
Chaperone Therapy in Fabry Disease

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Chaperone Therapy from Bench to Bedside
3. Chaperone Therapy Concept
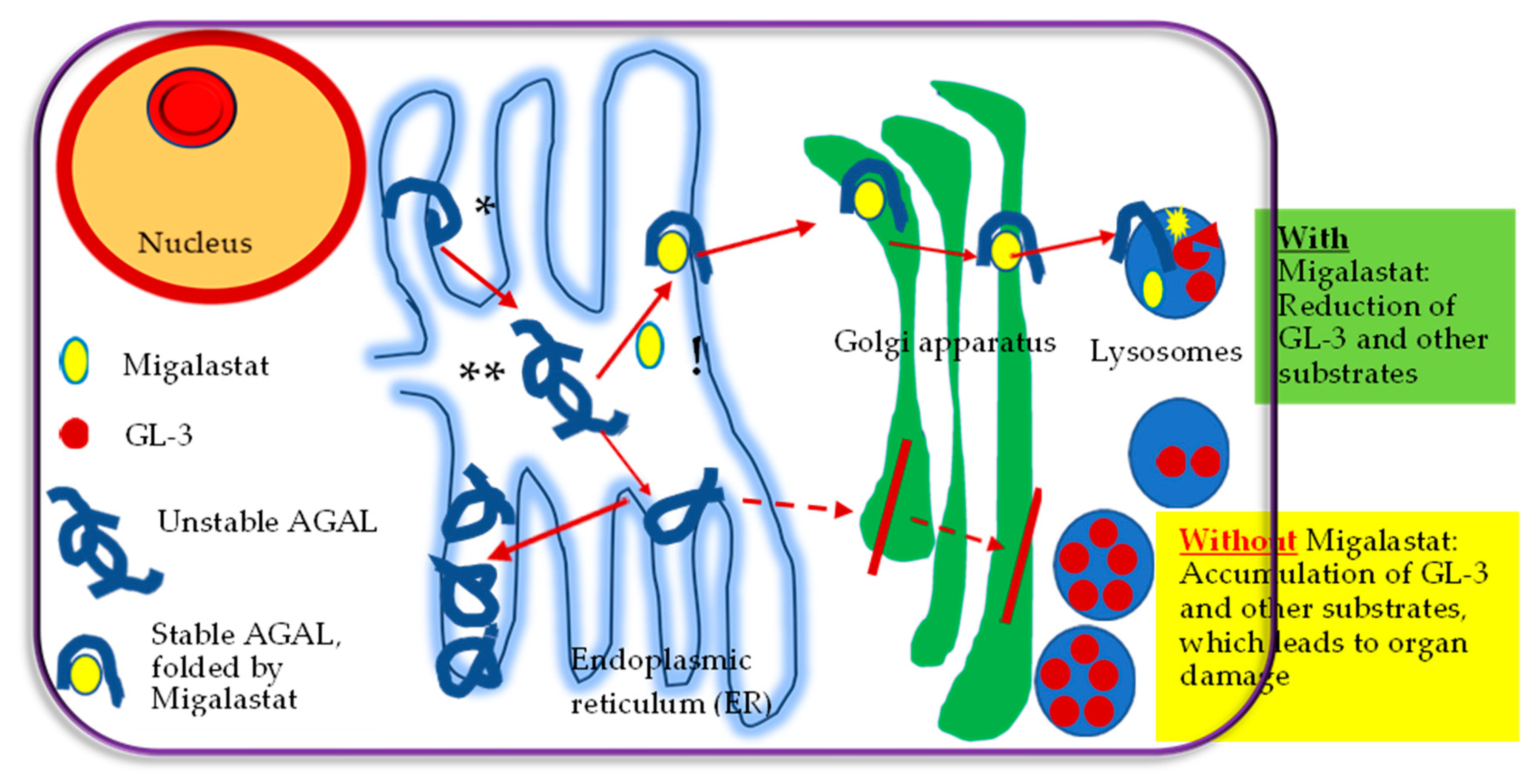
3.1. Migalastat
3.1.1. Amenability to Migalastat
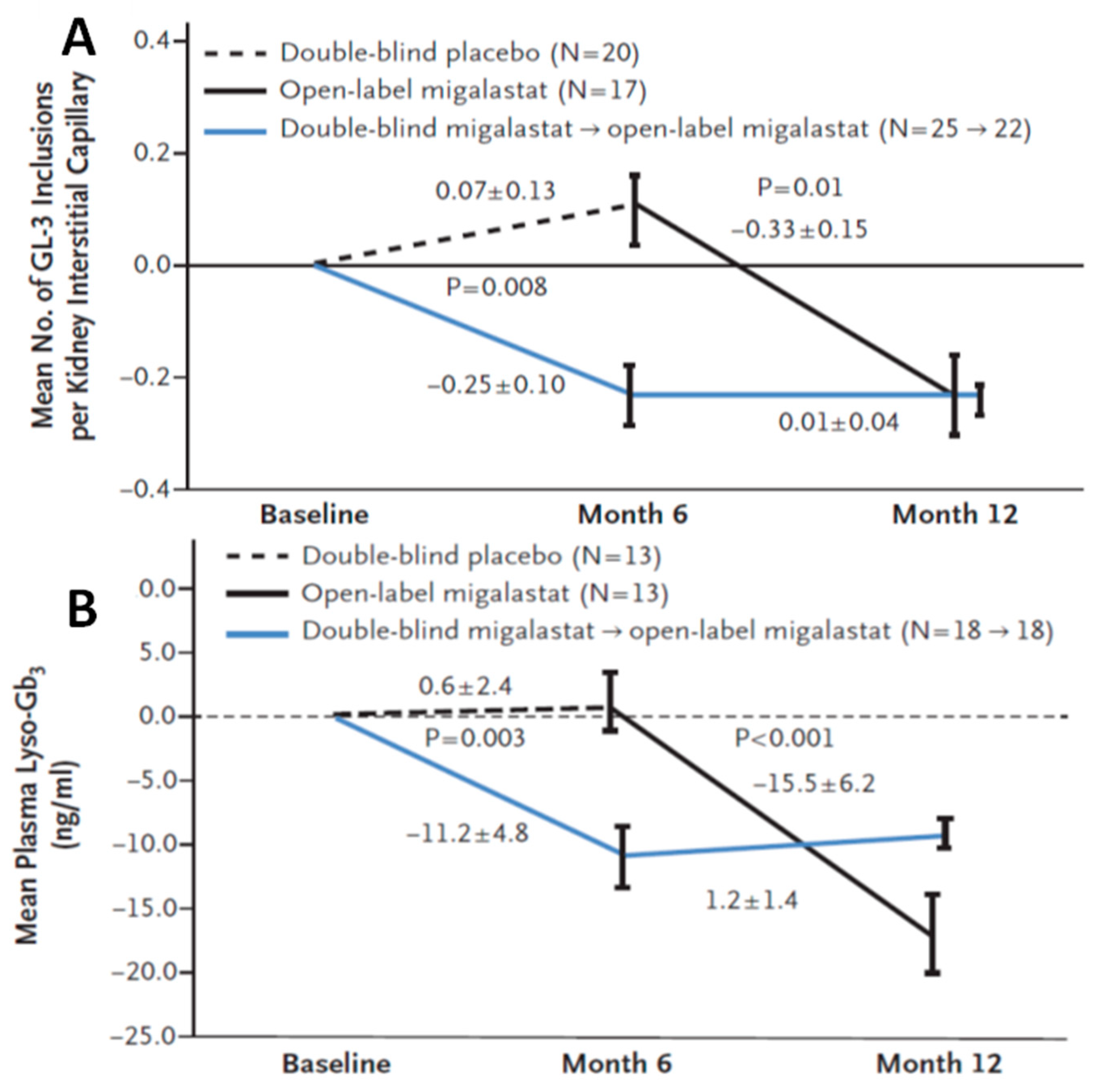
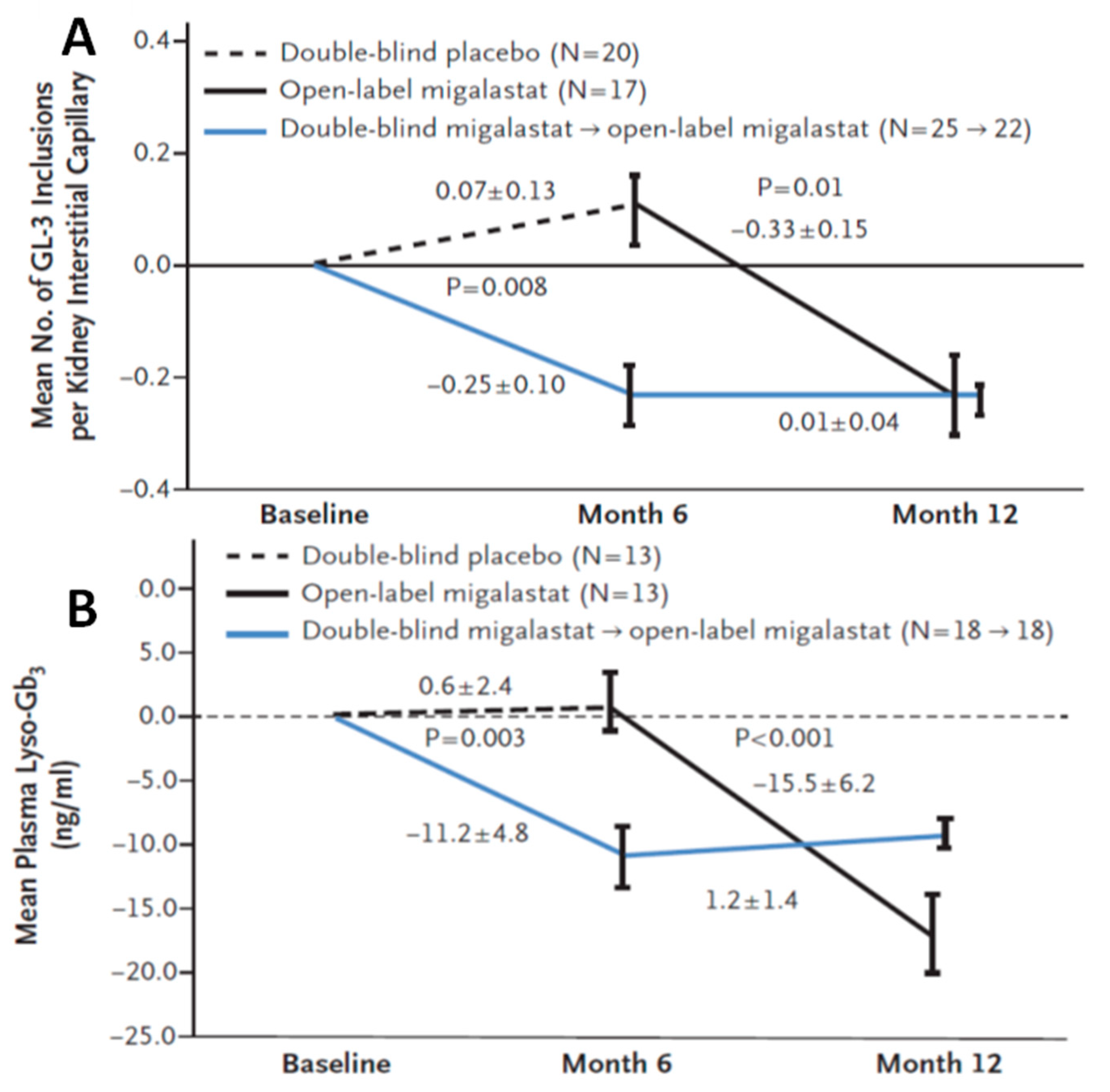
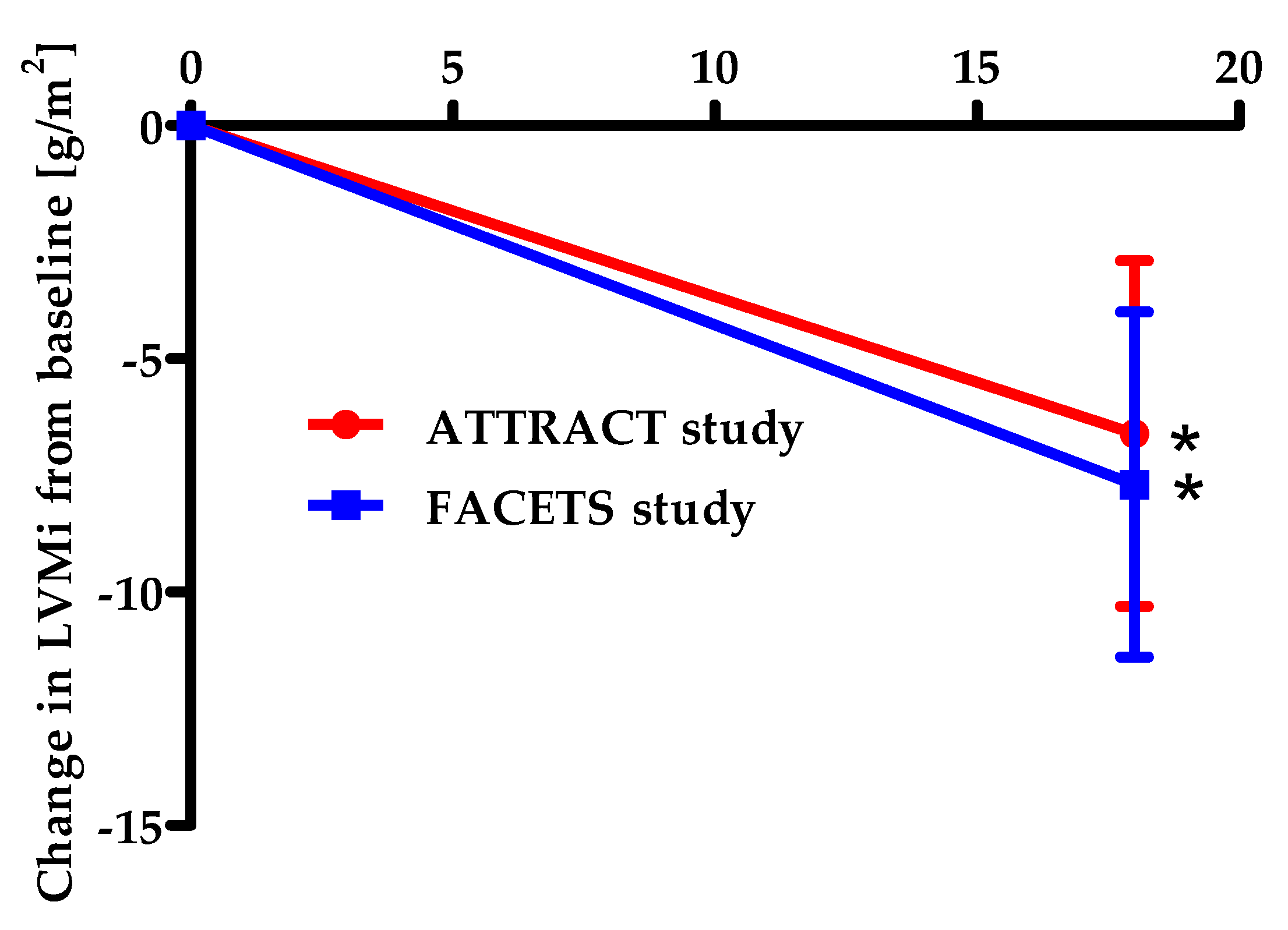
3.1.2. Efficacy of Migalastat
4. Clinical Workup during Chaperone Therapy
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brady, R.O.; Gal, A.E.; Bradley, R.M.; Martensson, E.; Warshaw, A.L.; Laster, L. Enzymatic defect in Fabry’s disease. Ceramidetrihexosidase deficiency. N. Engl. J. Med. 1967, 276, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Desnick, R.J.; Brady, R.; Barranger, J.; Collins, A.J.; Germain, D.P.; Goldman, M.; Grabowski, G.; Packman, S.; Wilcox, W.R. Fabry disease, an under-recognized multisystemic disorder: Expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann. Intern. Med. 2003, 138, 338–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Tol, L.; Smid, B.E.; Poorthuis, B.J.; Biegstraaten, M.; Deprez, R.H.; Linthorst, G.E.; Hollak, C.E. A systematic review on screening for Fabry disease: Prevalence of individuals with genetic variants of unknown significance. J. Med. Genet. 2014, 51, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Liguori, R.; Incensi, A.; de Pasqua, S.; Mignani, R.; Fileccia, E.; Santostefano, M.; Biagini, E.; Rapezzi, C.; Palmieri, S.; Romani, I.; et al. Skin globotriaosylceramide 3 deposits are specific to Fabry disease with classical mutations and associated with small fibre neuropathy. PLoS ONE 2017, 12, e0180581. [Google Scholar] [CrossRef] [Green Version]
- Wozniak, M.A.; Kittner, S.J.; Tuhrim, S.; Cole, J.W.; Stern, B.; Dobbins, M.; Grace, M.E.; Nazarenko, I.; Dobrovolny, R.; McDade, E.; et al. Frequency of unrecognized Fabry disease among young European-American and African-American men with first ischemic stroke. Stroke 2010, 41, 78–81. [Google Scholar] [CrossRef] [Green Version]
- Van der Tol, L.; Svarstad, E.; Ortiz, A.; Tondel, C.; Oliveira, J.P.; Vogt, L.; Waldek, S.; Hughes, D.A.; Lachmann, R.H.; Terryn, W.; et al. Chronic kidney disease and an uncertain diagnosis of Fabry disease: Approach to a correct diagnosis. Mol. Genet. Metab. 2015, 114, 242–247. [Google Scholar] [CrossRef]
- Nagueh, S.F. Anderson-Fabry disease and other lysosomal storage disorders. Circulation 2014, 130, 1081–1090. [Google Scholar] [CrossRef] [Green Version]
- Laney, D.A.; Peck, D.S.; Atherton, A.M.; Manwaring, L.P.; Christensen, K.M.; Shankar, S.P.; Grange, D.K.; Wilcox, W.R.; Hopkin, R.J. Fabry disease in infancy and early childhood: A systematic literature review. Genet. Med. 2015, 17, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Vardarli, I.; Rischpler, C.; Herrmann, K.; Weidemann, F. Diagnosis and Screening of Patients with Fabry Disease. Ther. Clin. Risk Manag. 2020, 16, 551–558. [Google Scholar] [CrossRef]
- Van der Veen, S.J.; Hollak, C.E.M.; van Kuilenburg, A.B.P.; Langeveld, M. Developments in the treatment of Fabry disease. J. Inherit. Metab. Dis. 2020, 43, 908–921. [Google Scholar] [CrossRef] [Green Version]
- Feriozzi, S.; Hughes, D.A. New drugs for the treatment of Anderson-Fabry disease. J. Nephrol. 2021, 34, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, A.; Germain, D.P.; Desnick, R.J.; Politei, J.; Mauer, M.; Burlina, A.; Eng, C.; Hopkin, R.J.; Laney, D.; Linhart, A.; et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol. Genet. Metab. 2018, 123, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Niemann, M.; Rolfs, A.; Stork, S.; Bijnens, B.; Breunig, F.; Beer, M.; Ertl, G.; Wanner, C.; Weidemann, F. Gene mutations versus clinically relevant phenotypes: Lyso-Gb3 defines Fabry disease. Circ. Cardiovasc. Genet. 2014, 7, 8–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidemann, F.; Beer, M.; Kralewski, M.; Siwy, J.; Kampmann, C. Early detection of organ involvement in Fabry disease by biomarker assessment in conjunction with LGE cardiac MRI: Results from the SOPHIA study. Mol. Genet. Metab. 2019, 126, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Linhart, A.; Kampmann, C.; Zamorano, J.L.; Sunder-Plassmann, G.; Beck, M.; Mehta, A.; Elliott, P.M.; European, F.O.S.I. Cardiac manifestations of Anderson-Fabry disease: Results from the international Fabry outcome survey. Eur. Heart J. 2007, 28, 1228–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidemann, F.; Breunig, F.; Beer, M.; Sandstede, J.; Stork, S.; Voelker, W.; Ertl, G.; Knoll, A.; Wanner, C.; Strotmann, J.M. The variation of morphological and functional cardiac manifestation in Fabry disease: Potential implications for the time course of the disease. Eur. Heart J. 2005, 26, 1221–1227. [Google Scholar] [CrossRef]
- McCafferty, E.H.; Scott, L.J. Migalastat: A Review in Fabry Disease. Drugs 2019, 79, 543–554. [Google Scholar] [CrossRef] [Green Version]
- Di Nora, C.; Livi, U. Heart transplantation in cardiac storage diseases: Data on Fabry disease and cardiac amyloidosis. Curr. Opin. Organ Transpl. 2020, 25, 211–217. [Google Scholar] [CrossRef]
- Beck, M. Agalsidase alfa for the treatment of Fabry disease: New data on clinical efficacy and safety. Expert Opin. Biol. Ther. 2009, 9, 255–261. [Google Scholar] [CrossRef]
- Ishii, S.; Kase, R.; Sakuraba, H.; Suzuki, Y. Characterization of a mutant alpha-galactosidase gene product for the late-onset cardiac form of Fabry disease. Biochem. Biophys. Res. Commun. 1993, 197, 1585–1589. [Google Scholar] [CrossRef]
- Romeo, G.; D’Urso, M.; Pisacane, A.; Blum, E.; De Falco, A.; Ruffilli, A. Residual activity of alpha-galactosidase A in Fabry’s disease. Biochem. Genet. 1975, 13, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Okumiya, T.; Ishii, S.; Takenaka, T.; Kase, R.; Kamei, S.; Sakuraba, H.; Suzuki, Y. Galactose stabilizes various missense mutants of alpha-galactosidase in Fabry disease. Biochem. Biophys. Res. Commun. 1995, 214, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Frustaci, A.; Chimenti, C.; Ricci, R.; Natale, L.; Russo, M.A.; Pieroni, M.; Eng, C.M.; Desnick, R.J. Improvement in cardiac function in the cardiac variant of Fabry’s disease with galactose-infusion therapy. N. Engl. J. Med. 2001, 345, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Liguori, L.; Monticelli, M.; Allocca, M.; Hay Mele, B.; Lukas, J.; Cubellis, M.V.; Andreotti, G. Pharmacological Chaperones: A Therapeutic Approach for Diseases Caused by Destabilizing Missense Mutations. Int. J. Mol. Sci. 2020, 21, 489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.Q.; Ishii, S.; Asano, N.; Suzuki, Y. Accelerated transport and maturation of lysosomal alpha-galactosidase A in Fabry lymphoblasts by an enzyme inhibitor. Nat. Med. 1999, 5, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Moran, N. FDA approves Galafold, a triumph for Amicus. Nat. Biotechnol. 2018, 36, 913. [Google Scholar] [CrossRef]
- Yam, G.H.; Zuber, C.; Roth, J. A synthetic chaperone corrects the trafficking defect and disease phenotype in a protein misfolding disorder. FASEB J. 2005, 19, 12–18. [Google Scholar] [CrossRef] [Green Version]
- Asano, N.; Ishii, S.; Kizu, H.; Ikeda, K.; Yasuda, K.; Kato, A.; Martin, O.R.; Fan, J.Q. In vitro inhibition and intracellular enhancement of lysosomal alpha-galactosidase A activity in Fabry lymphoblasts by 1-deoxygalactonojirimycin and its derivatives. Eur. J. Biochem. 2000, 267, 4179–4186. [Google Scholar] [CrossRef]
- Yam, G.H.; Bosshard, N.; Zuber, C.; Steinmann, B.; Roth, J. Pharmacological chaperone corrects lysosomal storage in Fabry disease caused by trafficking-incompetent variants. Am. J. Physiol. Cell Physiol. 2006, 290, C1076–C1082. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, E.R.; Della Valle, M.C.; Wu, X.; Katz, E.; Pruthi, F.; Bond, S.; Bronfin, B.; Williams, H.; Yu, J.; Bichet, D.G.; et al. The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat. Genet. Med. 2017, 19, 430–438. [Google Scholar] [CrossRef] [Green Version]
- Germain, D.P.; Hughes, D.A.; Nicholls, K.; Bichet, D.G.; Giugliani, R.; Wilcox, W.R.; Feliciani, C.; Shankar, S.P.; Ezgu, F.; Amartino, H.; et al. Treatment of Fabry’s Disease with the Pharmacologic Chaperone Migalastat. N. Engl. J. Med. 2016, 375, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.A.; Nicholls, K.; Shankar, S.P.; Sunder-Plassmann, G.; Koeller, D.; Nedd, K.; Vockley, G.; Hamazaki, T.; Lachmann, R.; Ohashi, T.; et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J. Med. Genet. 2017, 54, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Amicus Therapeutics. Galafold™ (Migalastat) Capsules, for Oral Use: US Prescribing Information. 2018. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/208623Orig1s000Approv.pdf (accessed on 2 November 2021).
- European Medicines Agency. Migalastat (Galafold). EU Summary of Product Characteristics. 2016. Available online: https://www.ema.europa.eu/en/documents/product-information/galafold-epar-product-information_en.pdf (accessed on 2 November 2021).
- Khanna, R.; Soska, R.; Lun, Y.; Feng, J.; Frascella, M.; Young, B.; Brignol, N.; Pellegrino, L.; Sitaraman, S.A.; Desnick, R.J.; et al. The pharmacological chaperone 1-deoxygalactonojirimycin reduces tissue globotriaosylceramide levels in a mouse model of Fabry disease. Mol. Ther. 2010, 18, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, O.; Gago, M.F.; Miltenberger-Miltenyi, G.; Sousa, N.; Cunha, D. Fabry Disease Therapy: State-of-the-Art and Current Challenges. Int. J. Mol. Sci. 2020, 22, 206. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.S.; Khanna, R.; Schmith, V.; Lun, Y.; Shen, J.S.; Garcia, A.; Dungan, L.; Perry, A.; Martin, L.; Tsai, P.C.; et al. Migalastat Tissue Distribution: Extrapolation From Mice to Humans Using Pharmacokinetic Modeling and Comparison With Agalsidase Beta Tissue Distribution in Mice. Clin. Pharmacol. Drug Dev. 2021, 10, 1075–1088. [Google Scholar] [CrossRef]
- Lenders, M.; Stappers, F.; Brand, E. In Vitro and In Vivo Amenability to Migalastat in Fabry Disease. Mol. Ther. Methods Clin. Dev. 2020, 19, 24–34. [Google Scholar] [CrossRef]
- Benjamin, E.R.; Flanagan, J.J.; Schilling, A.; Chang, H.H.; Agarwal, L.; Katz, E.; Wu, X.; Pine, C.; Wustman, B.; Desnick, R.J.; et al. The pharmacological chaperone 1-deoxygalactonojirimycin increases alpha-galactosidase A levels in Fabry patient cell lines. J. Inherit. Metab. Dis. 2009, 32, 424–440. [Google Scholar] [CrossRef]
- Shimotori, M.; Maruyama, H.; Nakamura, G.; Suyama, T.; Sakamoto, F.; Itoh, M.; Miyabayashi, S.; Ohnishi, T.; Sakai, N.; Wataya-Kaneda, M.; et al. Novel mutations of the GLA gene in Japanese patients with Fabry disease and their functional characterization by active site specific chaperone. Hum. Mutat. 2008, 29, 331. [Google Scholar] [CrossRef]
- Oommen, S.; Zhou, Y.; Meiyappan, M.; Gurevich, A.; Qiu, Y. Inter-assay variability influences migalastat amenability assessments among Fabry disease variants. Mol. Genet. Metab. 2019, 127, 74–85. [Google Scholar] [CrossRef]
- Lenders, M.; Nordbeck, P.; Kurschat, C.; Karabul, N.; Kaufeld, J.; Hennermann, J.B.; Patten, M.; Cybulla, M.; Muntze, J.; Uceyler, N.; et al. Treatment of Fabry’s Disease With Migalastat: Outcome From a Prospective Observational Multicenter Study (FAMOUS). Clin. Pharmacol. Ther. 2020, 108, 326–337. [Google Scholar] [CrossRef]
- Lenders, M.; Stappers, F.; Niemietz, C.; Schmitz, B.; Boutin, M.; Ballmaier, P.J.; Zibert, A.; Schmidt, H.; Brand, S.M.; Auray-Blais, C.; et al. Mutation-specific Fabry disease patient-derived cell model to evaluate the amenability to chaperone therapy. J. Med. Genet. 2019, 56, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Galafold Amenability Table. 2021. Available online: https://www.galafoldamenabilitytable.com/hcp (accessed on 4 November 2021).
- Mauer, M.; Sokolovskiy, A.; Barth, J.A.; Castelli, J.P.; Williams, H.N.; Benjamin, E.R.; Najafian, B. Reduction of podocyte globotriaosylceramide content in adult male patients with Fabry disease with amenable GLA mutations following 6 months of migalastat treatment. J. Med. Genet. 2017, 54, 781–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiffmann, R.; Bichet, D.G.; Jovanovic, A.; Hughes, D.A.; Giugliani, R.; Feldt-Rasmussen, U.; Shankar, S.P.; Barisoni, L.; Colvin, R.B.; Jennette, J.C.; et al. Migalastat improves diarrhea in patients with Fabry disease: Clinical-biomarker correlations from the phase 3 FACETS trial. Orphanet. J. Rare Dis. 2018, 13, 68. [Google Scholar] [CrossRef] [Green Version]
- Germain, D.P.; Nicholls, K.; Giugliani, R.; Bichet, D.G.; Hughes, D.A.; Barisoni, L.M.; Colvin, R.B.; Jennette, J.C.; Skuban, N.; Castelli, J.P.; et al. Efficacy of the pharmacologic chaperone migalastat in a subset of male patients with the classic phenotype of Fabry disease and migalastat-amenable variants: Data from the phase 3 randomized, multicenter, double-blind clinical trial and extension study. Genet. Med. 2019, 21, 1987–1997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldt-Rasmussen, U.; Hughes, D.; Sunder-Plassmann, G.; Shankar, S.; Nedd, K.; Olivotto, I.; Ortiz, D.; Ohashi, T.; Hamazaki, T.; Skuban, N.; et al. Long-term efficacy and safety of migalastat treatment in Fabry disease: 30-month results from the open-label extension of the randomized, phase 3 ATTRACT study. Mol. Genet. Metab. 2020, 131, 219–228. [Google Scholar] [CrossRef]
- Lenders, M.; Brand, E. Fabry Disease: The Current Treatment Landscape. Drugs 2021, 81, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Bichet, D.G.; Torra, R.; Wallace, E.; Hughes, D.; Giugliani, R.; Skuban, N.; Krusinska, E.; Feldt-Rasmussen, U.; Schiffmann, R.; Nicholls, K. Long-term follow-up of renal function in patients treated with migalastat for Fabry disease. Mol. Genet. Metab. Rep. 2021, 28, 100786. [Google Scholar] [CrossRef]
- Muntze, J.; Gensler, D.; Maniuc, O.; Liu, D.; Cairns, T.; Oder, D.; Hu, K.; Lorenz, K.; Frantz, S.; Wanner, C.; et al. Oral Chaperone Therapy Migalastat for Treating Fabry Disease: Enzymatic Response and Serum Biomarker Changes After 1 Year. Clin. Pharmacol. Ther. 2019, 105, 1224–1233. [Google Scholar] [CrossRef] [Green Version]
- Riccio, E.; Zanfardino, M.; Ferreri, L.; Santoro, C.; Cocozza, S.; Capuano, I.; Imbriaco, M.; Feriozzi, S.; Pisani, A.; Group, A. Switch from enzyme replacement therapy to oral chaperone migalastat for treating fabry disease: Real-life data. Eur. J. Hum. Genet. 2020, 28, 1662–1668. [Google Scholar] [CrossRef]
- Weidemann, F.; Sommer, C.; Duning, T.; Lanzl, I.; Mohrenschlager, M.; Naleschinski, D.; Baron, R.; Breunig, F.; Schaefer, R.; Strotmann, J.; et al. Division-related function and organ-related therapy in Fabry’s disease. An interdisciplinary challenge. Med. Klin. 2009, 104, 10–19. [Google Scholar] [CrossRef]
- Weidemann, F.; Niemann, M.; Warnock, D.G.; Ertl, G.; Wanner, C. The Fabry cardiomyopathy: Models for the cardiologist. Annu. Rev. Med. 2011, 62, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Wanner, C.; Arad, M.; Baron, R.; Burlina, A.; Elliott, P.M.; Feldt-Rasmussen, U.; Fomin, V.V.; Germain, D.P.; Hughes, D.A.; Jovanovic, A.; et al. European expert consensus statement on therapeutic goals in Fabry disease. Mol. Genet. Metab. 2018, 124, 189–203. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weidemann, F.; Jovanovic, A.; Herrmann, K.; Vardarli, I. Chaperone Therapy in Fabry Disease. Int. J. Mol. Sci. 2022, 23, 1887. https://doi.org/10.3390/ijms23031887
Weidemann F, Jovanovic A, Herrmann K, Vardarli I. Chaperone Therapy in Fabry Disease. International Journal of Molecular Sciences. 2022; 23(3):1887. https://doi.org/10.3390/ijms23031887
Chicago/Turabian StyleWeidemann, Frank, Ana Jovanovic, Ken Herrmann, and Irfan Vardarli. 2022. "Chaperone Therapy in Fabry Disease" International Journal of Molecular Sciences 23, no. 3: 1887. https://doi.org/10.3390/ijms23031887
APA StyleWeidemann, F., Jovanovic, A., Herrmann, K., & Vardarli, I. (2022). Chaperone Therapy in Fabry Disease. International Journal of Molecular Sciences, 23(3), 1887. https://doi.org/10.3390/ijms23031887