitter release at cultured Xenopus nerve-muscle synapses. J. Neurosci. 1997, 17, 2990–3001. [Google Scholar] [CrossRef] [Green Version]
- Vivas, O.; Moreno, C.M.; Santana, L.F.; Hille, B. Proximal clustering between BK and Cav1.3 channels promotes functional coupling and BK channel activation at low voltage. eLife 2017, 6, e28029. [Google Scholar] [CrossRef]
- Gutzmann, J.J.; Lin, L.; Hoffman, D.A. Functional coupling of Cav2.3 and BK potassium channels regulates action potential repolarization and short-term plasticity in the mouse hippocampus. Front. Cell Neurosci 2019, 13, 27. [Google Scholar] [CrossRef]
- Whitt, J.P.; McNally, B.A.; Meredith, A.L. Differential contribution of Ca2+ sources to day and night BK current activation in the circadian clock. J. Gen. Physiol. 2018, 150, 259–275. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, M.A.; Rivard, A.F.; Bachinger, H.P.; Adelman, J.P. Structure of the gating domain of a Ca2+-activated K+ channel complexed with Ca2+/calmodulin. Nature 2001, 410, 1120–1124. [Google Scholar] [CrossRef]
- King, B.; Rizwan, A.P.; Asmara, H.; Heath, N.C.; Engbers, J.D.; Dykstra, S.; Bartoletti, T.M.; Hameed, S.; Zamponi, G.W.; Turner, R.W. IKCa channels are a critical determinant of the slow AHP in CA1 pyramidal neurons. Cell Rep. 2015, 11, 175–182. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Mateos-Aparicio, P.; Honigsperger, C.; Raghuram, V.; Wu, W.W.; Ridder, M.C.; Sah, P.; Maylie, J.; Storm, J.F.; Adelman, J.P. IK1 channels do not contribute to the slow afterhyperpolarization in pyramidal neurons. eLife 2016, 5, e11206. [Google Scholar] [CrossRef] [Green Version]
- Sahu, G.; Asmara, H.; Zhang, F.X.; Zamponi, G.W.; Turner, R.W. Activity-dependent facilitation of Cav1.3 calcium channels promotes KCa3.1 activation in hippocampal neurons. J. Neurosci 2017, 37, 11255–11270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahu, G.; Wazen, R.M.; Colarusso, P.; Chen, S.R.W.; Zamponi, G.W.; Turner, R.W. Junctophilin proteins tether a Cav1-RyR2-KCa3.1 tripartite complex to regulate neuronal excitability. Cell Rep. 2019, 28, 2427–2442.e6. [Google Scholar] [CrossRef] [Green Version]
- Anderson, D.; Mehaffey, W.H.; Iftinca, M.; Rehak, R.; Engbers, J.D.; Hameed, S.; Zamponi, G.W.; Turner, R.W. Regulation of neuronal activity by Cav3-Kv4 channel signaling complexes. Nat. Neurosci. 2010, 13, 333–337. [Google Scholar] [CrossRef]
- Trimmer, J.S. Subcellular localization of K+ channels in mammalian brain neurons: Remarkable precision in the midst of extraordinary complexity. Neuron 2015, 85, 238–256. [Google Scholar] [CrossRef] [Green Version]
- An, W.F.; Bowlby, M.R.; Betty, M.; Cao, J.; Ling, H.P.; Mendoza, G.; Hinson, J.W.; Mattsson, K.I.; Strassle, B.W.; Trimmer, J.S.; et al. Modulation of A-type potassium channels by a family of calcium sensors. Nature 2000, 403, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, K.J.; Carroll, K.I.; Sung, M.A.; Doliveira, L.C.; Monaghan, M.M.; Burke, S.L.; Strassle, B.W.; Buchwalder, L.; Menegola, M.; Cao, J.; et al. KChIPs and Kv4 alpha subunits as integral components of A-type potassium channels in mammalian brain. J. Neurosci. 2004, 24, 7903–7915. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.; Engbers, J.D.; Heath, N.C.; Bartoletti, T.M.; Mehaffey, W.H.; Zamponi, G.W.; Turner, R.W. The Cav3-Kv4 complex acts as a calcium sensor to maintain inhibitory charge transfer during extracellular calcium fluctuations. J. Neurosci. 2013, 33, 7811–7824. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Lin, M.T.; Adelman, J.P.; Maylie, J. Distinct Ca2+ sources in dendritic spines of hippocampal CA1 neurons couple to SK and Kv4 channels. Neuron 2014, 81, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Kimm, T.; Bean, B.P. Inhibition of A-type potassium current by the peptide toxin SNX-482. J. Neurosci 2014, 34, 9182–9189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngo-Anh, T.J.; Bloodgood, B.L.; Lin, M.; Sabatini, B.L.; Maylie, J.; Adelman, J.P. SK channels and NMDA receptors form a Ca2+-mediated feedback loop in dendritic spines. Nat. Neurosci. 2005, 8, 642–649. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Lujan, R.; Watanabe, M.; Adelman, J.P.; Maylie, J. SK2 channel plasticity contributes to LTP at Schaffer collateral-CA1 synapses. Nat. Neurosci. 2008, 11, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Parajuli, L.K.; Nakajima, C.; Kulik, A.; Matsui, K.; Schneider, T.; Shigemoto, R.; Fukazawa, Y. Quantitative regional and ultrastructural localization of the Ca(v)2.3 subunit of R-type calcium channel in mouse brain. J. Neurosci. 2012, 32, 13555–13567. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Jung, S.C.; Clemens, A.M.; Petralia, R.S.; Hoffman, D.A. Regulation of dendritic excitability by activity-dependent trafficking of the A-type K+ channel subunit Kv4.2 in hippocampal neurons. Neuron 2007, 54, 933–947. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Kelley, M.H.; Wu, W.W.; Adelman, J.P.; Maylie, J. Apamin boosting of synaptic potentials in Cav2.3 R-type Ca2+ channel null mice. PLoS ONE 2015, 10, e0139332. [Google Scholar] [CrossRef]
- Giessel, A.J.; Sabatini, B.L. Boosting of synaptic potentials and spine Ca transients by the peptide toxin SNX-482 requires alpha-1E-encoded voltage-gated Ca channels. PLoS ONE 2011, 6, e20939. [Google Scholar] [CrossRef]
- Murphy, J.G.; Gutzmann, J.J.; Lin, L.; Hu, J.; Petralia, R.S.; Wang, Y.X.; Hoffman, D.A. R-type voltage-gated Ca2+ channels mediate A-type K+ current regulation of synaptic input in hippocampal dendrites. Cell Rep. 2022, 38, 110264. [Google Scholar] [CrossRef]
- Hoffman, D.A.; Magee, J.C.; Colbert, C.M.; Johnston, D. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature 1997, 387, 869–875. [Google Scholar] [CrossRef]
- Heath, N.C.; Rizwan, A.P.; Engbers, J.D.; Anderson, D.; Zamponi, G.W.; Turner, R.W. The expression pattern of a Cav3-Kv4 complex differentially regulates spike output in cerebellar granule cells. J. Neurosci. 2014, 34, 8800–8812. [Google Scholar] [CrossRef] [Green Version]
- Serodio, P.; Rudy, B. Differential expression of Kv4 K+ channel subunits mediating subthreshold transient K+ (A-type) currents in rat brain. J. Neurophysiol. 1998, 79, 1081–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strassle, B.W.; Menegola, M.; Rhodes, K.J.; Trimmer, J.S. Light and electron microscopic analysis of KChIP and Kv4 localization in rat cerebellar granule cells. J. Comp. Neurol. 2005, 484, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Amarillo, Y.; De Santiago-Castillo, J.A.; Dougherty, K.; Maffie, J.; Kwon, E.; Covarrubias, M.; Rudy, B. Ternary Kv4.2 channels recapitulate voltage-dependent inactivation kinetics of A-type K+ channels in cerebellar granule neurons. J. Physiol. 2008, 586, 2093–2106. [Google Scholar] [CrossRef]
- Dayal, A.; Schrotter, K.; Pan, Y.; Fohr, K.; Melzer, W.; Grabner, M. The Ca2+ influx through the mammalian skeletal muscle dihydropyridine receptor is irrelevant for muscle performance. Nat. Commun. 2017, 8, 475. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Tao-Cheng, J.H.; Zerfas, P.; McBain, C.J. The K+ channel, Kv2.1, is apposed to astrocytic processes and is associated with inhibitory postsynaptic membranes in hippocampal and cortical principal neurons and inhibitory interneurons. Neuroscience 1998, 84, 37–48. [Google Scholar] [CrossRef]
- Antonucci, D.E.; Lim, S.T.; Vassanelli, S.; Trimmer, J.S. Dynamic localization and clustering of dendritic Kv2.1 voltage-dependent potassium channels in developing hippocampal neurons. Neuroscience 2001, 108, 69–81. [Google Scholar] [CrossRef]
- Misonou, H.; Mohapatra, D.P.; Trimmer, J.S. Kv2.1: A voltage-gated K+ channel critical to dynamic control of neuronal excitability. Neurotoxicology 2005, 26, 743–752. [Google Scholar] [CrossRef]
- King, A.N.; Manning, C.F.; Trimmer, J.S. A unique ion channel clustering domain on the axon initial segment of mammalian neurons. J. Comp. Neurol. 2014, 522, 2594–2608. [Google Scholar] [CrossRef] [Green Version]
- Fox, P.D.; Haberkorn, C.J.; Akin, E.J.; Seel, P.J.; Krapf, D.; Tamkun, M.M. Induction of stable ER-plasma-membrane junctions by Kv2.1 potassium channels. J. Cell Sci. 2015, 128, 2096–2105. [Google Scholar] [CrossRef] [Green Version]
- Johnson, B.; Leek, A.N.; Sole, L.; Maverick, E.E.; Levine, T.P.; Tamkun, M.M. Kv2 potassium channels form endoplasmic reticulum/plasma membrane junctions via interaction with VAPA and VAPB. Proc. Natl. Acad. Sci. USA 2018, 115, E7331–E7340. [Google Scholar] [CrossRef] [Green Version]
- Kirmiz, M.; Vierra, N.C.; Palacio, S.; Trimmer, J.S. Identification of VAPA and VAPB as Kv2 channel-interacting proteins defining endoplasmic reticulum-plasma membrane junctions in mammalian brain neurons. J. Neurosci 2018, 38, 7562–7584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirmiz, M.; Palacio, S.; Thapa, P.; King, A.N.; Sack, J.T.; Trimmer, J.S. Remodeling neuronal ER-PM junctions is a conserved nonconducting function of Kv2 plasma membrane ion channels. Mol. Biol Cell 2018, 29, 2410–2432. [Google Scholar] [CrossRef]
- Lim, S.T.; Antonucci, D.E.; Scannevin, R.H.; Trimmer, J.S. A novel targeting signal for proximal clustering of the Kv2.1 K+ channel in hippocampal neurons. Neuron 2000, 25, 385–397. [Google Scholar] [CrossRef] [Green Version]
- O’Dwyer, S.C.; Palacio, S.; Matsumoto, C.; Guarina, L.; Klug, N.R.; Tajada, S.; Rosati, B.; McKinnon, D.; Trimmer, J.S.; Santana, L.F. Kv2.1 channels play opposing roles in regulating membrane potential, Ca2+ channel function, and myogenic tone in arterial smooth muscle. Proc. Natl. Acad. Sci. USA 2020, 117, 3858–3866. [Google Scholar] [CrossRef] [Green Version]
- Vierra, N.C.; O’Dwyer, S.C.; Matsumoto, C.; Santana, L.F.; Trimmer, J.S. Regulation of neuronal excitation-transcription coupling by Kv2.1-induced clustering of somatic L-type Ca2+ channels at ER-PM junctions. Proc. Natl. Acad. Sci. USA 2021, 118, e2110094118. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, S.; Gullberg, M.; Jarvius, J.; Olsson, C.; Pietras, K.; Gustafsdottir, S.M.; Ostman, A.; Landegren, U. Protein detection using proximity-dependent DNA ligation assays. Nat. Biotechnol. 2002, 20, 473–477. [Google Scholar] [CrossRef]
- Soderberg, O.; Gullberg, M.; Jarvius, M.; Ridderstrale, K.; Leuchowius, K.J.; Jarvius, J.; Wester, K.; Hydbring, P.; Bahram, F.; Larsson, L.G.; et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 2006, 3, 995–1000. [Google Scholar] [CrossRef]
- Wild, A.R.; Sinnen, B.L.; Dittmer, P.J.; Kennedy, M.J.; Sather, W.A.; Dell’Acqua, M.L. Synapse-to-nucleus communication through NFAT Is mediated by L-type Ca2+ channel Ca2+ spike propagation to the soma. Cell Rep. 2019, 26, 3537–3550.e4. [Google Scholar] [CrossRef] [Green Version]
- Grundemann, J.; Clark, B.A. Calcium-activated potassium channels at nodes of Ranvier secure axonal spike propagation. Cell Rep. 2015, 12, 1715–1722. [Google Scholar] [CrossRef] [Green Version]
- Hirono, M.; Ogawa, Y.; Misono, K.; Zollinger, D.R.; Trimmer, J.S.; Rasband, M.N.; Misonou, H. BK channels localize to the paranodal junction and regulate action potentials in myelinated axons of cerebellar Purkinje cells. J. Neurosci. 2015, 35, 7082–7094. [Google Scholar] [CrossRef] [Green Version]
- Kerti, K.; Lorincz, A.; Nusser, Z. Unique somato-dendritic distribution pattern of Kv4.2 channels on hippocampal CA1 pyramidal cells. Eur. J. Neurosci. 2012, 35, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Prybylowski, K.; Chang, K.; Sans, N.; Kan, L.; Vicini, S.; Wenthold, R.J. The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron 2005, 47, 845–857. [Google Scholar] [CrossRef] [Green Version]
- Allen, D.; Bond, C.T.; Lujan, R.; Ballesteros-Merino, C.; Lin, M.T.; Wang, K.; Klett, N.; Watanabe, M.; Shigemoto, R.; Stackman, R.W., Jr.; et al. The SK2-long isoform directs synaptic localization and function of SK2-containing channels. Nat. Neurosci. 2011, 14, 744–749. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.; Lujan, R.; Schwenk, J.; Kelley, M.H.; Aguado, C.; Watanabe, M.; Fakler, B.; Maylie, J.; Adelman, J.P. Membrane palmitoylated protein 2 is a synaptic scaffold protein.n required for synaptic SK2-containing channel function. eLife 2016, 5, e12637. [Google Scholar] [CrossRef]
- Misonou, H.; Mohapatra, D.P.; Park, E.W.; Leung, V.; Zhen, D.; Misonou, K.; Anderson, A.E.; Trimmer, J.S. Regulation of ion channel localization and phosphorylation by neuronal activity. Nat. Neurosci. 2004, 7, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Misonou, H.; Menegola, M.; Mohapatra, D.P.; Guy, L.K.; Park, K.S.; Trimmer, J.S. Bidirectional activity-dependent regulation of neuronal ion channel phosphorylation. J. Neurosci. 2006, 26, 13505–13514. [Google Scholar] [CrossRef] [Green Version]
- Hage, T.A.; Salkoff, L. Sodium-activated potassium channels are functionally coupled to persistent sodium currents. J. Neurosci. 2012, 32, 2714–2721. [Google Scholar] [CrossRef] [Green Version]
- Pryce, K.D.; Powell, R.; Agwa, D.; Evely, K.M.; Sheehan, G.D.; Nip, A.; Tomasello, D.L.; Gururaj, S.; Bhattacharjee, A. Magi-1 scaffolds NaV1.8 and Slack KNa channels in dorsal root ganglion neurons regulating excitability and pain. FASEB J. 2019, 33, 7315–7330. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.J.; Amazu, C.; Puga-Molina, L.C.; Ma, X.; England, S.K.; Santi, C.M. SLO2.1/NALCN a sodium signaling complex that regulates uterine activity. iScience 2021, 24, 103210. [Google Scholar] [CrossRef]
- Harvey, J.R.M.; Plante, A.E.; Meredith, A.L. Ion channels controlling circadian rhythms in suprachiasmatic nucleus excitability. Physiol. Rev. 2020, 100, 1415–1454. [Google Scholar] [CrossRef]
- Qin, W.; Cho, K.F.; Cavanagh, P.E.; Ting, A.Y. Deciphering molecular interactions by proximity labeling. Nat. Methods 2021, 18, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Bar, D.Z.; Atkatsh, K.; Tavarez, U.; Erdos, M.R.; Gruenbaum, Y.; Collins, F.S. Biotinylation by antibody recognition-a method for proximity labeling. Nat. Methods 2018, 15, 127–133. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cav α1 Subunit | Ca2+ Current |
|---|---|
| Cav1.1 | L-type |
| Cav1.2 | L-type |
| Cav1.3 | L-type |
| Cav1.4 | L-type |
| Cav2.1 | P/Q-type |
| Cav2.2 | N-type |
| Cav2.3 | R-type |
| Cav3.1 | T-type |
| Cav3.2 | T-type |
| Cav3.3 | T-type |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vierra, N.C.; Trimmer, J.S. Ion Channel Partnerships: Odd and Not-So-Odd Couples Controlling Neuronal Ion Channel Function. Int. J. Mol. Sci. 2022, 23, 1953. https://doi.org/10.3390/ijms23041953
Vierra NC, Trimmer JS. Ion Channel Partnerships: Odd and Not-So-Odd Couples Controlling Neuronal Ion Channel Function. International Journal of Molecular Sciences. 2022; 23(4):1953. https://doi.org/10.3390/ijms23041953
Chicago/Turabian StyleVierra, Nicholas C., and James S. Trimmer. 2022. "Ion Channel Partnerships: Odd and Not-So-Odd Couples Controlling Neuronal Ion Channel Function" International Journal of Molecular Sciences 23, no. 4: 1953. https://doi.org/10.3390/ijms23041953
APA StyleVierra, N. C., & Trimmer, J. S. (2022). Ion Channel Partnerships: Odd and Not-So-Odd Couples Controlling Neuronal Ion Channel Function. International Journal of Molecular Sciences, 23(4), 1953. https://doi.org/10.3390/ijms23041953