
Development of Planar Illumination Strategies for Solving Mysteries in the Sub-Cellular Realm


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
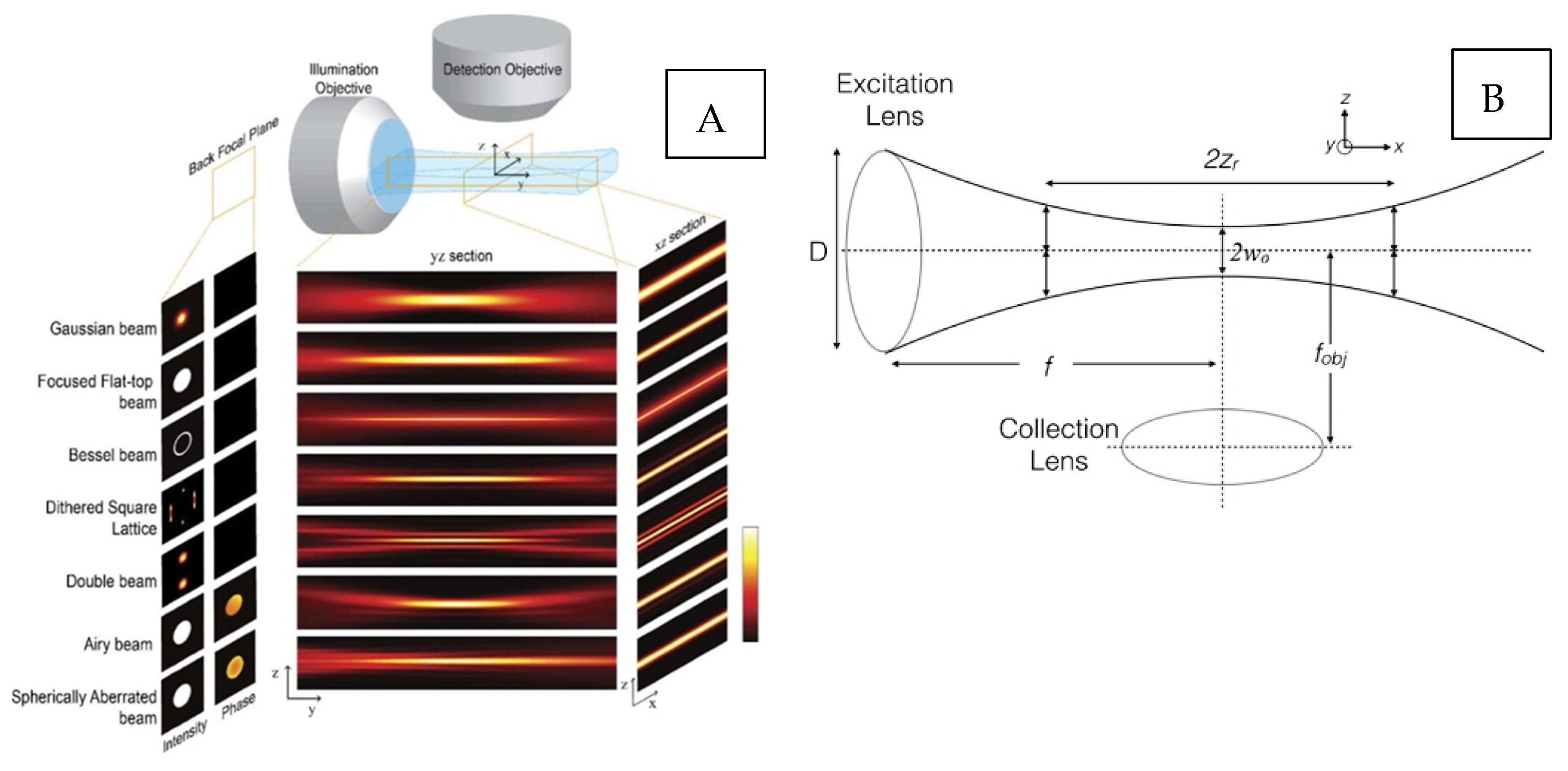
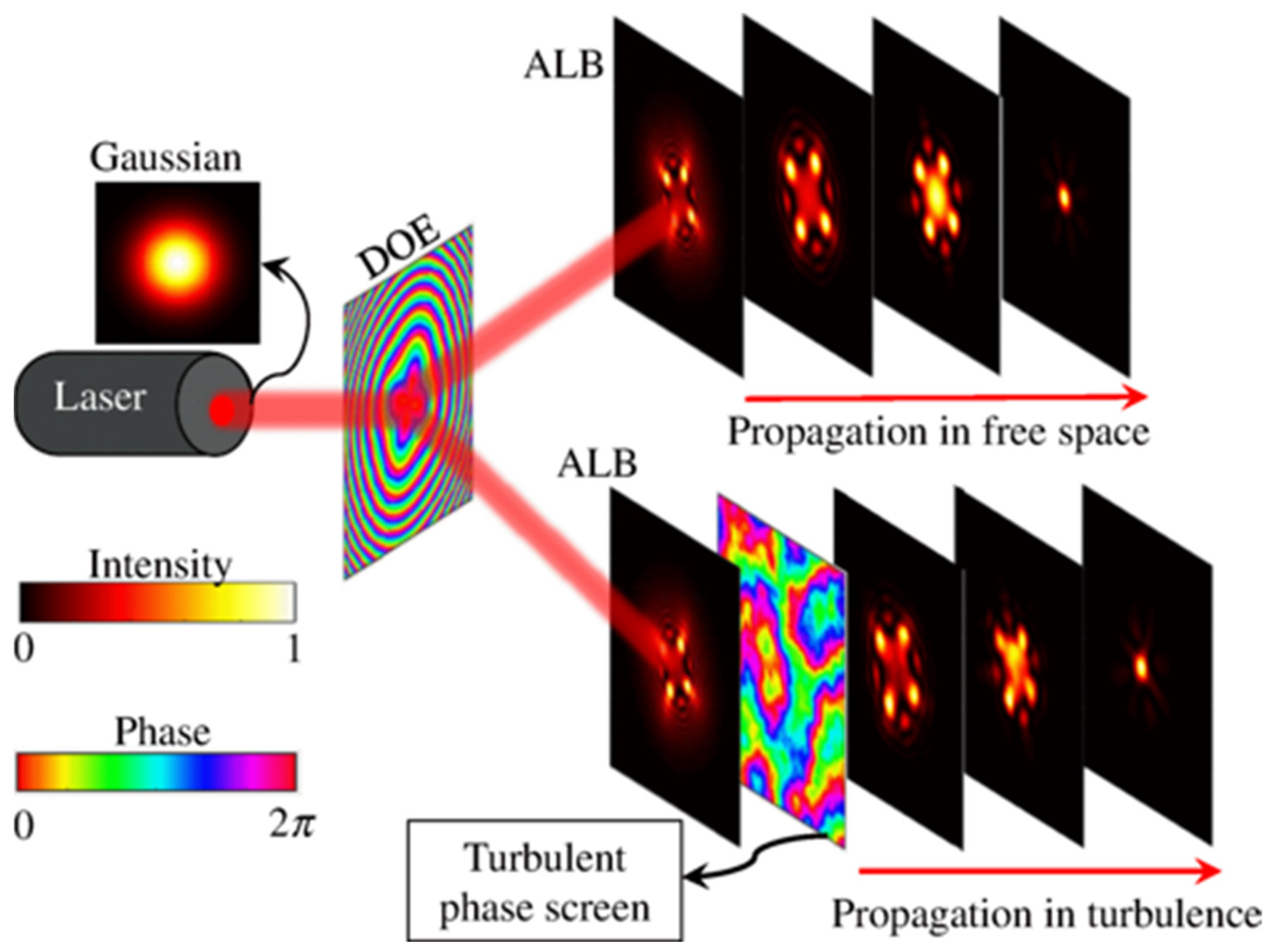
2. Light Sheet Modality Considerations
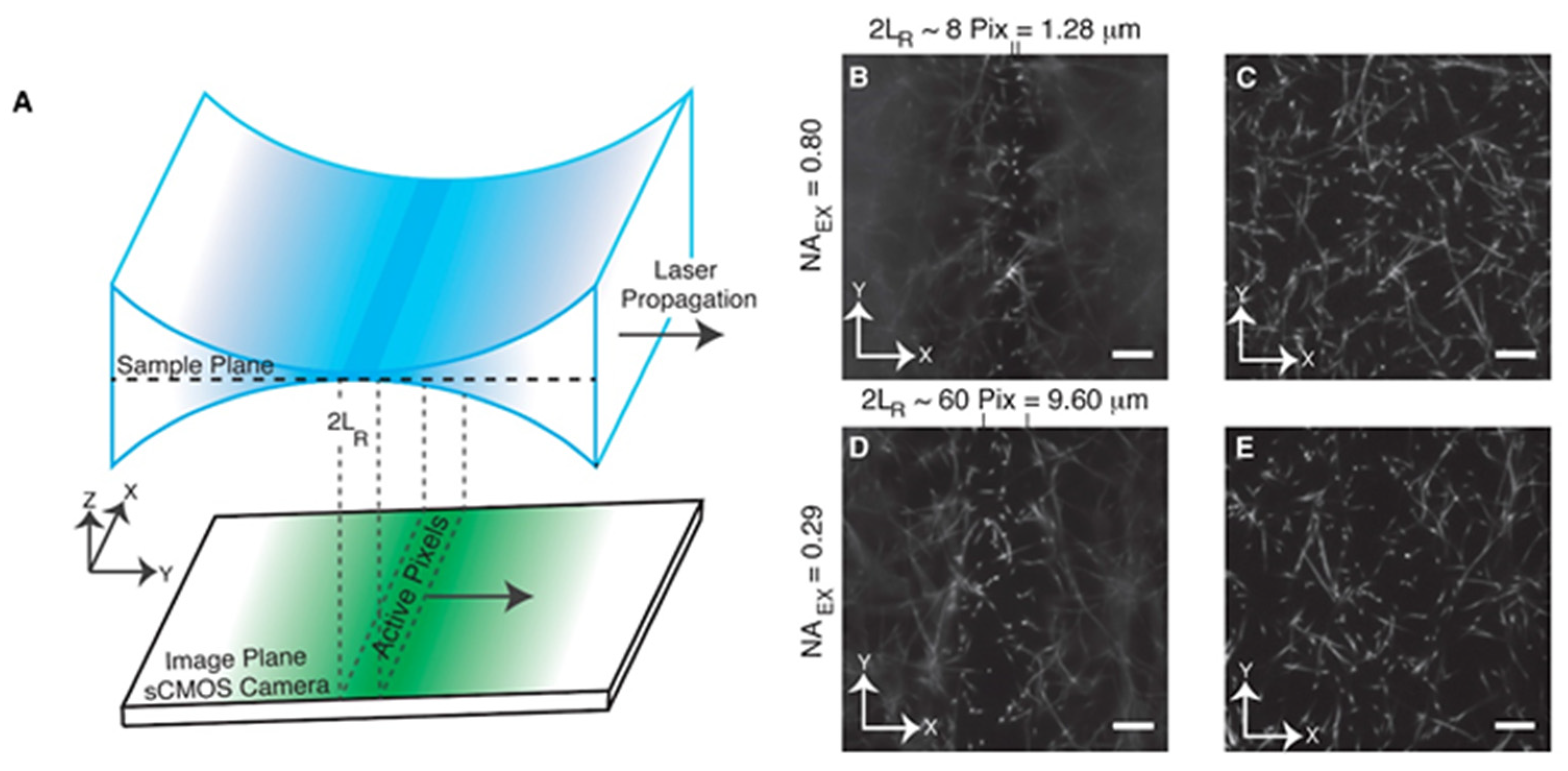
3. Axially Sweeping Light Sheet Microscopy
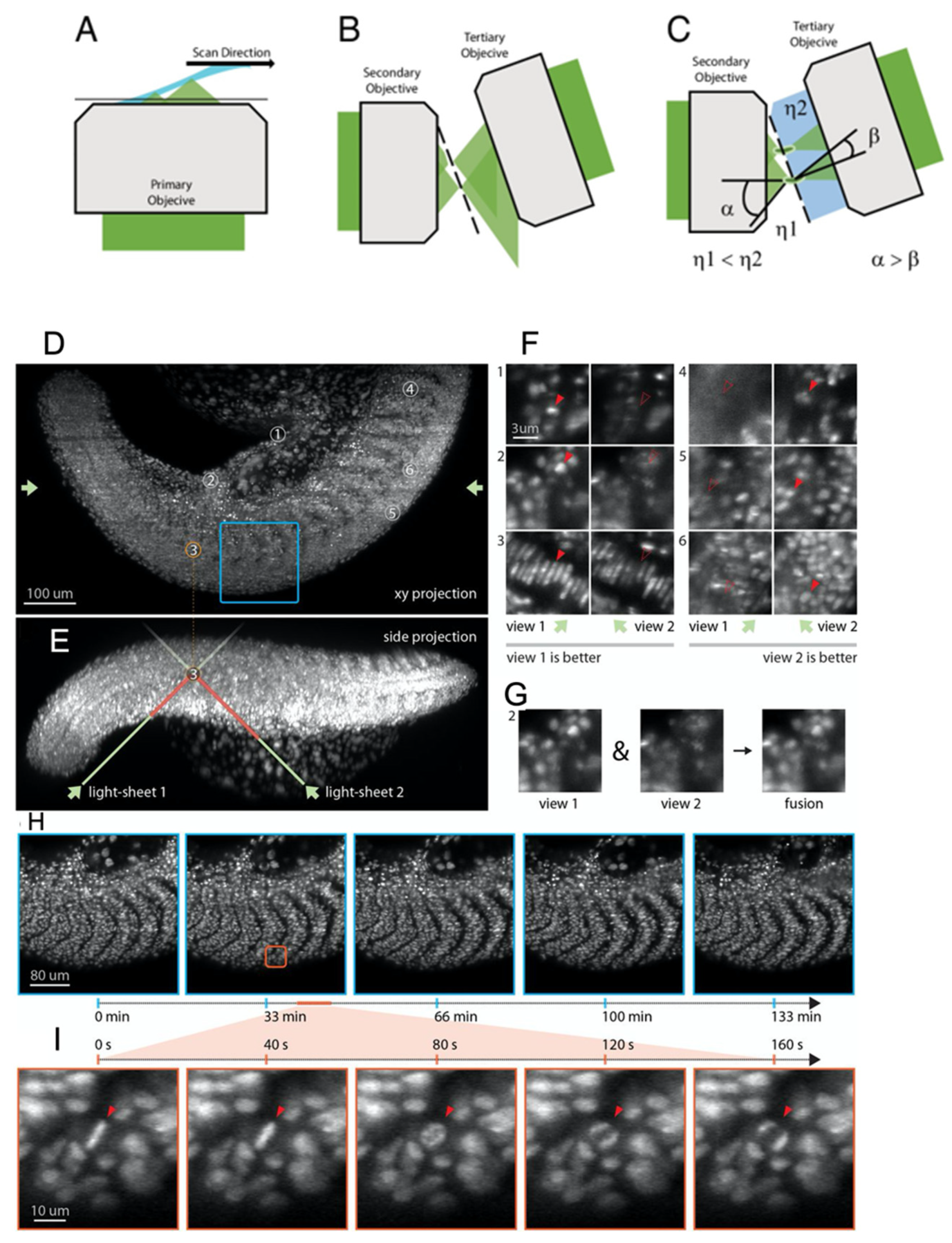
4. Oblique Plane Microscopy
5. Lattice Light Sheet Microscopy
6. Sub-Voxel-Resolving Light Sheet Microscopy
7. Single-Molecule-Localization Light Sheet Microscopy
8. Conclusions
Funding
Conflicts of Interest
References
- Agocs, E.; Attota, R.K. Enhancing optical microscopy illumination to enable quantitative imaging. Sci. Rep. 2018, 8, 4782. [Google Scholar] [CrossRef]
- Reigoto, A.M.; Andrade, S.A.; Seixas, M.C.R.R.; Costa, M.L.; Mermelstein, C. A comparative study on the use of microscopy in pharmacology and cell biology research. PLoS ONE 2021, 16, e0245795. [Google Scholar] [CrossRef]
- Karzbrun, E.; Kshirsagar, A.; Cohen, S.R.; Hanna, J.H.; Reiner, O. Human brain organoids on a chip reveal the physics of folding. Nat. Phys. 2018, 14, 515–522. [Google Scholar] [CrossRef]
- Rios, A.C.; Clevers, H. Imaging organoids: A bright future ahead. Nat. Methods 2018, 15, 24–26. [Google Scholar] [CrossRef]
- Figueroa, B.; Xu, F.X.; Hu, R.; Men, S.; Fu, D. Quantitative Imaging of Intracellular Density with Ratiometric Stimulated Raman Scattering Microscopy. bioRxiv 2021. [Google Scholar]
- Yang, W.; Carrillo-Reid, L.; Bando, Y.; Peterka, D.S.; Yuste, R. Simultaneous two-photon imaging and two-photon optogenetics of cortical circuits in three dimensions. ELife 2018, 7, e32671. [Google Scholar] [CrossRef]
- Joshi, J.; Rubart, M.; Zhu, W. Optogenetics: Background, methodological advances and potential applications for cardiovascular research and medicine. Front. Bioeng. Biotechnol. 2020, 7, 466. [Google Scholar] [CrossRef] [Green Version]
- Power, R.M.; Huisken, J. A guide to light-sheet fluorescence microscopy for multiscale imaging. Nat. Methods 2017, 14, 360–373. [Google Scholar] [CrossRef]
- Thorn, K. A quick guide to light microscopy in cell biology. Mol. Biol. Cell 2016, 27, 219–222. [Google Scholar] [CrossRef]
- Thorn, K. Genetically encoded fluorescent tags. Mol. Biol. Cell 2017, 28, 848–857. [Google Scholar] [CrossRef]
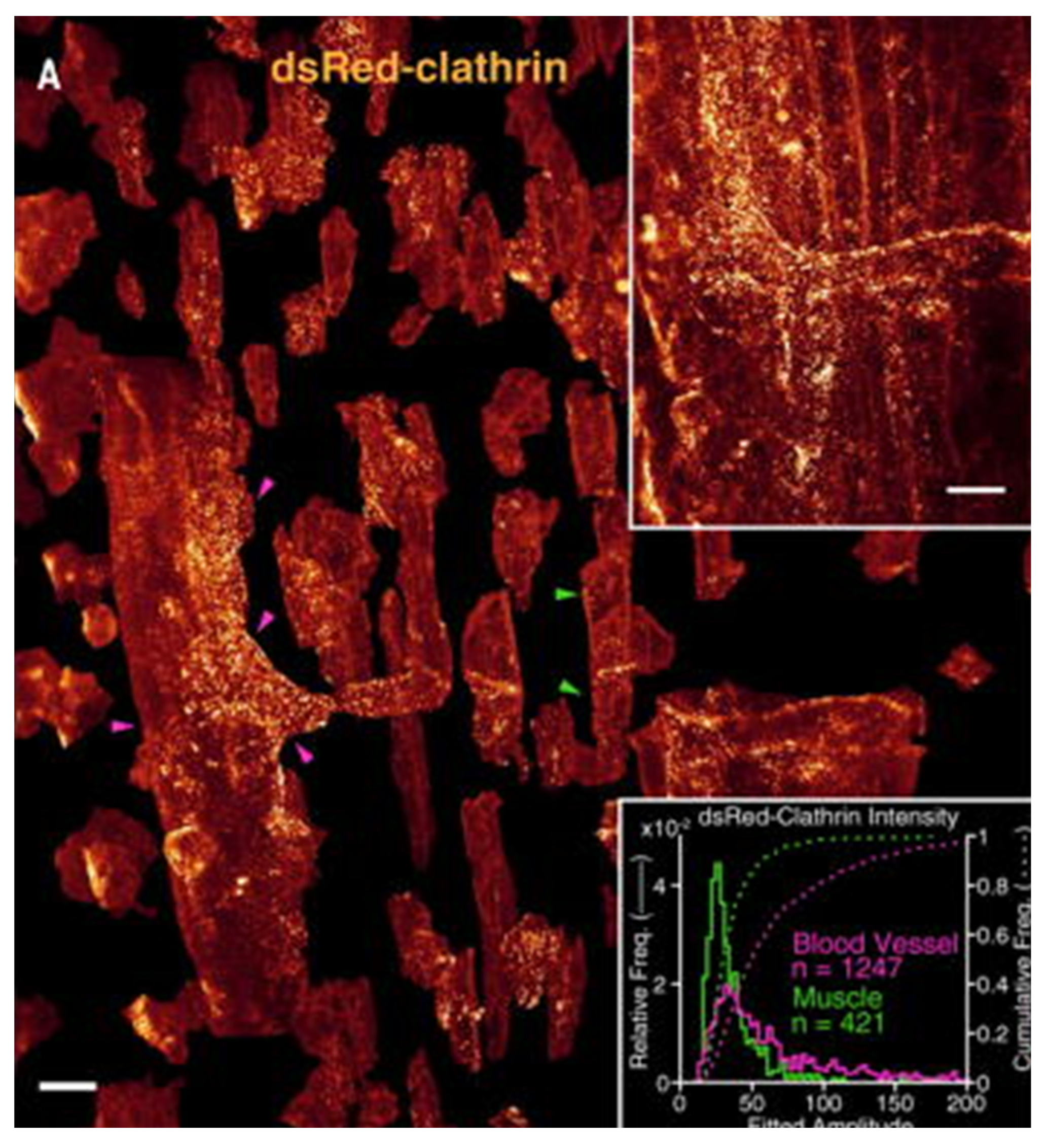
- Chakraborty, T.; Driscoll, M.K.; Jeffery, E.; Murphy, M.M.; Roudot, P.; Chang, B.-J.; Vora, S.; Wong, W.M.; Nielson, C.D.; Zhang, H.; et al. Light-sheet microscopy of cleared tissues with isotropic, subcellular resolution. Nat. Methods 2019, 16, 1109–1113. [Google Scholar] [CrossRef]
- Dean, K.M.; Roudot, P.; Reis, C.R.; Welf, E.S.; Mettlen, M.; Fiolka, R. Diagonally scanned light-sheet microscopy for fast volumetric imaging of adherent cells. Biophys. J. 2016, 110, 1456–1465. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Wojcik, M.; Wang, Y.; Moon, S.; Zin, E.A.; Marnani, N.; Newman, Z.L.; Flannery, J.G.; Xu, K.; Zhang, X. Oblique-plane single-molecule localization microscopy for tissues and small intact animals. Nat. Methods 2019, 16, 853–857. [Google Scholar] [CrossRef]
- Sapoznik, E.; Chang, B.-J.; Huh, J.; Ju, R.J.; Azarova, E.V.; Pohlkamp, T.; Welf, E.S.; Broadbent, D.; Carisey, A.F.; Stehbens, S.J.; et al. A versatile oblique plane microscope for large-scale and high-resolution imaging of subcellular dynamics. ELife 2020, 9, e57681. [Google Scholar] [CrossRef]
- Watkins, S.C.; St Croix, C.M. Light sheet imaging comes of age. J. Cell Biol. 2018, 217, 1567–1569. [Google Scholar] [CrossRef]
- Zaqout, S.; Becker, L.-L.; Kaindl, A.M. Immunofluorescence staining of paraffin sections step by step. Front. Neuroanat. 2020, 14, 83. [Google Scholar] [CrossRef]
- Li, N.; Zhao, R.; Sun, Y.; Ye, Z.; He, K.; Fang, X. Single-molecule imaging and tracking of molecular dynamics in living cells. Natl. Sci. Rev. 2017, 4, 739–760. [Google Scholar] [CrossRef] [Green Version]
- Kohl, J.; Ng, J.; Cachero, S.; Ciabatti, E.; Dolan, M.-J.; Sutcliffe, B.; Tozer, A.; Ruehle, S.; Krueger, D.; Frechter, S.; et al. Ultrafast tissue staining with chemical tags. Proc. Natl. Acad. Sci. USA 2014, 111, E3805. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Zhang, Z.-T.; Tang, S.-W.; Zhao, B.-S.; Li, H.; Song, J.-Z.; Li, D.; Xie, Z.; Glick, B.; Emr Scott, D. A validated set of fluorescent-protein-based markers for major organelles in yeast (saccharomyces cerevisiae). MBio 2019, 10, e01691-19. [Google Scholar] [CrossRef]
- Crivat, G.; Taraska, J.W. Imaging proteins inside cells with fluorescent tags. Trends Biotechnol. 2012, 30, 8–16. [Google Scholar] [CrossRef] [Green Version]
- Abramson, R.G.; Burton, K.R.; Yu, J.-P.J.; Scalzetti, E.M.; Yankeelov, T.E.; Rosenkrantz, A.B.; Mendiratta-Lala, M.; Bartholmai, B.J.; Ganeshan, D.; Lenchik, L.; et al. Methods and challenges in quantitative imaging biomarker development. Acad. Radiol. 2015, 22, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Meseguer, E.; Barberá-Tomás, D.; Benito-Amat, C.; Díaz-Faes, A.A.; Martí-Bonmatí, L. What do biomarkers add: Mapping quantitative imaging biomarkers research. Eur. J. Radiol. 2022, 146, 110052. [Google Scholar] [CrossRef]
- Waterhouse, D.J.; Fitzpatrick, C.R.M.; Pogue, B.W.; O’Connor, J.P.B.; Bohndiek, S.E. A roadmap for the clinical implementation of optical-imaging biomarkers. Nat. Biomed. Eng. 2019, 3, 339–353. [Google Scholar] [CrossRef]
- Selleck, M.J.; Senthil, M.; Wall, N.R. Making meaningful clinical use of biomarkers. Biomark. Insights 2017, 12, 1177271917715236. [Google Scholar] [CrossRef] [Green Version]
- Carvajal-Hausdorf, D.E.; Schalper, K.A.; Neumeister, V.M.; Rimm, D.L. Quantitative measurement of cancer tissue biomarkers in the lab and in the clinic. Lab. Investig. 2015, 95, 385–396. [Google Scholar] [CrossRef] [Green Version]
- Tandon, I.; Johns, S.; Woessner, A.; Perez, J.; Cross, D.; Ozkizilcik, A.; Muldoon, T.J.; Vallurupalli, S.; Padala, M.; Quinn, K.P.; et al. Label-free optical biomarkers detect early calcific aortic valve disease in a wild-type mouse model. BMC Cardiovasc. Disord. 2020, 20, 521. [Google Scholar] [CrossRef]
- Zhou, H.; Nguyen, L.; Arnesano, C.; Ando, Y.; Raval, M.; Rodgers, J.T.; Fraser, S.; Lu, R.; Shen, K. Non-invasive optical biomarkers distinguish and track the metabolic status of single hematopoietic stem cells. iScience 2020, 23, 100831. [Google Scholar] [CrossRef] [Green Version]
- Jun, Y.W.; Cho, S.W.; Jung, J.; Huh, Y.; Kim, Y.; Kim, D.; Ahn, K.H. Frontiers in probing alzheimer’s disease biomarkers with fluorescent small molecules. ACS Cent. Sci. 2019, 5, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Reshma, V.; Varsha, B.K.; Rakesh, P.; Radhika, M.B.; Soumya, M.; D’Mello, S. Aggrandizing oral submucous fibrosis grading using an adjunct special stain: A pilot study. J. Oral Maxillofac. Pathol. 2016, 20, 36–46. [Google Scholar] [CrossRef] [Green Version]
- Lahiani, A.; Klaiman, E.; Grimm, O. Enabling histopathological annotations on immunofluorescent images through virtualization of hematoxylin and eosin. J. Pathol. Inform. 2018, 9, 1. [Google Scholar] [CrossRef]
- Azevedo Tosta, T.A.; Neves, L.A.; do Nascimento, M.Z. Segmentation methods of H&E-stained histological images of lymphoma: A review. Inform. Med. Unlocked 2017, 9, 35–43. [Google Scholar] [CrossRef]
- Chlipala, E.; Bendzinski, C.M.; Chu, K.; Johnson, J.I.; Brous, M.; Copeland, K.; Bolon, B. Optical density-based image analysis method for the evaluation of hematoxylin and eosin staining precision. J. Histotechnol. 2020, 43, 29–37. [Google Scholar] [CrossRef]
- Kleczek, P.; Jaworek-Korjakowska, J.; Gorgon, M. A novel method for tissue segmentation in high-resolution h&e-stained histopathological whole-slide images. Comput. Med. Imaging Graph. 2020, 79, 101686. [Google Scholar] [CrossRef] [PubMed]
- Hinton, J.P.; Dvorak, K.; Roberts, E.; French, W.J.; Grubbs, J.C.; Cress, A.E.; Tiwari, H.A.; Nagle, R.B. A method to reuse archived H&E stained histology slides for a multiplex protein biomarker analysis. Methods Protoc. 2019, 2, 86. [Google Scholar] [CrossRef] [Green Version]
- Boyd, A.; Cain, O.; Chauhan, A.; Webb, G.J. Medical liver biopsy: Background, indications, procedure and histopathology. Frontline Gastroenterol. 2020, 11, 40. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.; Kwok, S.J.J.; Yun, S.H. In vivo fluorescence microscopy: Lessons from observing cell behavior in their native environment. Physiology 2015, 30, 40–49. [Google Scholar] [CrossRef] [Green Version]
- Dean, K.M.; Palmer, A.E. Advances in fluorescence labeling strategies for dynamic cellular imaging. Nat. Chem. Biol. 2014, 10, 512–523. [Google Scholar] [CrossRef]
- Djuric, U.; Zadeh, G.; Aldape, K.; Diamandis, P. Precision histology: How deep learning is poised to revitalize histomorphology for personalized cancer care. NPJ Precis. Oncol. 2017, 1, 22. [Google Scholar] [CrossRef]
- Cui, M.; Zhang, D.Y. Artificial intelligence and computational pathology. Lab. Investig. 2021, 101, 412–422. [Google Scholar] [CrossRef]
- Azam, A.S.; Miligy, I.M.; Kimani, P.K.-U.; Maqbool, H.; Hewitt, K.; Rajpoot, N.M.; Snead, D.R.J. Diagnostic concordance and discordance in digital pathology: A systematic review and meta-analysis. J. Clin. Pathol. 2021, 74, 448. [Google Scholar] [CrossRef]
- Taqi, S.A.; Sami, S.A.; Sami, L.B.; Zaki, S.A. A review of artifacts in histopathology. J. Oral Maxillofac. Pathol. 2018, 22, 279. [Google Scholar] [CrossRef]
- Bindhu, P.; Krishnapillai, R.; Thomas, P.; Jayanthi, P. Facts in artifacts. J. Oral Maxillofac. Pathol. 2013, 17, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Hui, H.; Zhang, Y.; Tong, W.; Tian, F.; Yang, X.; Liu, J.; Chen, Y.; Tian, J. Deep learning for virtual histological staining of bright-field microscopic images of unlabeled carotid artery tissue. Mol. Imaging Biol. 2020, 22, 1301–1309. [Google Scholar] [CrossRef]
- Kuru, K. Optimization and enhancement of H&E stained microscopical images by applying bilinear interpolation method on lab color mode. Theor. Biol. Med. Model. 2014, 11, 9. [Google Scholar] [CrossRef] [Green Version]
- Rivenson, Y.; de Haan, K.; Wallace, W.D.; Ozcan, A. Emerging advances to transform histopathology using virtual staining. BME Front. 2020, 2020, 9647163. [Google Scholar] [CrossRef]
- Treuting, P.M.; Snyder, J.M.; Ikeno, Y.; Schofield, P.N.; Ward, J.M.; Sundberg, J.P. The vital role of pathology in improving reproducibility and translational relevance of aging studies in rodents. Vet. Pathol. 2016, 53, 244–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husain, S.A.; King, K.L.; Batal, I.; Dube, G.K.; Hall, I.E.; Brennan, C.; Stokes, M.B.; Crew, R.J.; Carpenter, D.; Alvarado Verduzco, H.; et al. Reproducibility of deceased donor kidney procurement biopsies. Clin. J. Am. Soc. Nephrol. 2020, 15, 257. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.M.; Schofield, P.N.; Sundberg, J.P. Reproducibility of histopathological findings in experimental pathology of the mouse: A sorry tail. Lab. Anim. 2017, 46, 146–151. [Google Scholar] [CrossRef] [Green Version]
- Lauber, D.T.; Fülöp, A.; Kovács, T.; Szigeti, K.; Máthé, D.; Szijártó, A. State of the art in vivo imaging techniques for laboratory animals. Lab. Anim. 2017, 51, 465–478. [Google Scholar] [CrossRef] [Green Version]
- Dunst, S.; Tomancak, P. Imaging flies by fluorescence microscopy: Principles, technologies, and applications. Genetics 2019, 211, 15–34. [Google Scholar] [CrossRef] [Green Version]
- Vedula, V.; Lee, J.; Xu, H.; Kuo, C.-C.J.; Hsiai, T.K.; Marsden, A.L. A method to quantify mechanobiologic forces during zebrafish cardiac development using 4-d light sheet imaging and computational modeling. PLoS Comput. Biol. 2017, 13, e1005828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertz, J. Optical sectioning microscopy with planar or structured illumination. Nat. Methods 2011, 8, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.J. Comparison of widefield/deconvolution and confocal microscopy for three-dimensional imaging. In Handbook of Biological Confocal Microscopy; Pawley, J.B., Ed.; Springer: Boston, MA, USA, 2006; pp. 453–467. ISBN 978-0-387-45524-2. [Google Scholar]
- Myers, K.A.; Janetopoulos, C. Recent advances in imaging subcellular processes. F1000Research 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Schermelleh, L.; Heintzmann, R.; Leonhardt, H. A guide to super-resolution fluorescence microscopy. J. Cell Biol. 2010, 190, 165–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, R.S.; Wu, Y.; Kanchanawong, P.; Shroff, H.; Waterman, C.M. Microscopy in 3D: A biologist’s toolbox. Trends Cell Biol. 2011, 21, 682–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huszka, G.; Gijs, M.A.M. Super-resolution optical imaging: A comparison. Micro Nano Eng. 2019, 2, 7–28. [Google Scholar] [CrossRef]
- Vangindertael, J.; Camacho, R.; Sempels, W.; Mizuno, H.; Dedecker, P.; Janssen, K.P.F. An introduction to optical super-resolution microscopy for the adventurous biologist. Methods Appl. Fluoresc. 2018, 6, 022003. [Google Scholar] [CrossRef]
- Wolenski, J.S.; Julich, D. Fluorescence microscopy gets faster and clearer: Roles of photochemistry and selective illumination. Yale J. Biol. Med. 2014, 87, 21–32. [Google Scholar]
- Teranikar, T.; Messerschmidt, V.; Lim, J.; Bailey, Z.; Chiao, J.-C.; Cao, H.; Liu, J.; Lee, J. Correcting anisotropic intensity in light sheet images using dehazing and image morphology. APL Bioeng. 2020, 4, 036103. [Google Scholar] [CrossRef]
- Teranikar, T.; Villarreal, C.; Salehin, N.; Lim, J.; Ijaseun, T.; Cao, H.; Chuong, C.; Lee, J. Feature detection to segment cardiomyocyte nuclei for investigating cardiac contractility. bioRxiv 2021. [Google Scholar] [CrossRef]
- Ding, Y.; Lee, J.; Ma, J.; Sung, K.; Yokota, T.; Singh, N.; Dooraghi, M.; Abiri, P.; Wang, Y.; Kulkarni, R.P.; et al. Light-sheet fluorescence imaging to localize cardiac lineage and protein distribution. Sci. Rep. 2017, 7, 42209. [Google Scholar] [CrossRef] [PubMed]
- Ponsetto, J.L.; Bezryadina, A.; Wei, F.; Onishi, K.; Shen, H.; Huang, E.; Ferrari, L.; Ma, Q.; Zou, Y.; Liu, Z. Experimental demonstration of localized plasmonic structured illumination microscopy. ACS Nano 2017, 11, 5344–5350. [Google Scholar] [CrossRef] [PubMed]
- Richter, V.; Piper, M.; Wagner, M.; Schneckenburger, H. Increasing resolution in live cell microscopy by structured illumination (SIM). Appl. Sci. 2019, 9, 1188. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Shroff, H. Faster, sharper, and deeper: Structured illumination microscopy for biological imaging. Nat. Methods 2018, 15, 1011–1019. [Google Scholar] [CrossRef]
- Gohn-Kreuz, C.; Rohrbach, A. Light needles in scattering media using self-reconstructing beams and the STED principle. Optica 2017, 4, 1134–1142. [Google Scholar] [CrossRef]
- Spahn, C.; Grimm, J.B.; Lavis, L.D.; Lampe, M.; Heilemann, M. Whole-cell, 3D, and multicolor STED imaging with exchangeable fluorophores. Nano Lett. 2019, 19, 500–505. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Dong, P.; Chen, X.; Hsieh, T.-H.S.; Banala, S.; De Marzio, M.; English, B.P.; Qi, Y.; Jung, S.K.; Kieffer-Kwon, K.-R.; et al. 3D ATAC-PALM: Super-resolution imaging of the accessible genome. Nat. Methods 2020, 17, 430–436. [Google Scholar] [CrossRef]
- Wäldchen, F.; Schlegel, J.; Götz, R.; Luciano, M.; Schnermann, M.; Doose, S.; Sauer, M. Whole-cell imaging of plasma membrane receptors by 3D lattice light-sheet DSTORM. Nat. Commun. 2020, 11, 887. [Google Scholar] [CrossRef]
- Xu, H.; Tong, Z.; Ye, Q.; Sun, T.; Hong, Z.; Zhang, L.; Bortnick, A.; Cho, S.; Beuzer, P.; Axelrod, J.; et al. Molecular organization of mammalian meiotic chromosome axis revealed by expansion STORM microscopy. Proc. Natl. Acad. Sci. USA 2019, 116, 18423–18428. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Ma, H.; Liu, Y. Stochastic optical reconstruction microscopy (STORM). Curr. Protoc. Cytom. 2017, 81, 12–46. [Google Scholar] [CrossRef]
- Lambert, T.J.; Waters, J.C. Navigating challenges in the application of superresolution microscopy. J. Cell Biol. 2016, 216, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zhou, J.; Wang, L.; Wang, M.; Wu, W.; Chen, J.; Qu, J.; Gao, B.Z.; Shao, Y. Current challenges and solutions of super-resolution structured illumination microscopy. APL Photonics 2021, 6, 020901. [Google Scholar] [CrossRef]
- Möckl, L.; Moerner, W.E. Super-resolution microscopy with single molecules in biology and beyond–essentials, current trends, and future challenges. J. Am. Chem. Soc. 2020, 142, 17828–17844. [Google Scholar] [CrossRef]
- Bishop, K.W.; Glaser, A.K.; Liu, J.T.C. Performance tradeoffs for single- and dual-objective open-top light-sheet microscope designs: A simulation-based analysis. Biomed. Opt. Express 2020, 11, 4627–4650. [Google Scholar] [CrossRef] [PubMed]
- Tosheva, K.L.; Yuan, Y.; Matos Pereira, P.; Culley, S.; Henriques, R. Between life and death: Strategies to reduce phototoxicity in super-resolution microscopy. J. Phys. D Appl. Phys. 2020, 53, 163001. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.-C.; Legant, W.R.; Wang, K.; Shao, L.; Milkie, D.E.; Davidson, M.W.; Janetopoulos, C.; Wu, X.S.; Hammer, J.A., 3rd; Liu, Z.; et al. Lattice light-sheet microscopy: Imaging molecules to embryos at high spatiotemporal resolution. Science 2014, 346, 1257998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, B.-J.; Dean, K.M.; Fiolka, R. Systematic and quantitative comparison of lattice and gaussian light-sheets. Opt. Express 2020, 28, 27052–27077. [Google Scholar] [CrossRef]
- Santi, P.A. Light sheet fluorescence microscopy: A review. J. Histochem. Cytochem. 2011, 59, 129–138. [Google Scholar] [CrossRef] [Green Version]
- Gustavsson, A.-K.; Petrov, P.N.; Lee, M.Y.; Shechtman, Y.; Moerner, W.E. Tilted light sheet microscopy with 3D point spread functions for single-molecule super-resolution imaging in mammalian cells. Int. Soc. Opt. Photonics 2018, 10500, 105000M. [Google Scholar] [CrossRef]
- Fei, P.; Nie, J.; Lee, J.; Ding, Y.; Li, S.; Yu, Z.; Zhang, H.; Hagiwara, M.; Yu, T.; Segura, T.; et al. Sub-voxel light-sheet microscopy for high-resolution, high-throughput volumetric imaging of large biomedical specimens. bioRxiv 2018. [Google Scholar] [CrossRef] [Green Version]
- Olarte, O.E.; Andilla, J.; Gualda, E.J.; Loza-Alvarez, P. Light-sheet microscopy: A tutorial. Adv. Opt. Photon. 2018, 10, 111–179. [Google Scholar] [CrossRef]
- Remacha, E.; Friedrich, L.; Vermot, J.; Fahrbach, F.O. How to define and optimize axial resolution in light-sheet microscopy: A simulation-based approach. Biomed. Opt. Express 2019, 11, 8–26. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.; Saghafi, S.; Pende, M.; Sabdyusheva-Litschauer, I.; Hahn, C.M.; Foroughipour, M.; Jährling, N.; Dodt, H.-U. Deconvolution of light sheet microscopy recordings. Sci. Rep. 2019, 9, 17625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huisken, J.; Swoger, J.; Del Bene, F.; Wittbrodt, J.; Stelzer, E.H. Optical sectioning deep inside live embryos by selective plane illumination microscopy. Science 2004, 305, 1007–1009. [Google Scholar] [CrossRef] [Green Version]
- Dean, K.M.; Roudot, P.; Welf, E.S.; Danuser, G.; Fiolka, R. Deconvolution-free subcellular imaging with axially swept light sheet microscopy. Biophys. J. 2015, 108, 2807–2815. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Chandris, P.; Winter, P.W.; Kim, E.Y.; Jaumouillé, V.; Kumar, A.; Guo, M.; Leung, J.M.; Smith, C.; Rey-Suarez, I.; et al. Simultaneous multiview capture and fusion improves spatial resolution in wide-field and light-sheet microscopy. Optica 2016, 3, 897–910. [Google Scholar] [CrossRef]
- Yu, T.; Zhu, J.; Li, D.; Zhu, D. Physical and chemical mechanisms of tissue optical clearing. iScience 2021, 24, 102178. [Google Scholar] [CrossRef]
- Jing, D.; Zhang, S.; Luo, W.; Gao, X.; Men, Y.; Ma, C.; Liu, X.; Yi, Y.; Bugde, A.; Zhou, B.O.; et al. Tissue Clearing of both hard and soft tissue organs with the PEGASOS method. Cell Res. 2018, 28, 803–818. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Chen, M.; Zhu, L.; Wu, J.Y.; Du, S.; Li, Y. Measure and model a 3-D space-variant psf for fluorescence microscopy image deblurring. Opt. Express 2018, 26, 14375–14391. [Google Scholar] [CrossRef]
- Chakraborty, T.; Driscoll, M.; Murphy, M.; Roudot, P.; Chang, B.-J.; Vora, S.; Wong, W.M.; Nielson, C.; Zhang, H.; Zhemkov, V.; et al. Light-sheet microscopy with isotropic, sub-micron resolution and solvent-independent large-scale imaging. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Landry, J.; Hamann, S.; Solgaard, O. High-speed axially swept light sheet microscopy using a linear MEMS phased array for isotropic resolution. J. Biomed. Opt. 2020, 25, 106504. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, A.-K.; Petrov, P.N.; Lee, M.Y.; Shechtman, Y.; Moerner, W.E. 3D single-molecule super-resolution microscopy with a tilted light sheet. Nat. Commun. 2018, 9, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Lange, M.; Millett-Sikking, A.; Solak, A.C.; Kumar, S.V.; Wang, W.; Kobayashi, H.; McCarroll, M.N.; Whitehead, L.W.; Fiolka, R.P.; et al. High-resolution, large imaging volume, and multi-view single objective light-sheet microscopy. bioRxiv 2021. [Google Scholar] [CrossRef]
- Kumar, M.; Kozorovitskiy, Y. Tilt (in)variant lateral scan in oblique plane microscopy: A geometrical optics approach. Biomed. Opt. Express 2020, 11, 3346–3359. [Google Scholar] [CrossRef] [PubMed]
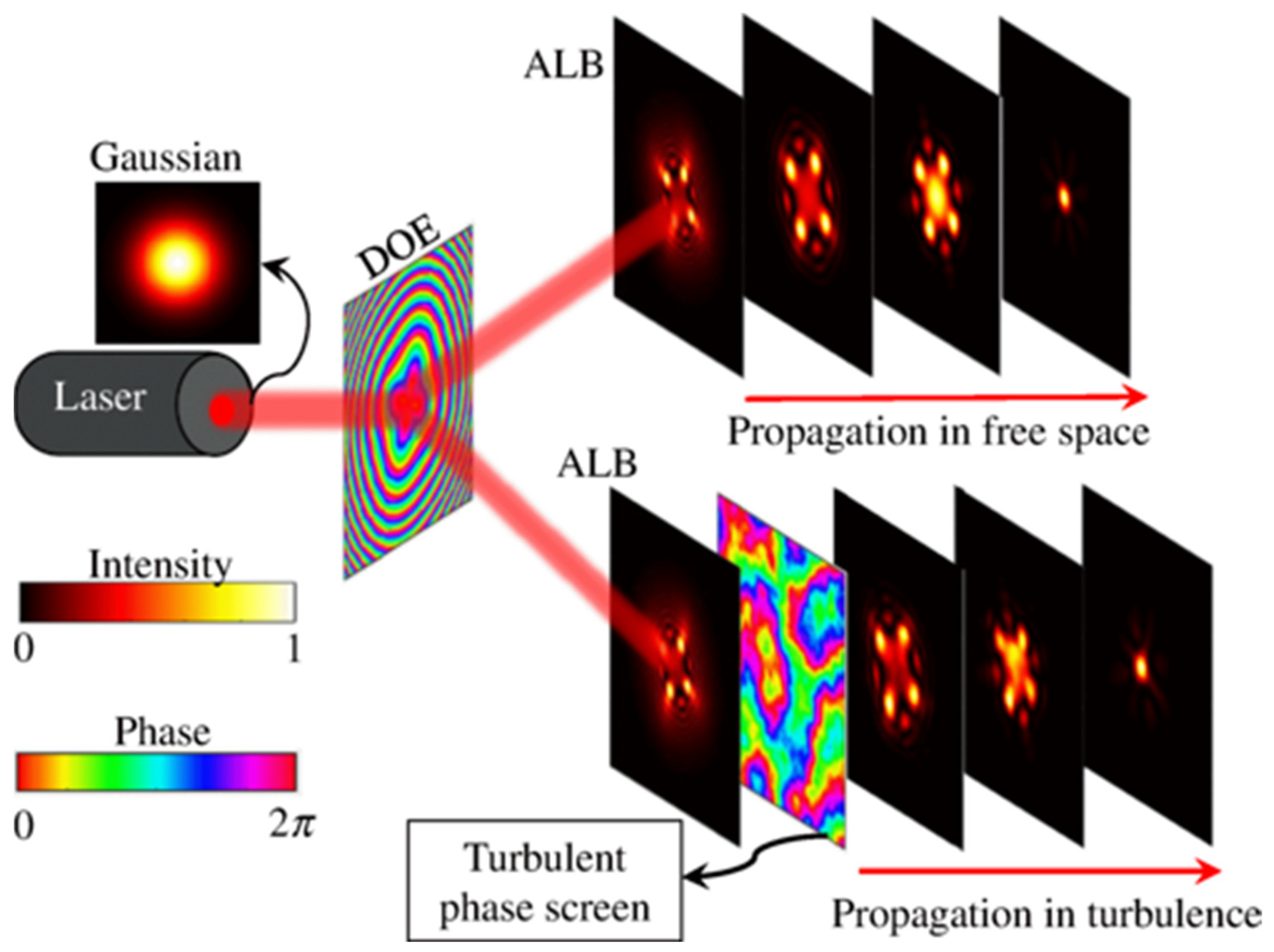
- Dev, V.; Reddy, A.N.K.; Ustinov, A.V.; Khonina, S.N.; Pal, V. Autofocusing and self-healing properties of aberration laser beams in a turbulent media. Phys. Rev. Appl. 2021, 16, 014061. [Google Scholar] [CrossRef]
- Mimori-Kiyosue, Y. Imaging mitotic processes in three dimensions with lattice light-sheet microscopy. Chromosome Res. 2021, 29, 37–50. [Google Scholar] [CrossRef]
- Liu, T.-L.; Upadhyayula, S.; Milkie, D.E.; Singh, V.; Wang, K.; Swinburne, I.A.; Mosaliganti, K.R.; Collins, Z.M.; Hiscock, T.W.; Shea, J.; et al. Observing the cell in its native state: Imaging subcellular dynamics in multicellular organisms. Science 2018, 360, eaaq1392. [Google Scholar] [CrossRef] [Green Version]
- Planchon, T.A.; Gao, L.; Milkie, D.E.; Davidson, M.W.; Galbraith, J.A.; Galbraith, C.G.; Betzig, E. Rapid three-dimensional isotropic imaging of living cells using bessel beam plane illumination. Nat. Methods 2011, 8, 417–423. [Google Scholar] [CrossRef] [Green Version]
- Gao, R.; Asano, S.M.; Upadhyayula, S.; Pisarev, I.; Milkie, D.E.; Liu, T.-L.; Singh, V.; Graves, A.; Huynh, G.H.; Zhao, Y.; et al. Cortical column and whole-brain imaging with molecular contrast and nanoscale resolution. Science 2019, 363, eaau8302. [Google Scholar] [CrossRef]
- Chen, F.; Tillberg, P.W.; Boyden, E.S. Optical imaging. expansion microscopy. Science 2015, 347, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Kunz, T.C.; Götz, R.; Gao, S.; Sauer, M.; Kozjak-Pavlovic, V. Using expansion microscopy to visualize and characterize the morphology of mitochondrial cristae. Front. Cell Dev. Biol. 2020, 8, 617. [Google Scholar] [CrossRef] [PubMed]
- Parra-Damas, A.; Saura, C.A. Tissue clearing and expansion methods for imaging brain pathology in neurodegeneration: From circuits to synapses and beyond. Front. Neurosci. 2020, 14, 914. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Liu, S.; Yu, T.; Li, Y.; Ping, J.; Peng, W.; Zhao, F.; Huang, Y.; Mei, W.; Zeng, S.; et al. Fast, 3D Isotropic imaging of whole mouse brain using multiangle-resolved subvoxel SPIM. Adv. Sci. 2020, 7, 1901891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, F.; Yang, Y.; Li, Y.; Jiang, H.; Xie, X.; Yu, T.; Liu, Q.; Zhang, H.; Jia, H.; Liu, S.; et al. Efficient and cost-effective 3D cellular imaging by sub-voxel-resolving light-sheet add-on microscopy. J. Biophotonics 2020, 13, e201960243. [Google Scholar] [CrossRef] [PubMed]
- Guan, Z.; Lee, J.; Jiang, H.; Dong, S.; Jen, N.; Hsiai, T.; Ho, C.-M.; Fei, P. Compact plane illumination plugin device to enable light sheet fluorescence imaging of multi-cellular organisms on an inverted wide-field microscope. Biomed. Opt. Express 2016, 7, 194–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.S.; Zimmerley, M.; Li, Y.; Watters, R.; Cang, H. Single-molecule Super-resolution Light-sheet Microscopy. Chem. Phys. Chem. 2014, 15, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.-H.; Tang, W.-C.; Liu, Y.-T.; Chang, S.-W.; Wu, F.C.M.; Chen, C.-Y.; Tsai, Y.-C.; Yang, S.-M.; Kuo, C.-W.; Okada, Y.; et al. Lightsheet localization microscopy enables fast, large-scale, and Three-Dimensional super-resolution imaging. Commun. Biol. 2019, 2, 177. [Google Scholar] [CrossRef]
- Cella Zanacchi, F.; Lavagnino, Z.; Perrone Donnorso, M.; Del Bue, A.; Furia, L.; Faretta, M.; Diaspro, A. Live-cell 3D super-resolution imaging in thick biological samples. Nat. Methods 2011, 8, 1047–1049. [Google Scholar] [CrossRef]
- Tokunaga, M.; Imamoto, N.; Sakata-Sogawa, K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat. Methods 2008, 5, 159–161. [Google Scholar] [CrossRef]
- Konopka, C.; Bednarek, S. Variable-angle epifluorescence microscopy: A new way to look at protein dynamics in the plant cell cortex. Plant J. Cell Mol. Biol. 2008, 53, 186–196. [Google Scholar] [CrossRef]
- Gebhardt, J.C.M.; Suter, D.M.; Roy, R.; Zhao, Z.W.; Chapman, A.R.; Basu, S.; Maniatis, T.; Xie, X.S. Single-molecule imaging of transcription factor binding to DNA in live mammalian cells. Nat. Methods 2013, 10, 421–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.S.; Zhu, Q.; Elkins, K.; Tse, K.; Li, Y.; Fitzpatrick, J.A.J.; Verma, I.M.; Cang, H. Light-sheet bayesian microscopy enables deep-cell super-resolution imaging of heterochromatin in live human embryonic stem cells. Opt. Nanoscopy 2013, 2, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teranikar, T.; Lim, J.; Ijaseun, T.; Lee, J. Development of Planar Illumination Strategies for Solving Mysteries in the Sub-Cellular Realm. Int. J. Mol. Sci. 2022, 23, 1643. https://doi.org/10.3390/ijms23031643
Teranikar T, Lim J, Ijaseun T, Lee J. Development of Planar Illumination Strategies for Solving Mysteries in the Sub-Cellular Realm. International Journal of Molecular Sciences. 2022; 23(3):1643. https://doi.org/10.3390/ijms23031643
Chicago/Turabian StyleTeranikar, Tanveer, Jessica Lim, Toluwani Ijaseun, and Juhyun Lee. 2022. "Development of Planar Illumination Strategies for Solving Mysteries in the Sub-Cellular Realm" International Journal of Molecular Sciences 23, no. 3: 1643. https://doi.org/10.3390/ijms23031643
APA StyleTeranikar, T., Lim, J., Ijaseun, T., & Lee, J. (2022). Development of Planar Illumination Strategies for Solving Mysteries in the Sub-Cellular Realm. International Journal of Molecular Sciences, 23(3), 1643. https://doi.org/10.3390/ijms23031643