
Phosphorylation of Eukaryotic Initiation Factor 4G1 (eIF4G1) at Ser1147 Is Specific for eIF4G1 Bound to eIF4E in Delayed Neuronal Death after Ischemia

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Rat eIF4G1 Sequence Reports Phosphorylation Sites Homologous to Human eIF4G1 in the IDL Region
2.2. eIF4G1 Shows a Similar Level of Expression in the Cerebral Cortex and Hippocampal CA1 Region under Ischemia-Reperfusion (IR) Stress
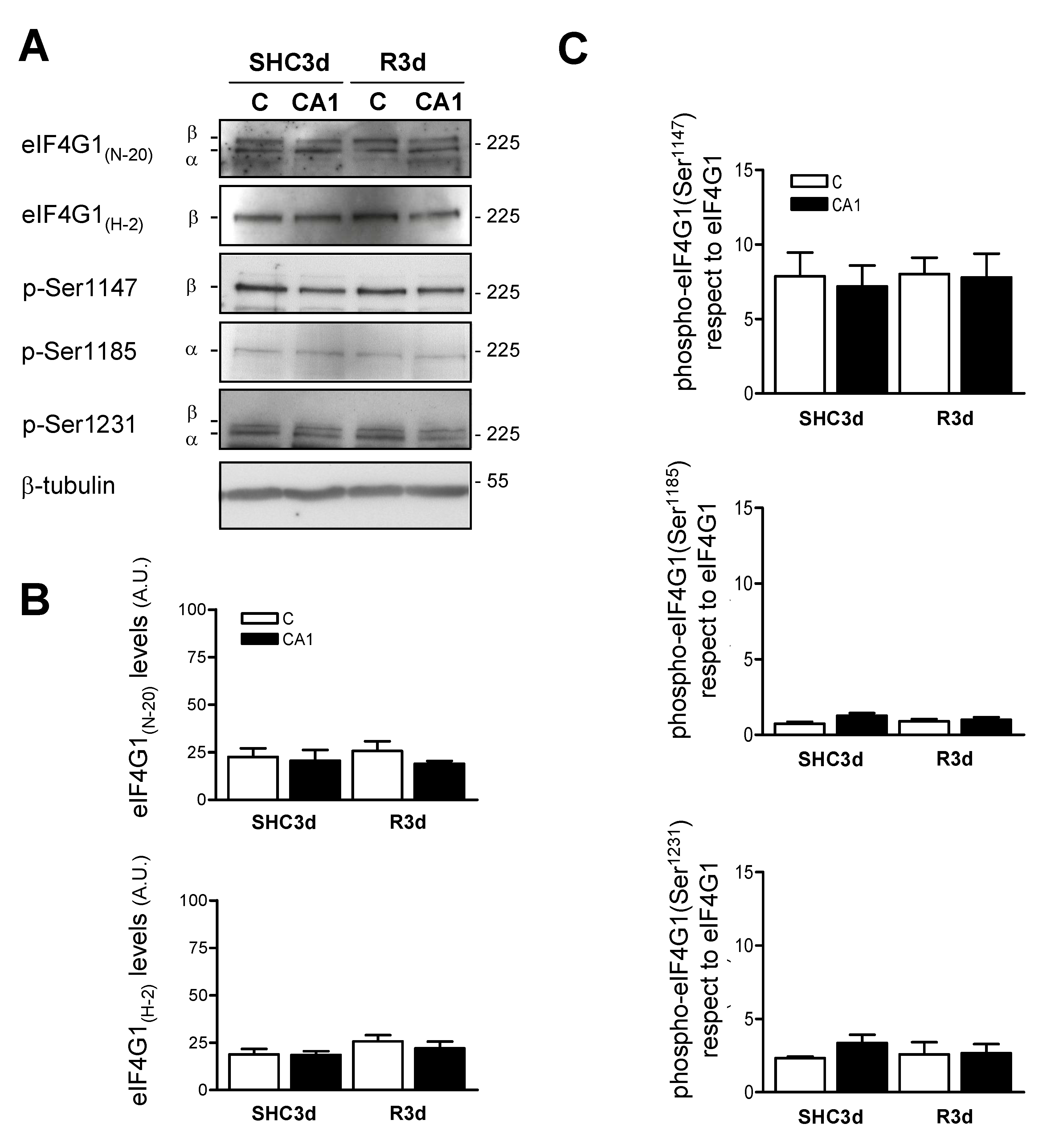
2.3. Characterization of the Phosphorylation Status of eIF4G1 at Ser1147, Ser1185, and Ser1231 Phospho-Sites in the Cortical and CA1 Brain Regions
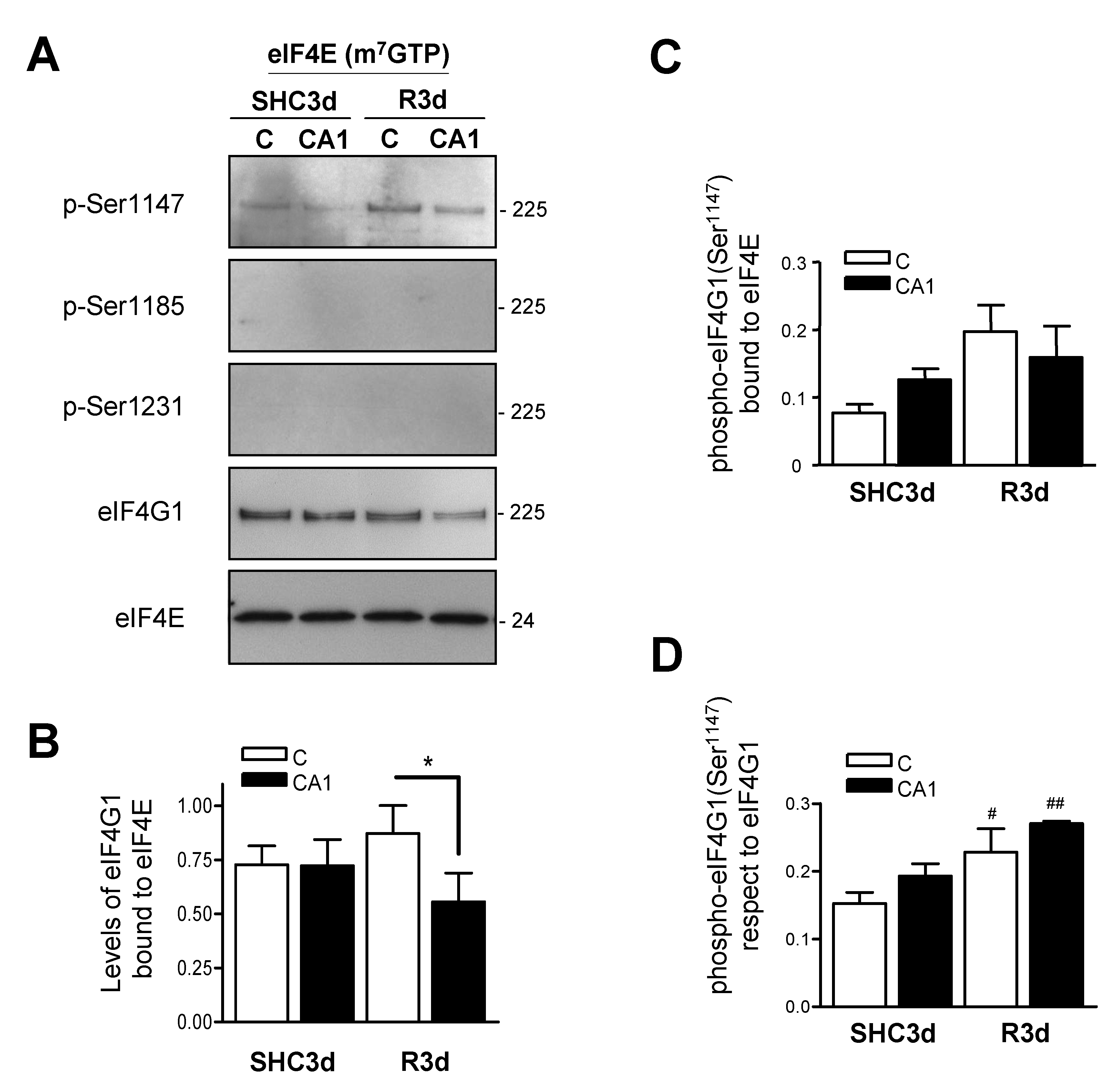
2.4. eIF4G1 Bound to eIF4E is Phosphorylated at Ser1147
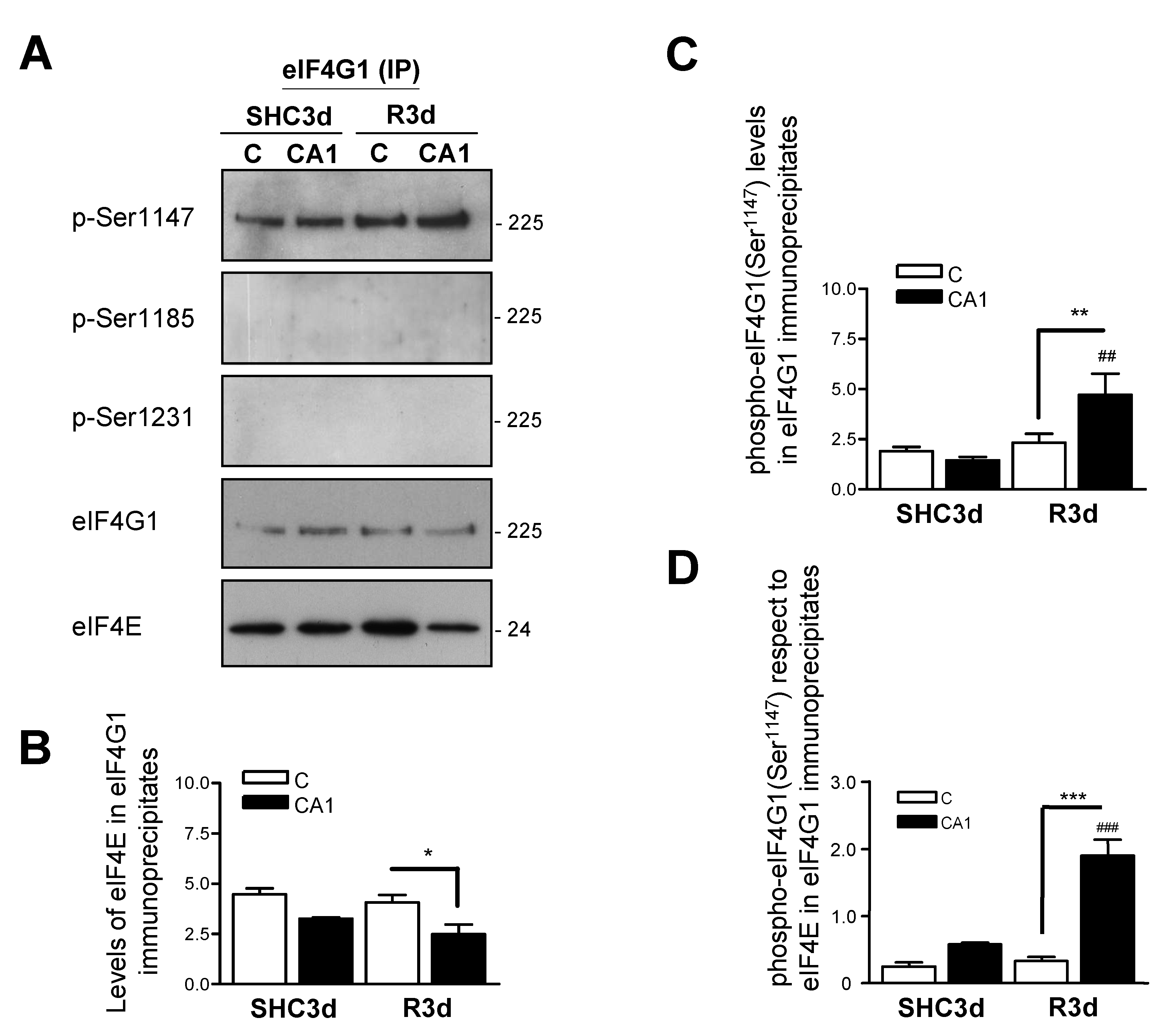
2.5. Association of eIF4E to eIF4G1 Phosphorylated at Ser1147 in eIF4G1 Immunoprecipitates
2.6. Increased Colocalization of eIF4G1 Phosphorylated at Ser1147 with eIF4E in the CA1 Region under Ischemia-Reperfusion (IR) Stress
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Sequence Analysis
4.3. Animal Model of Cerebral Ischemia and Reperfusion
4.4. Sample Preparation
4.5. eIF4G and eIF4E Binding Assay
4.6. Immunoprecipitation
4.7. Western Blot Analysis
4.8. Immunohistochemistry
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Feigin, V.L.; Forouzanfar, M.H.; Krishnamurthi, R.; Mensah, G.A.; Connor, M.; Bennett, D.A.; Moran, A.E.; Sacco, R.L.; Anderson, L.; Truelsen, T.; et al. Global and regional burden of stroke during 1990–2010: Findings from the Global Burden of Disease Study 2010. Lancet 2014, 383, 245–254. [Google Scholar] [CrossRef]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Das, S.R.; de Ferranti, S.; Despres, J.P.; Fullerton, H.J.; et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation 2016, 133, e38–360. [Google Scholar] [CrossRef]
- GBD 2016 Neurology Collaborators. Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef]
- Lipton, P. Ischemic cell death in brain neurons. Physiol. Rev. 1999, 79, 1431–1568. [Google Scholar] [CrossRef] [PubMed]
- Hossmann, K.A. Viability thresholds and the penumbra of focal ischemia. Ann. Neurol. 1994, 36, 557–565. [Google Scholar] [CrossRef] [PubMed]
- White, B.C.; Sullivan, J.M.; DeGracia, D.J.; O’Neil, B.J.; Neumar, R.W.; Grossman, L.I.; Rafols, J.A.; Krause, G.S. Brain ischemia and reperfusion: Molecular mechanisms of neuronal injury. J. Neurol. Sci. 2000, 179, 1–33. [Google Scholar] [CrossRef]
- Kirino, T. Delayed neuronal death. Neuropatholy 2000, 20, S95–S97. [Google Scholar] [CrossRef]
- Pulsinelli, W.A.; Brierley, J.B.; Plum, F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982, 11, 491–498. [Google Scholar] [CrossRef]
- Hossmann, K.A. Disturbances of cerebral protein synthesis and ischemic cell death. Prog. Brain Res. 1993, 96, 161–177. [Google Scholar] [PubMed]
- Harukuni, I.; Bhardwaj, A. Mechanisms of brain injury after global cerebral ischemia. Neuro. Clin. 2006, 24, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Ayuso, M.I.; Martinez-Alonso, E.; Cid, C.; Alonso de Lecinana, M.; Alcazar, A. The translational repressor eIF4E-binding protein 2 (4E-BP2) correlates with selective delayed neuronal death after ischemia. J. Cereb. Blood Flow Metab. 2013, 33, 1173–1181. [Google Scholar] [CrossRef]
- Ayuso, M.I.; Martinez-Alonso, E.; Regidor, I.; Alcazar, A. Stress granule induction after brain ischemia is independent of eukaryotic translation initiation factor (eIF) 2α phosphorylation and is correlated with a decrease in eIF4B and eIF4E proteins. J. Biol. Chem. 2016, 291, 27252–27264. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.J.; Hellen, C.U.; Pestova, T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef] [PubMed]
- Shatkin, A.J. Capping of eucaryotic mRNAs. Cell 1976, 9, 645–653. [Google Scholar] [CrossRef]
- Kozak, M. Rethinking some mechanisms invoked to explain translational regulation in eukaryotes. Gene 2006, 382, 1–11. [Google Scholar] [CrossRef]
- Lin, C.J.; Cencic, R.; Mills, J.R.; Robert, F.; Pelletier, J. c-Myc and eIF4F are components of a feedforward loop that links transcription and translation. Cancer Res. 2008, 68, 5326–5334. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Valle, F.; Braunstein, S.; Zavadil, J.; Formenti, S.C.; Schneider, R.J. eIF4GI links nutrient sensing by mTOR to cell proliferation and inhibition of autophagy. J. Cell Biol. 2008, 181, 293–307. [Google Scholar] [CrossRef]
- Marcotrigiano, J.; Lomakin, I.B.; Sonenberg, N.; Pestova, T.V.; Hellen, C.U.; Burley, S.K. A conserved HEAT domain within eIF4G directs assembly of the translation initiation machinery. Mol. Cell 2001, 7, 193–203. [Google Scholar] [CrossRef]
- Hinton, T.M.; Coldwell, M.J.; Carpenter, G.A.; Morley, S.J.; Pain, V.M. Functional analysis of individual binding activities of the scaffold protein eIF4G. J. Biol. Chem. 2007, 282, 1695–1708. [Google Scholar] [CrossRef]
- Lewis, S.M.; Cerquozzi, S.; Graber, T.E.; Ungureanu, N.H.; Andrews, M.; Holcik, M. The eIF4G homolog DAP5/p97 supports the translation of select mRNAs during endoplasmic reticulum stress. Nucleic Acids Res. 2008, 36, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Virgili, G.; Frank, F.; Feoktistova, K.; Sawicki, M.; Sonenberg, N.; Fraser, C.S.; Nagar, B. Structural analysis of the DAP5 MIF4G domain and its interaction with eIF4A. Structure 2013, 21, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Raught, B.; Gingras, A.C.; Gygi, S.P.; Imataka, H.; Morino, S.; Gradi, A.; Aebersold, R.; Sonenberg, N. Serum-stimulated, rapamycin-sensitive phosphorylation sites in the eukaryotic translation initiation factor 4GI. EMBO J. 2000, 19, 434–444. [Google Scholar] [CrossRef]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Martin de la Vega, C.; Burda, J.; Nemethova, M.; Quevedo, C.; Alcazar, A.; Martin, M.E.; Danielisova, V.; Fando, J.L.; Salinas, M. Possible mechanisms involved in the down-regulation of translation during transient global ischaemia in the rat brain. Biochem. J. 2001, 357, 819–826. [Google Scholar] [CrossRef]
- Ayuso, M.I.; Hernandez-Jimenez, M.; Martin, M.E.; Salinas, M.; Alcazar, A. New hierarchical phosphorylation pathway of the translational repressor eIF4E-binding protein 1 (4E-BP1) in ischemia-reperfusion stress. J. Biol. Chem. 2010, 285, 34355–34363. [Google Scholar] [CrossRef] [PubMed]
- Chong, Z.Z.; Yao, Q.; Li, H.H. The rationale of targeting mammalian target of rapamycin for ischemic stroke. Cell. Signal. 2013, 25, 1598–1607. [Google Scholar] [CrossRef]
- Ayuso, M.I.; Martinez-Alonso, E.; Salvador, N.; Bonova, P.; Regidor, I.; Alcazar, A. Dissociation of eIF4E-binding protein 2 (4E-BP2) from eIF4E independent of Thr37/Thr46 phosphorylation in the ischemic stress response. PLoS ONE 2015, 10, e0121958. [Google Scholar] [CrossRef]
- Dobrikov, M.I.; Dobrikova, E.Y.; Gromeier, M. Dynamic regulation of the translation initiation helicase complex by mitogenic signal transduction to eukaryotic translation initiation factor 4G. Mol. Cell Biol. 2013, 33, 937–946. [Google Scholar] [CrossRef]
- Sun, J.; Nan, G. The Mitogen-Activated Protein Kinase (MAPK) Signaling Pathway as a Discovery Target in Stroke. J. Mol. Neurosci. 2016, 59, 90–98. [Google Scholar] [CrossRef]
- Dobrikov, M.; Dobrikova, E.; Shveygert, M.; Gromeier, M. Phosphorylation of eukaryotic translation initiation factor 4G1 (eIF4G1) by protein kinase Cα regulates eIF4G1 binding to Mnk1. Mol. Cell Biol. 2011, 31, 2947–2959. [Google Scholar] [CrossRef] [PubMed]
- Dobrikov, M.I.; Shveygert, M.; Brown, M.C.; Gromeier, M. Mitotic phosphorylation of eukaryotic initiation factor 4G1 (eIF4G1) at Ser1232 by Cdk1:cyclin B inhibits eIF4A helicase complex binding with RNA. Mol. Cell Biol. 2014, 34, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.B.; Ye, Q.; Tu, J.W.; Yu, X.Y. Transient cerebral ischemia/reperfusion-induced acute lung injury in rats associated with protein kinase C alpha expression. Int. J. Clin. Exp. Pathol. 2018, 11, 3487–3493. [Google Scholar] [PubMed]
- Thilmann, R.; Xie, Y.; Kleihues, P.; Kiessling, M. Persistent inhibition of protein synthesis precedes delayed neuronal death in postischemic gerbil hippocampus. Acta Neuropathol. 1986, 71, 88–93. [Google Scholar] [CrossRef]
- DeGracia, D.J.; Neumar, R.W.; White, B.C.; Krause, G.S. Global brain ischemia and reperfusion: Modifications in eukaryotic initiation factors associated with inhibition of translation initiation. J. Neurochem. 1996, 67, 2005–2012. [Google Scholar] [CrossRef] [PubMed]
- Raval, A.P.; Dave, K.R.; Perez-Pinzon, M.A. Resveratrol mimics ischemic preconditioning in the brain. J. Cereb. Blood Flow Metab. 2006, 26, 1141–1147. [Google Scholar] [CrossRef] [PubMed]
- DeGracia, D.J.; Hu, B.R. Irreversible translation arrest in the reperfused brain. J. Cereb. Blood Flow Metab. 2007, 27, 875–893. [Google Scholar] [CrossRef]
- Brito Querido, J.; Sokabe, M.; Kraatz, S.; Gordiyenko, Y.; Skehel, J.M.; Fraser, C.S.; Ramakrishnan, V. Structure of a human 48S translational initiation complex. Science 2020, 369, 1220–1227. [Google Scholar] [CrossRef]
- Haghighat, A.; Mader, S.; Pause, A.; Sonenberg, N. Repression of cap-dependent translation by 4E-binding protein 1: Competition with p220 for binding to eukaryotic initiation factor-4E. EMBO J. 1995, 14, 5701–5709. [Google Scholar] [CrossRef]
- Svitkin, Y.V.; Herdy, B.; Costa-Mattioli, M.; Gingras, A.C.; Raught, B.; Sonenberg, N. Eukaryotic translation initiation factor 4E availability controls the switch between cap-dependent and internal ribosomal entry site-mediated translation. Mol. Cell Biol. 2005, 25, 10556–10565. [Google Scholar] [CrossRef]
- Martínez-Alonso, E.; Guerra-Pérez, N.; Escobar-Peso, A.; Regidor, I.; Masjuan, J.; Alcázar, A. Differential association of 4E-BP2-interacting proteins is related to selective delayed neuronal death after ischemia. Int. J. Mol. Sci. 2021, 22, 10327. [Google Scholar] [CrossRef]
- Bovee, M.L.; Marissen, W.E.; Zamora, M.; Lloyd, R.E. The predominant elF4G-specific cleavage activity in poliovirus-infected HeLa cells is distinct from 2A protease. Virology 1998, 245, 229–240. [Google Scholar] [CrossRef][Green Version]
- Bellsolell, L.; Cho-Park, P.F.; Poulin, F.; Sonenberg, N.; Burley, S.K. Two structurally atypical HEAT domains in the C-terminal portion of human eIF4G support binding to eIF4A and Mnk1. Structure 2006, 14, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Gkogkas, C.G.; Khoutorsky, A.; Ran, I.; Rampakakis, E.; Nevarko, T.; Weatherill, D.B.; Vasuta, C.; Yee, S.; Truitt, M.; Dallaire, P.; et al. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 2013, 493, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Raught, B.; Gingras, A.C. Signaling to translation initiation. In Translational Control in Biology and Medicine; Mathews, M.B., Sonenberg, N., Hershey, J.W.B., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2007; pp. 369–401. [Google Scholar]
- Hartman, N.W.; Lin, T.V.; Zhang, L.; Paquelet, G.E.; Feliciano, D.M.; Bordey, A. mTORC1 targets the translational repressor 4E-BP2, but not S6 kinase 1/2, to regulate neural stem cell self-renewal in vivo. Cell Rep. 2013, 5, 433–444. [Google Scholar] [CrossRef]
- Cuesta, R.; Xi, Q.; Schneider, R.J. Adenovirus-specific translation by displacement of kinase Mnk1 from cap-initiation complex eIF4F. EMBO J. 2000, 19, 3465–3474. [Google Scholar] [CrossRef] [PubMed]
- DeGracia, D.J.; Rafols, J.A.; Morley, S.J.; Kayali, F. Immunohistochemical mapping of total and phosphorylated eukaryotic initiation factor 4G in rat hippocampus following global brain ischemia and reperfusion. Neuroscience 2006, 139, 1235–1248. [Google Scholar] [CrossRef]
- Bolster, D.R.; Vary, T.C.; Kimball, S.R.; Jefferson, L.S. Leucine regulates translation initiation in rat skeletal muscle via enhanced eIF4G phosphorylation. J. Nutr. 2004, 134, 1704–1710. [Google Scholar] [CrossRef]
- DeGracia, D.J.; Kumar, R.; Owen, C.R.; Krause, G.S.; White, B.C. Molecular pathways of protein synthesis inhibition during brain reperfusion: Implications for neuronal survival or death. J. Cereb. Blood Flow Metab. 2002, 22, 127–141. [Google Scholar] [CrossRef]
- Rami, A.; Bechmann, I.; Stehle, J.H. Exploiting endogenous anti-apoptotic proteins for novel therapeutic strategies in cerebral ischemia. Prog. Neurobiol. 2008, 85, 273–296. [Google Scholar] [CrossRef]
- Morley, S.J.; Coldwell, M.J. Matter of Life and Death: Translation Initiation during Apoptosis. In Translational Control in Biology and Medicine; Mathews, M.B., Sonenberg, N., Hershey, J.W.B., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2007; pp. 433–458. [Google Scholar]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Bonilla, L.; Cid, C.; Alcazar, A.; Burda, J.; Ayuso, I.; Salinas, M. Regulatory proteins of eukaryotic initiation factor 2-alpha subunit (eIF2α) phosphatase, under ischemic reperfusion and tolerance. J. Neurochem. 2007, 103, 1368–1380. [Google Scholar] [CrossRef] [PubMed]
- Garcia, L.; Burda, J.; Hrehorovska, M.; Burda, R.; Martin, M.E.; Salinas, M. Ischaemic preconditioning in the rat brain: Effect on the activity of several initiation factors, Akt and extracellular signal-regulated protein kinase phosphorylation, and GRP78 and GADD34 expression. J. Neurochem. 2004, 88, 136–147. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Alonso, E.; Guerra-Pérez, N.; Escobar-Peso, A.; Peracho, L.; Vera-Lechuga, R.; Cruz-Culebras, A.; Masjuan, J.; Alcázar, A. Phosphorylation of Eukaryotic Initiation Factor 4G1 (eIF4G1) at Ser1147 Is Specific for eIF4G1 Bound to eIF4E in Delayed Neuronal Death after Ischemia. Int. J. Mol. Sci. 2022, 23, 1830. https://doi.org/10.3390/ijms23031830
Martínez-Alonso E, Guerra-Pérez N, Escobar-Peso A, Peracho L, Vera-Lechuga R, Cruz-Culebras A, Masjuan J, Alcázar A. Phosphorylation of Eukaryotic Initiation Factor 4G1 (eIF4G1) at Ser1147 Is Specific for eIF4G1 Bound to eIF4E in Delayed Neuronal Death after Ischemia. International Journal of Molecular Sciences. 2022; 23(3):1830. https://doi.org/10.3390/ijms23031830
Chicago/Turabian StyleMartínez-Alonso, Emma, Natalia Guerra-Pérez, Alejandro Escobar-Peso, Lorena Peracho, Rocío Vera-Lechuga, Antonio Cruz-Culebras, Jaime Masjuan, and Alberto Alcázar. 2022. "Phosphorylation of Eukaryotic Initiation Factor 4G1 (eIF4G1) at Ser1147 Is Specific for eIF4G1 Bound to eIF4E in Delayed Neuronal Death after Ischemia" International Journal of Molecular Sciences 23, no. 3: 1830. https://doi.org/10.3390/ijms23031830
APA StyleMartínez-Alonso, E., Guerra-Pérez, N., Escobar-Peso, A., Peracho, L., Vera-Lechuga, R., Cruz-Culebras, A., Masjuan, J., & Alcázar, A. (2022). Phosphorylation of Eukaryotic Initiation Factor 4G1 (eIF4G1) at Ser1147 Is Specific for eIF4G1 Bound to eIF4E in Delayed Neuronal Death after Ischemia. International Journal of Molecular Sciences, 23(3), 1830. https://doi.org/10.3390/ijms23031830