
Exploring the Structural Rearrangements of the Human Insulin-Degrading Enzyme through Molecular Dynamics Simulations


Abstract
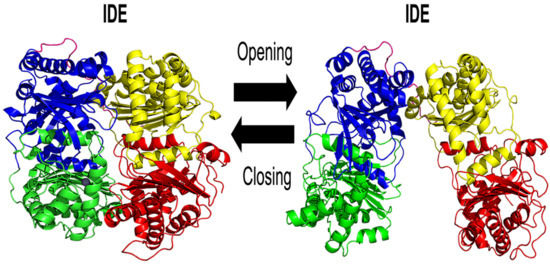
1. Introduction
2. Results and Discussion
2.1. MD Simulations Analysis
2.1.1. Root Mean Square Deviation (RMSD) Evaluation of IDE Structure
2.1.2. IDE Cavity Volume and Hydration Analysis
2.1.3. Root Mean Square Fluctuation (RMSF) of IDE Structure
2.1.4. IDE Hinge Dynamics Analysis
2.1.5. Gibbs Free Energy Landscape Analysis
2.1.6. Non-Covalent Interactions
3. Materials and Methods
3.1. Protein Modeling
3.2. Molecular Dynamics Simulations
3.3. Gibbs Free Energy Landscapes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rawlings, N.D.; Barrett, A.J. Homologues of insulinase, a new superfamily of metalloendopeptidases. Biochem. J. 1991, 275, 389–391. [Google Scholar] [CrossRef] [PubMed]
- Pivovarova, O.; Höhn, A.; Grune, T.; Pfeiffer, A.F.H.; Rudovich, N. Insulin-degrading enzyme: New therapeutic target for diabetes and Alzheimer’s disease? Ann. Med. 2016, 48, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Hulse, R.E.; Ralat, L.A.; Wei-Jen, T. Chapter 22 Structure, Function, and Regulation of Insulin-Degrading Enzyme. In Vitamins & Hormones; Elsevier: Amsterdam, The Netherlands, 2009; pp. 635–648. [Google Scholar]
- Tundo, G.R.; Sbardella, D.; Ciaccio, C.; Bianculli, A.; Orlandi, A.; Desimio, M.G.; Arcuri, G.; Coletta, M.; Marini, S. Insulin-degrading enzyme (IDE): A novel heat shock-like protein. J. Biol. Chem. 2013, 288, 2281–2289. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Gamba, A.; Leal, M.; Morelli, L.; Castano, E. Insulin-Degrading Enzyme: Structure-Function Relationship and its Possible Roles in Health and Disease. Curr. Pharm. Des. 2009, 15, 3644–3655. [Google Scholar] [CrossRef]
- Tang, W.-J. Targeting Insulin-Degrading Enzyme to Treat Type 2 Diabetes Mellitus. Trends Endocrinol. Metab. 2016, 27, 24–34. [Google Scholar] [CrossRef]
- González-Casimiro, C.; Merino, B.; Casanueva-Álvarez, E.; Postigo-Casado, T.; Cámara-Torres, P.; Fernández-Díaz, C.; Leissring, M.; Cózar-Castellano, I.; Perdomo, G. Modulation of Insulin Sensitivity by Insulin-Degrading Enzyme. Biomedicines 2021, 9, 86. [Google Scholar] [CrossRef]
- Duckworth, W.C.; Bennett, R.G.; Hamel, F.G. Insulin Degradation: Progress and Potential. Endocr. Rev. 1998, 19, 608–624. [Google Scholar] [CrossRef]
- Farris, W.; Mansourian, S.; Leissring, M.A.; Eckman, E.A.; Bertram, L.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J. Partial Loss-of-Function Mutations in In-sulin-Degrading Enzyme that Induce Diabetes also Impair Degradation of Amyloid β-Protein. Am. J. Pathol. 2004, 164, 1425–1434. [Google Scholar] [CrossRef]
- Kurochkin, I.V.; Guarnera, E.; Berezovsky, I.N. Insulin-Degrading Enzyme in the Fight against Alzheimer’s Disease. Trends Pharmacol. Sci. 2018, 39, 49–58. [Google Scholar] [CrossRef]
- Zhao, L. Insulin-Degrading Enzyme as a Downstream Target of Insulin Receptor Signaling Cascade: Implications for Alz-heimer’s Disease Intervention. J. Neurosci. 2004, 24, 11120–11126. [Google Scholar] [CrossRef]
- Nalivaeva, N.N.; Belyaev, N.D.; Kerridge, C.; Turner, A.J. Amyloid-clearing proteins and their epigenetic regulation as a thera-peutic target in Alzheimer’s disease. Front. Aging Neurosci. 2014, 6, 235. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wu, J.; Zhu, L.; Sha, L.; Yang, S.; Wei, J.; Ji, L.; Tang, X.; Mao, K.; Cao, L.; et al. Insulin degrading enzyme contributes to the pathology in a mixed model of Type 2 diabetes and Alzheimer’s disease: Possible mechanisms of IDE in T2D and AD. Biosci. Rep. 2018, 38, bsr20170862. [Google Scholar] [CrossRef] [PubMed]
- Carrasquillo, M.M.; Belbin, O.; Zou, F.; Allen, M.; Ertekin-Taner, N.; Ansari, M.; Wilcox, S.L.; Kashino, M.R.; Ma, L.; Younkin, L.H.; et al. Concordant Association of Insulin Degrading Enzyme Gene (IDE) Variants with IDE mRNA, Aß, and Alzheimer’s Disease. PLoS ONE 2010, 5, e8764. [Google Scholar] [CrossRef] [PubMed]
- Edland, S.D. Insulin-Degrading Enzyme, Apolipoprotein E, and Alzheimer’s Disease. J. Mol. Neurosci. 2004, 23, 213–218. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, D.; Huang, H.; Zhao, Y.; Zhou, H. Characteristics of insulin-degrading enzyme in alzheimer’s disease: A meta-analysis. Curr. Alzheimer Res. 2018, 15, 610–617. [Google Scholar] [CrossRef]
- Durham, T.B.; Toth, J.L.; Klimkowski, V.J.; Cao, J.X.; Siesky, A.M.; Alexander-Chacko, J.; Wu, G.Y.; Dixon, J.T.; McGee, J.E.; Wang, Y.; et al. Dual Exosite-binding Inhibitors of Insulin-degrading Enzyme Challenge Its Role as the Primary Mediator of Insulin Clearance in Vivo. J. Biol. Chem. 2015, 290, 20044–20059. [Google Scholar] [CrossRef]
- Maianti, J.P.; McFedries, A.; Foda, Z.H.; Kleiner, R.E.; Du, X.Q.; Leissring, M.A.; Tang, W.-J.; Charron, M.J.; Seeliger, M.A.; Saghatelian, A.; et al. Anti-diabetic activity of insulin-degrading enzyme inhibitors mediated by multiple hormones. Nature 2014, 511, 94–98. [Google Scholar] [CrossRef]
- Guo, Q.; Manolopoulou, M.; Bian, Y.; Schilling, A.B.; Tang, W.-J. Molecular Basis for the Recognition and Cleavages of IGF-II, TGF-α, and Amylin by Human Insulin-Degrading Enzyme. J. Mol. Biol. 2010, 395, 430–443. [Google Scholar] [CrossRef]
- Malito, E.; Ralat, L.A.; Manolopoulou, M.; Tsay, J.L.; Wadlington, N.; Tang, W.-J. Molecular Bases for the Recognition of Short Peptide Substrates and Cysteine-Directed Modifications of Human Insulin-Degrading Enzyme. Biochemistry 2008, 47, 12822–12834. [Google Scholar] [CrossRef]
- Manolopoulou, M.; Guo, Q.; Malito, E.; Schilling, A.B.; Tang, W.-J. Molecular Basis of Catalytic Chamber-assisted Unfolding and Cleavage of Human Insulin by Human Insulin-degrading Enzyme. J. Biol. Chem. 2009, 284, 14177–14188. [Google Scholar] [CrossRef]
- Ralat, L.A.; Guo, Q.; Ren, M.; Funke, T.; Dickey, D.M.; Potter, L.R.; Tang, W.-J. Insulin-degrading Enzyme Modulates the Natriuretic Peptide-mediated Signaling Response. J. Biol. Chem. 2011, 286, 4670–4679. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Guo, Q.; Guo, L.; Lenz, M.; Qian, F.; Koenen, R.R.; Xu, H.; Schilling, A.B.; Weber, C.; Ye, R.D.; et al. Polymerization of MIP-1 chemokine (CCL3 and CCL4) and clearance of MIP-1 by insulin-degrading enzyme. EMBO J. 2010, 29, 3952–3966. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Joachimiak, A.; Rosner, M.R.; Tang, W.-J. Structures of human insulin-degrading enzyme reveal a new substrate recognition mechanism. Nature 2006, 443, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Song, E.S.; Rodgers, D.W.; Hersh, L.B. A Monomeric Variant of Insulin Degrading Enzyme (IDE) Loses Its Regulatory Properties. PLoS ONE 2010, 5, e9719. [Google Scholar] [CrossRef]
- Perlman, R.K.; Gehm, B.D.; Kuo, W.L.; Rosner, M.R. Functional analysis of conserved residues in the active site of insulin-degrading enzyme. J. Biol. Chem. 1993, 268, 21538–21544. [Google Scholar] [CrossRef]
- Gehm, B.D.; Kuo, W.L.; Perlman, R.K.; Rosner, M.R. Mutations in a zinc-binding domain of human insulin-degrading enzyme eliminate catalytic activity but not insulin binding. J. Biol. Chem. 1993, 268, 7943–7948. [Google Scholar] [CrossRef]
- Zhang, Z.; Liang, W.G.; Bailey, L.J.; Tan, Y.Z.; Wei, H.; Wang, A.; Farcasanu, M.; A Woods, V.; A McCord, L.; Lee, D.; et al. Ensemble cryoEM elucidates the mechanism of insulin capture and degradation by human insulin degrading enzyme. eLife 2018, 7, e33572. [Google Scholar] [CrossRef]
- Leissring, M.A. Insulin-Degrading Enzyme: Paradoxes and Possibilities. Cells 2021, 10, 2445. [Google Scholar] [CrossRef]
- McCord, L.A.; Liang, W.G.; Dowdell, E.; Kalas, V.; Hoey, R.J.; Koide, A.; Koide, S.; Tang, W.-J. Conformational states and recognition of amyloidogenic peptides of human insulin-degrading enzyme. Proc. Natl. Acad. Sci. USA 2013, 110, 13827–13832. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- King, J.V.; Liang, W.G.; Scherpelz, K.P.; Schilling, A.B.; Meredith, S.C.; Tang, W.J. Molecular basis of substrate recognition and degradation by human presequence protease. Structure 2014, 22, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Song, E.S.; Daily, A.; Fried, M.G.; Juliano, M.A.; Juliano, L.; Hersh, L.B. Mutation of Active Site Residues of Insulin-degrading Enzyme Alters Allosteric Interactions. J. Biol. Chem. 2005, 280, 17701–17706. [Google Scholar] [CrossRef] [PubMed]
- Noinaj, N.; Bhasin, S.K.; Song, E.S.; Scoggin, K.E.; Juliano, M.A.; Juliano, L.; Hersh, L.B.; Rodgers, D.W. Identification of the Allosteric Regulatory Site of Insulysin. PLoS ONE 2011, 6, e20864. [Google Scholar] [CrossRef]
- da Cruz, C.H.B.; Seabra, G. Molecular Dynamics Simulations Reveal a Novel Mechanism for ATP Inhibition of Insulin Degrading Enzyme. J. Chem. Inf. Model. 2014, 54, 1380–1390. [Google Scholar] [CrossRef] [PubMed]
- A Structural View of Biology. Available online: http://www.rcsb.org/ (accessed on 30 May 2020).
- Im, H.; Manolopoulou, M.; Malito, E.; Shen, Y.; Zhao, J.; Neant-Fery, M.; Sun, C.-Y.; Meredith, S.C.; Sisodia, S.S.; Leissring, M.A.; et al. Structure of Substrate-free Human Insulin-degrading Enzyme (IDE) and Biophysical Analysis of ATP-induced Conformational Switch of IDE. J. Biol. Chem. 2007, 282, 25453–25463. [Google Scholar] [CrossRef]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.S.; Eramian, D.; Shen, M.; Pieper, U.; Sali, A. Comparative Protein Structure Modeling Using Modeller. Curr. Protoc. Bioinform. 2006, 15, 5–6. [Google Scholar] [CrossRef]
- Shen, M.-Y.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef]
- Bowie, J.U.; Lüthy, R.; Eisenberg, D. A Method to Identify Protein Sequences That Fold into a Known Three-Dimensional Structure. Science 1991, 253, 164–170. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical p K a Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, G.N.M.; De Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2016, 14, 71–73. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1998, 18, 1463–1472. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System; Version 2.0 Schrödinger; LLC: New York, NY, USA.
- Williams, T.; Kelley, C. Gnuplot 5.0: An Interactive Plotting Program. 2015. Available online: http://www.gnuplot.info/ (accessed on 30 May 2020).
- Paramo, T.; East, A.; Garzón, D.; Ulmschneider, M.B.; Bond, P.J. Efficient Characterization of Protein Cavities within Molecular Simulation Trajectories: Trj_cavity. J. Chem. Theory Comput. 2014, 10, 2151–2164. [Google Scholar] [CrossRef]
- Song, E.S.; Juliano, M.A.; Juliano, L.; Fried, M.G.; Wagner, S.L.; Hersh, L.B. ATP Effects on Insulin-degrading Enzyme Are Mediated Primarily through Its Triphosphate Moiety. J. Biol. Chem. 2004, 279, 54216–54220. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Domain 1 | Domain 4 | Non-Covalent Interaction Type | Occupancy (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Run 1 | Run 2 | Run 3 | Run 4 | Run 5 | Run 6 | Run 7 | |||
| D84 (sd) | K898 (sd) | SB | 31.7 | 73.2 | 39.7 | 35.8 | 92.0 | 76.3 | 37.4 |
| E133 (sd) | K884 (sd) | SB | 100.0 | 98.0 | 100.0 | 73.1 | 59.7 | 100.0 | 100.0 |
| E182 (sd) | R824 (sd) | SB | 57.6 | 100.0 | 86.5 | 26.9 | 0.0 | 26.9 | 88.5 |
| S132 (sd) | E817 (sd) | HB | 82.3 | 39.4 | 34.1 | 0.0 | 0.0 | 35.8 | 27.1 |
| K85 (sd) | D895 (sd) | SB | 90.8 | 80.0 | 60.3 | 43.3 | 35.1 | 52.0 | 100.0 |
| N184 (sd) | Q828 (bb) | HB | 70.6 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| N184 (sd) | Q828 (sd) | HB | NA | 0.0 | 0.0 | 0.0 | 0.0 | 11.0 | 0.0 |
| R181 (sd) | Q828 (sd) | HB | NA | 0.0 | 12.9 | 0.0 | 0.0 | 0.0 | 0.0 |
| N184 (sd) | Q828 (sd) | HB | NA | 14.9 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghoula, M.; Janel, N.; Camproux, A.-C.; Moroy, G. Exploring the Structural Rearrangements of the Human Insulin-Degrading Enzyme through Molecular Dynamics Simulations. Int. J. Mol. Sci. 2022, 23, 1746. https://doi.org/10.3390/ijms23031746
Ghoula M, Janel N, Camproux A-C, Moroy G. Exploring the Structural Rearrangements of the Human Insulin-Degrading Enzyme through Molecular Dynamics Simulations. International Journal of Molecular Sciences. 2022; 23(3):1746. https://doi.org/10.3390/ijms23031746
Chicago/Turabian StyleGhoula, Mariem, Nathalie Janel, Anne-Claude Camproux, and Gautier Moroy. 2022. "Exploring the Structural Rearrangements of the Human Insulin-Degrading Enzyme through Molecular Dynamics Simulations" International Journal of Molecular Sciences 23, no. 3: 1746. https://doi.org/10.3390/ijms23031746
APA StyleGhoula, M., Janel, N., Camproux, A.-C., & Moroy, G. (2022). Exploring the Structural Rearrangements of the Human Insulin-Degrading Enzyme through Molecular Dynamics Simulations. International Journal of Molecular Sciences, 23(3), 1746. https://doi.org/10.3390/ijms23031746