
De Novo Assembly and Annotation of the Vaginal Metatranscriptome Associated with Bacterial Vaginosis



Abstract
:1. Introduction
2. Results
2.1. Summary of RNA Sequencing Data Associated with BV for Transcriptome Analyses
2.2. De Novo Assembly of Vaginal Transcriptome
2.3. Identification of Ribosomal RNAs and Transfer RNAs from Vaginal Transcriptome
2.4. Prediction of Open Reading Frames (ORFs)
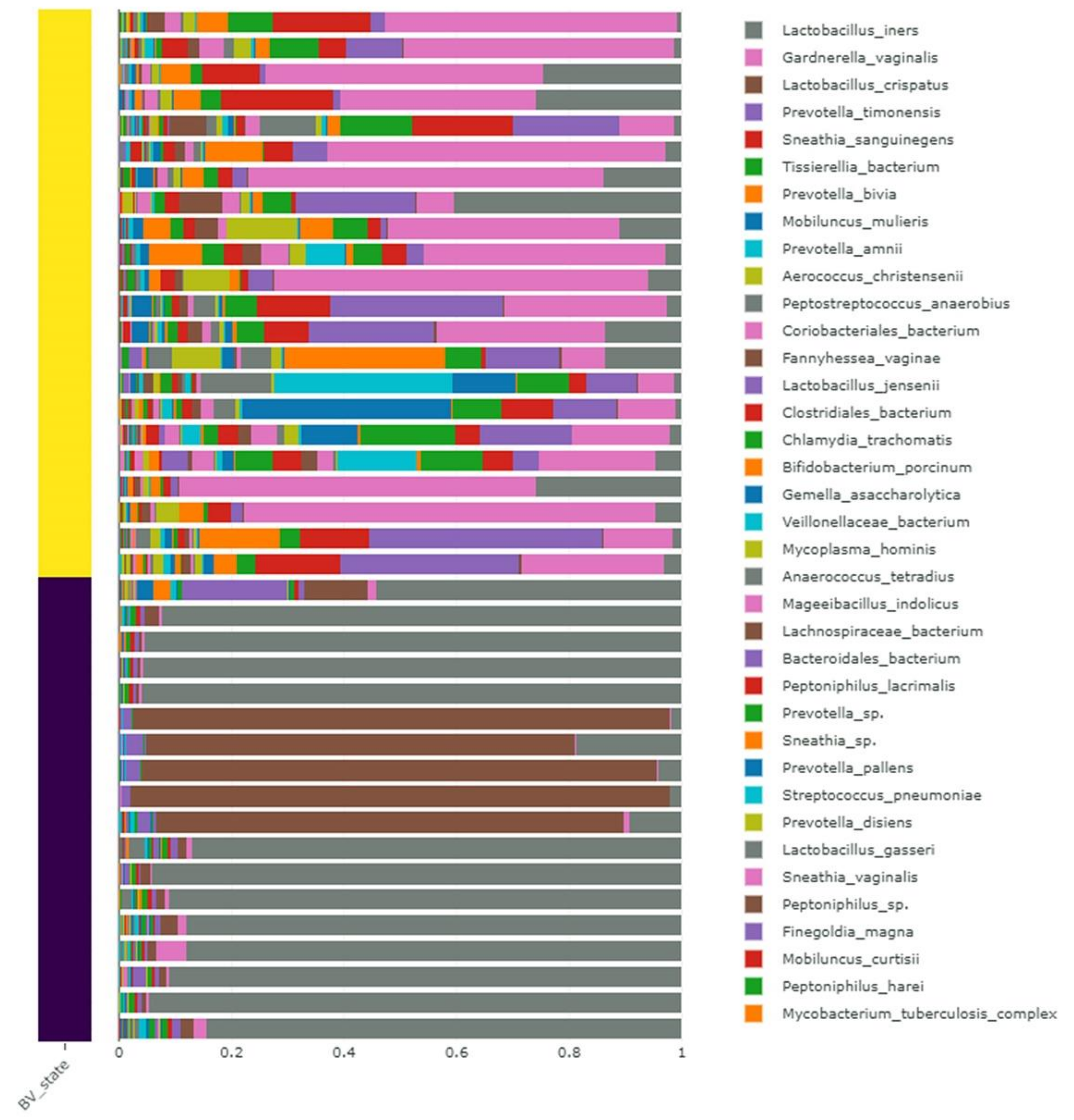
2.5. Taxonomical Classification of Identified Microorganisms
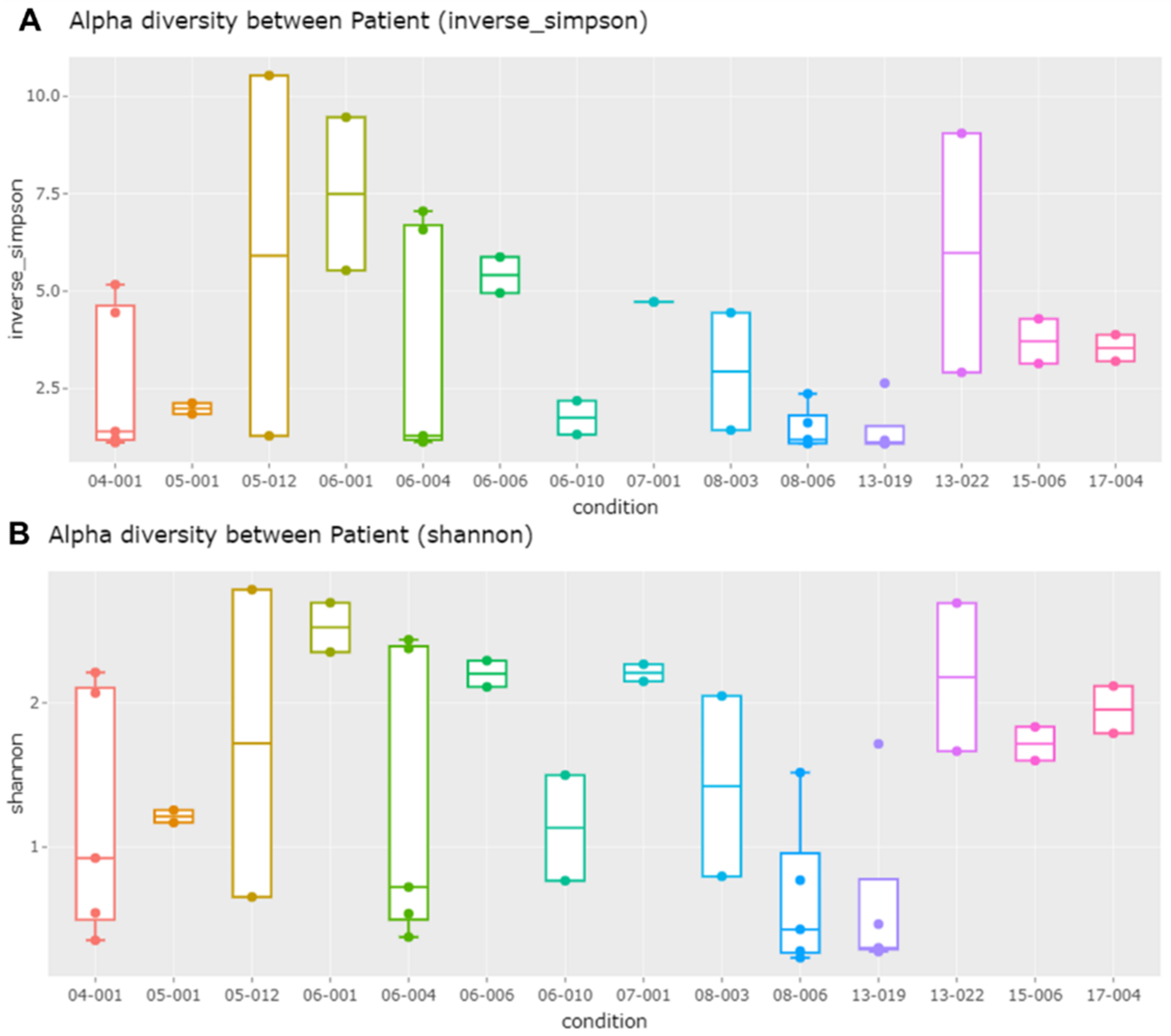
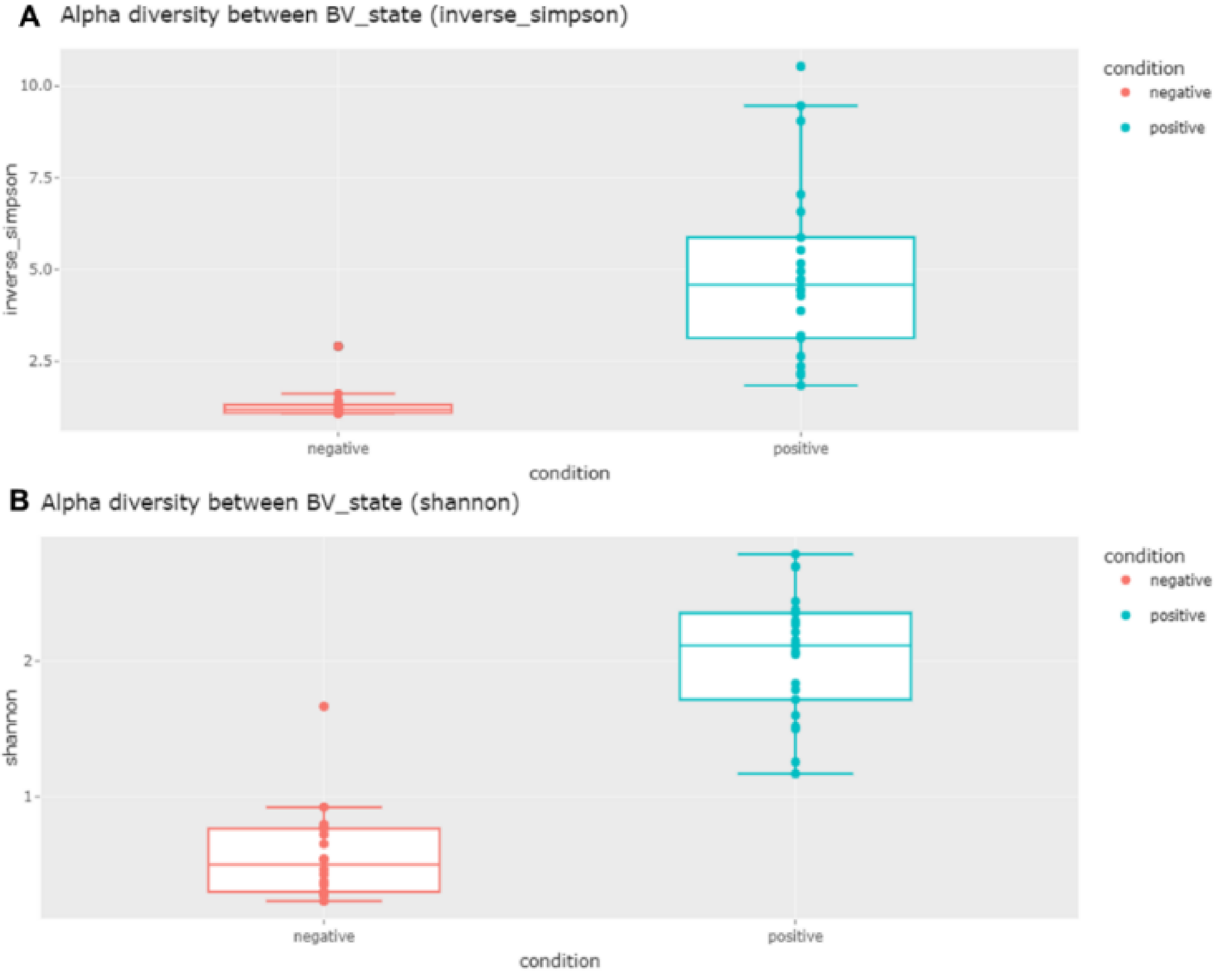
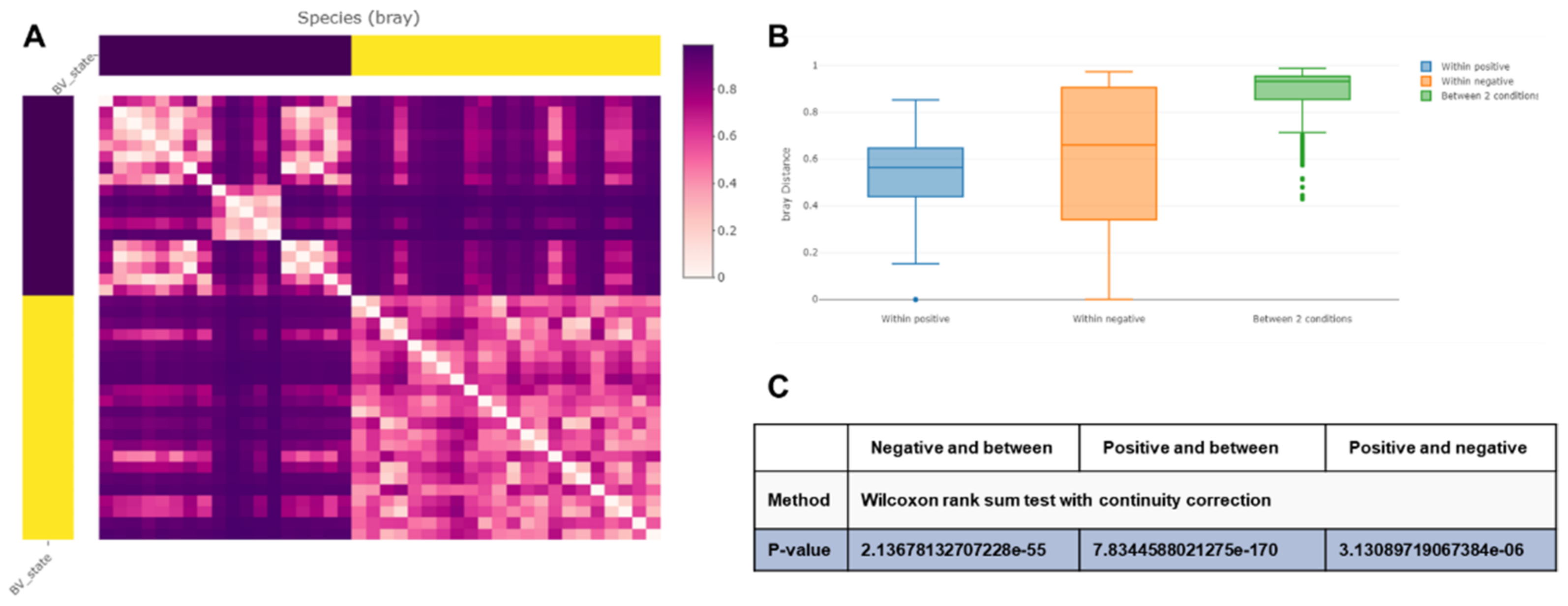
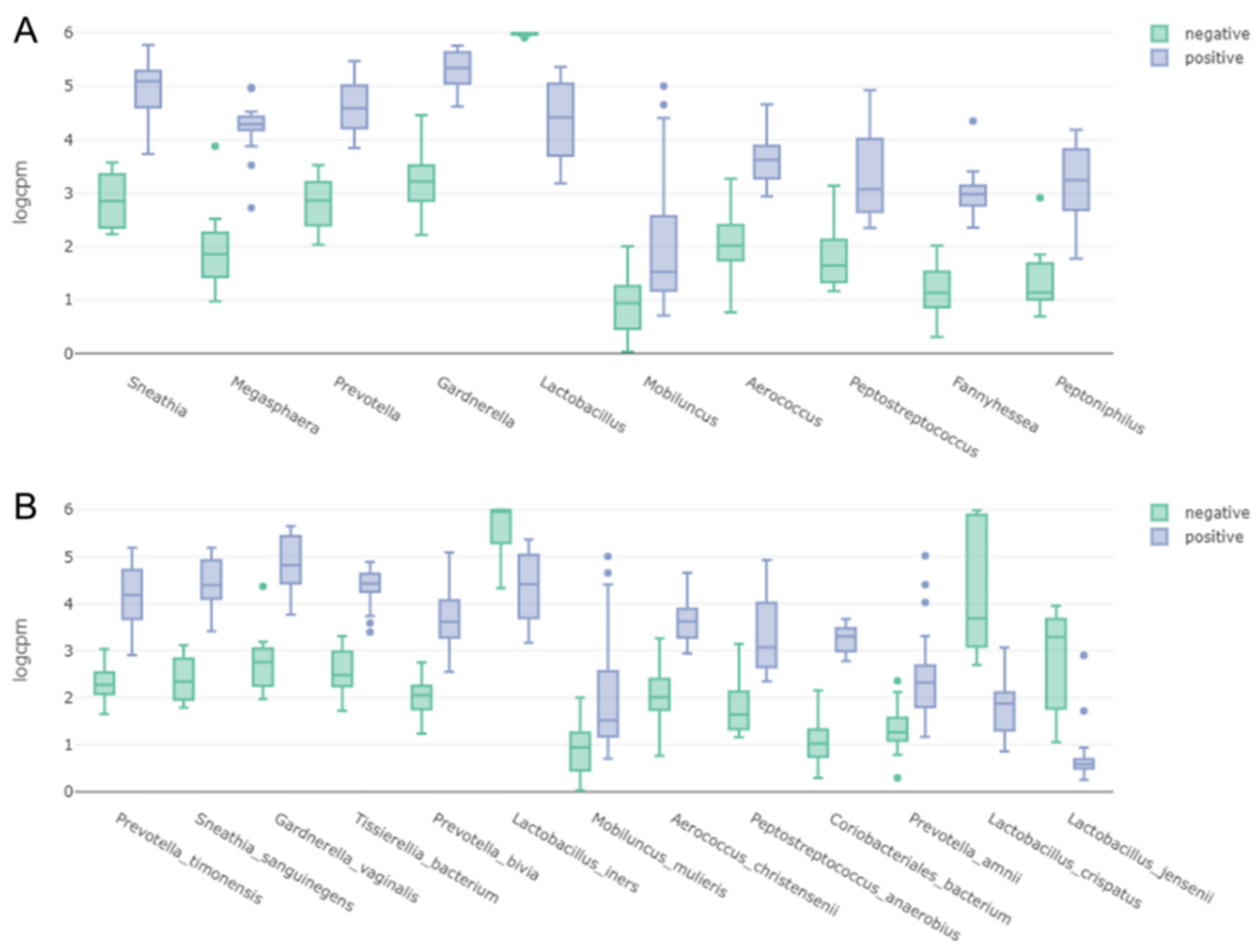
2.6. Alpha and Beta Diversity of Identified Microorganisms in Different Subjects and According to BV State
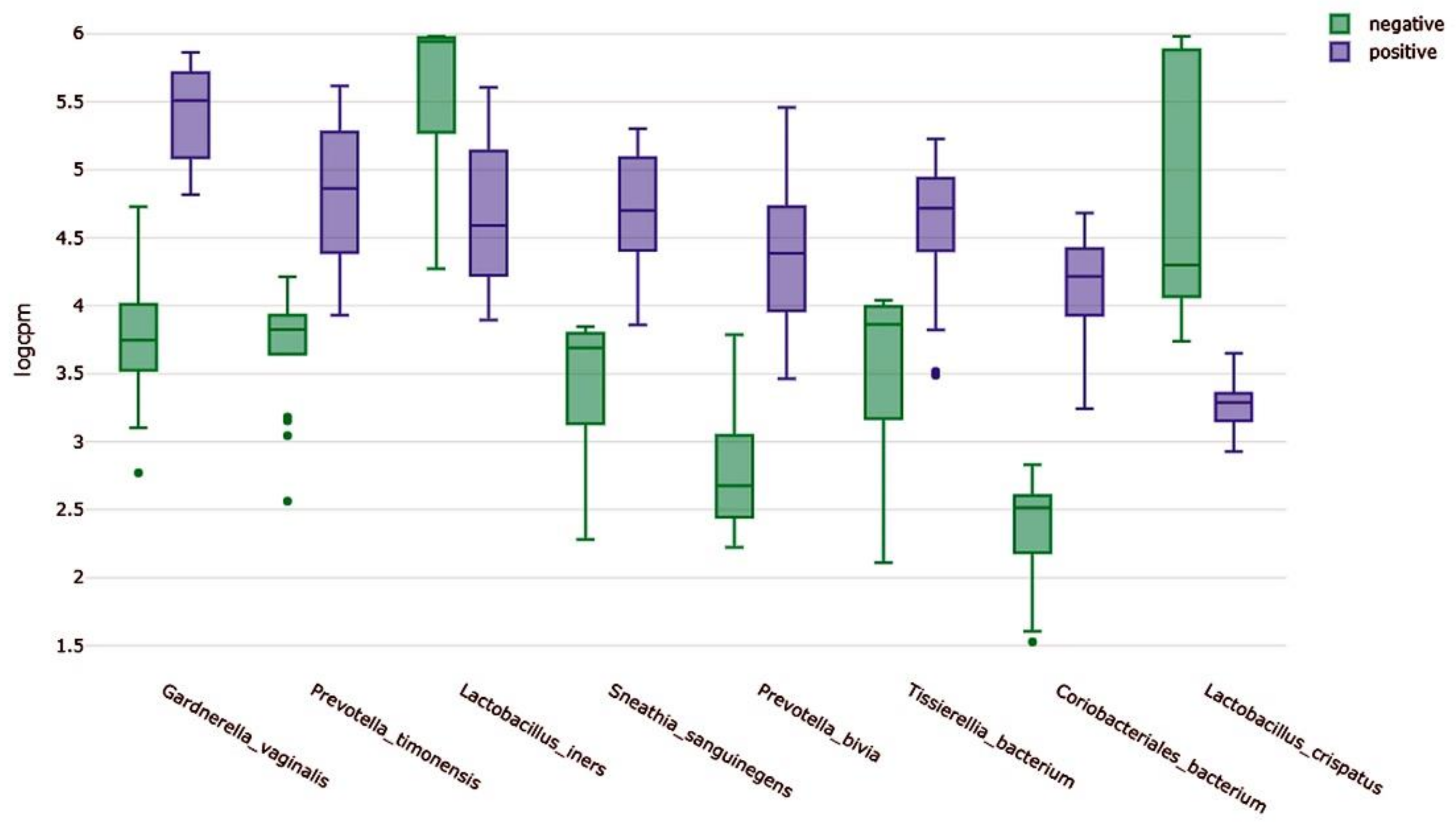
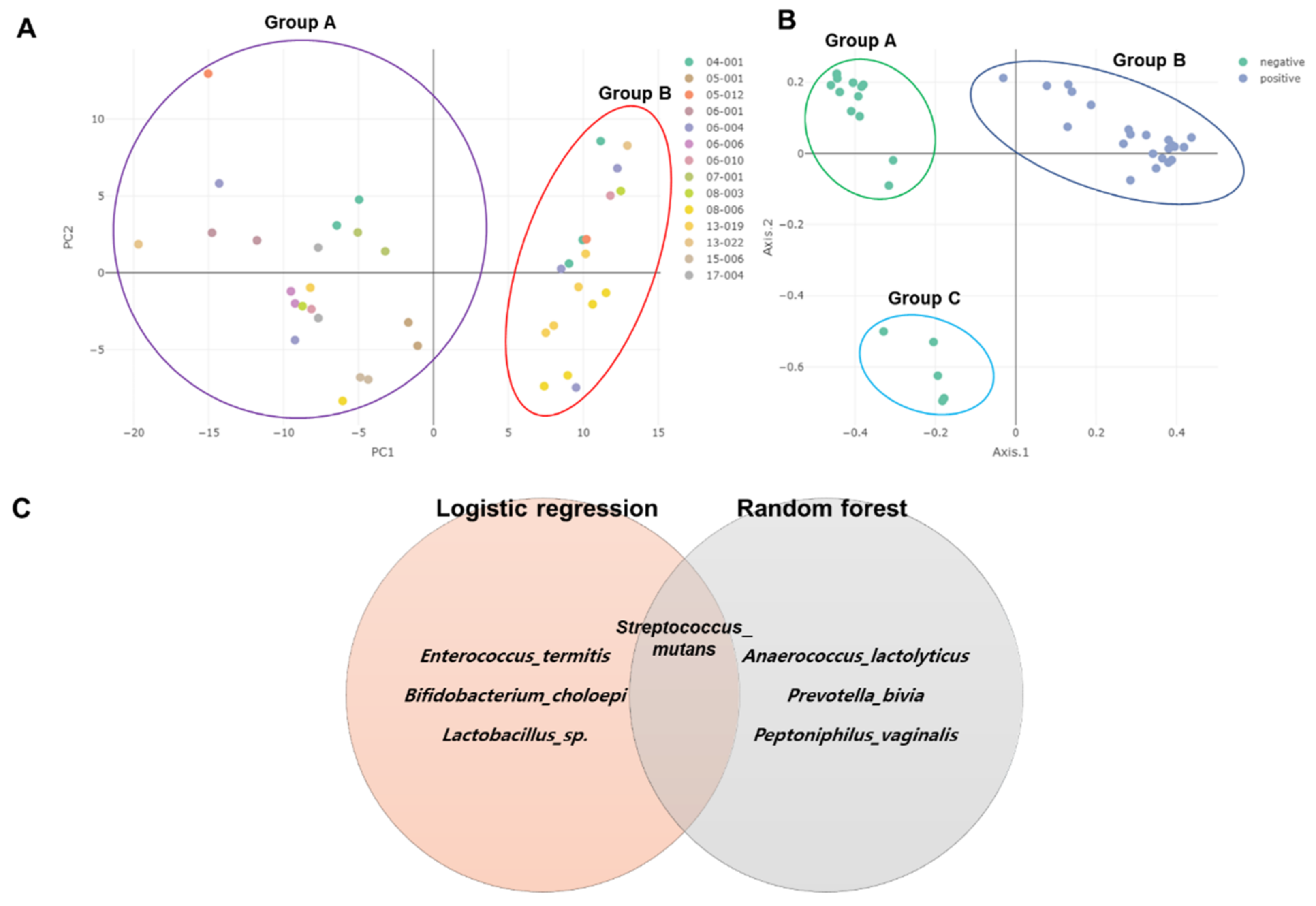
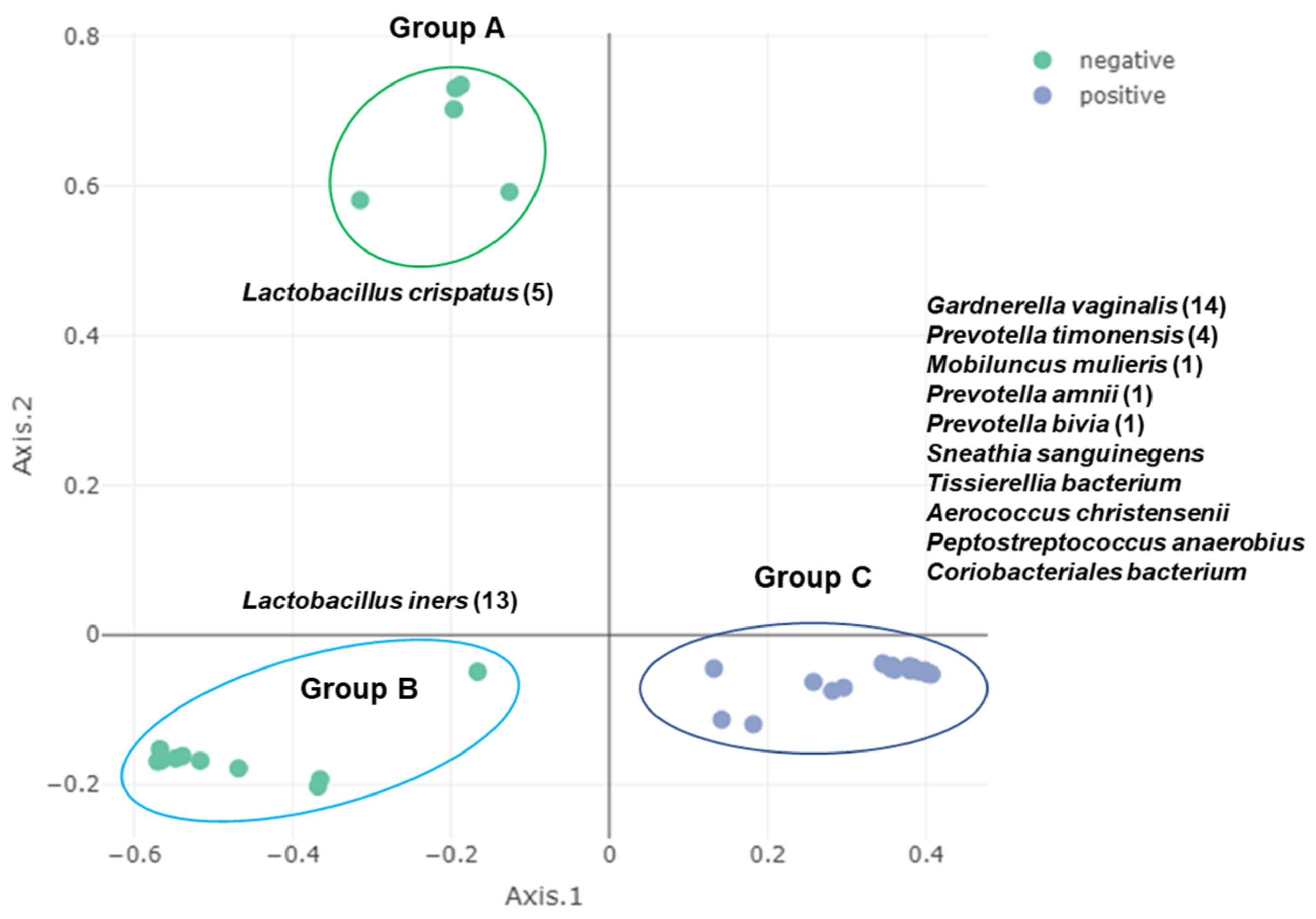
2.7. Clustering of Samples and Identification of Biomarkers
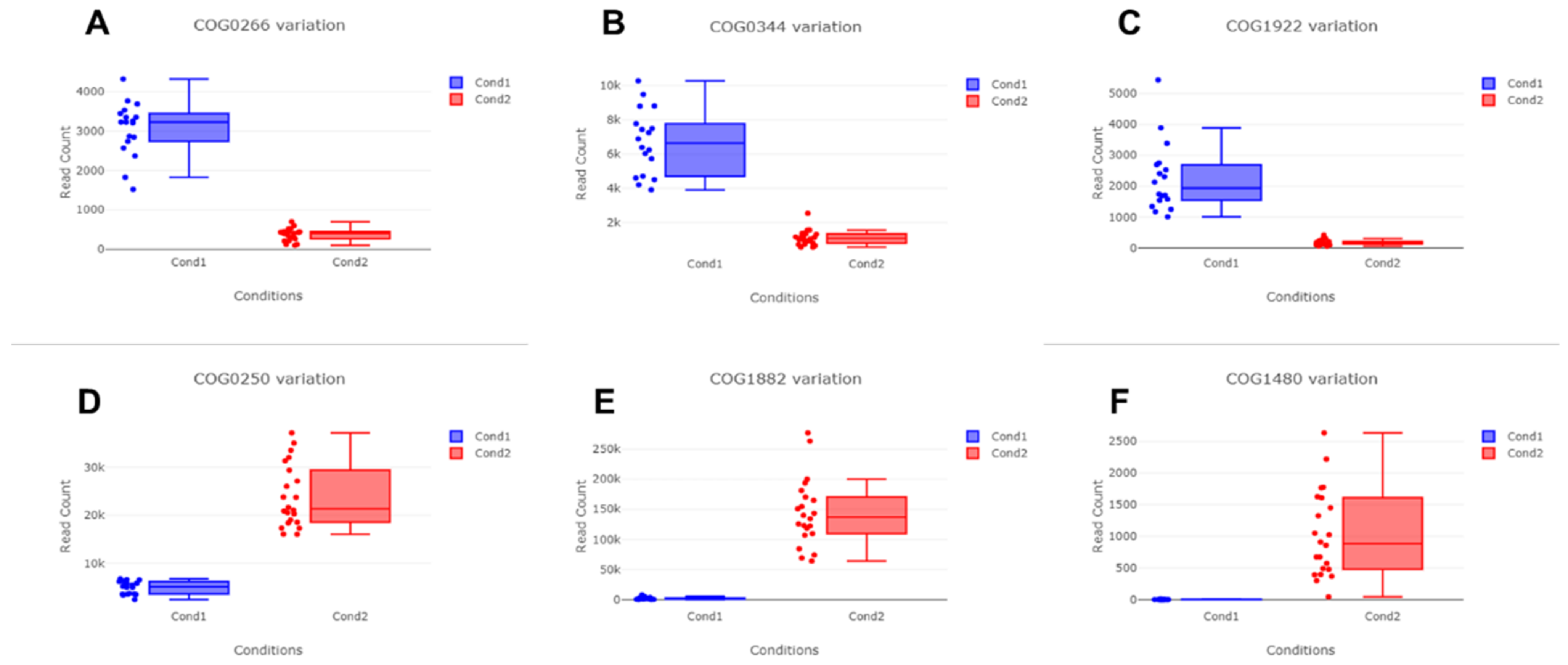
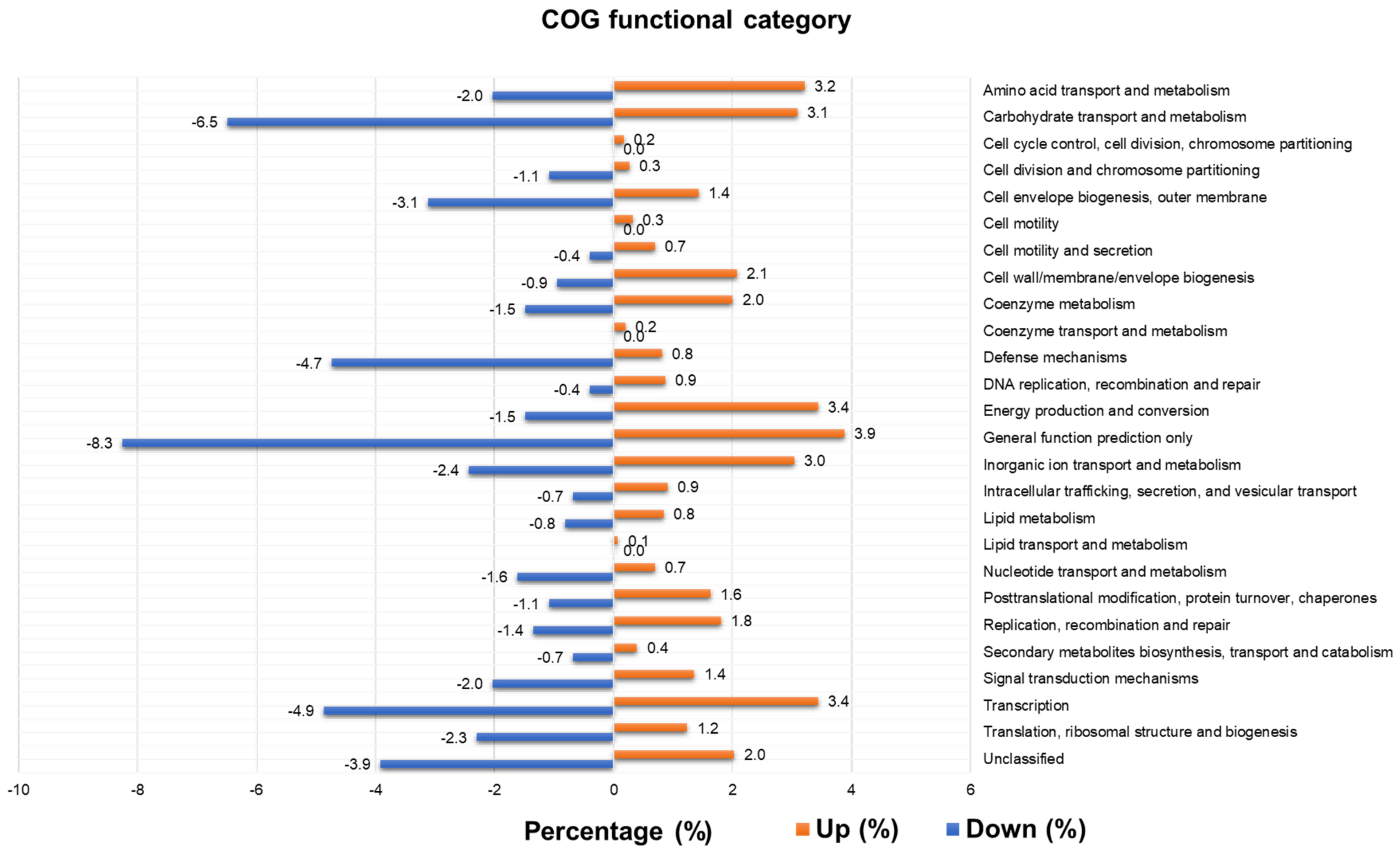
2.8. Functional Annotation of Identified Contigs According to Clusters of Orthologous Genes (COGs) and Identification of Differentially Expressed COGs
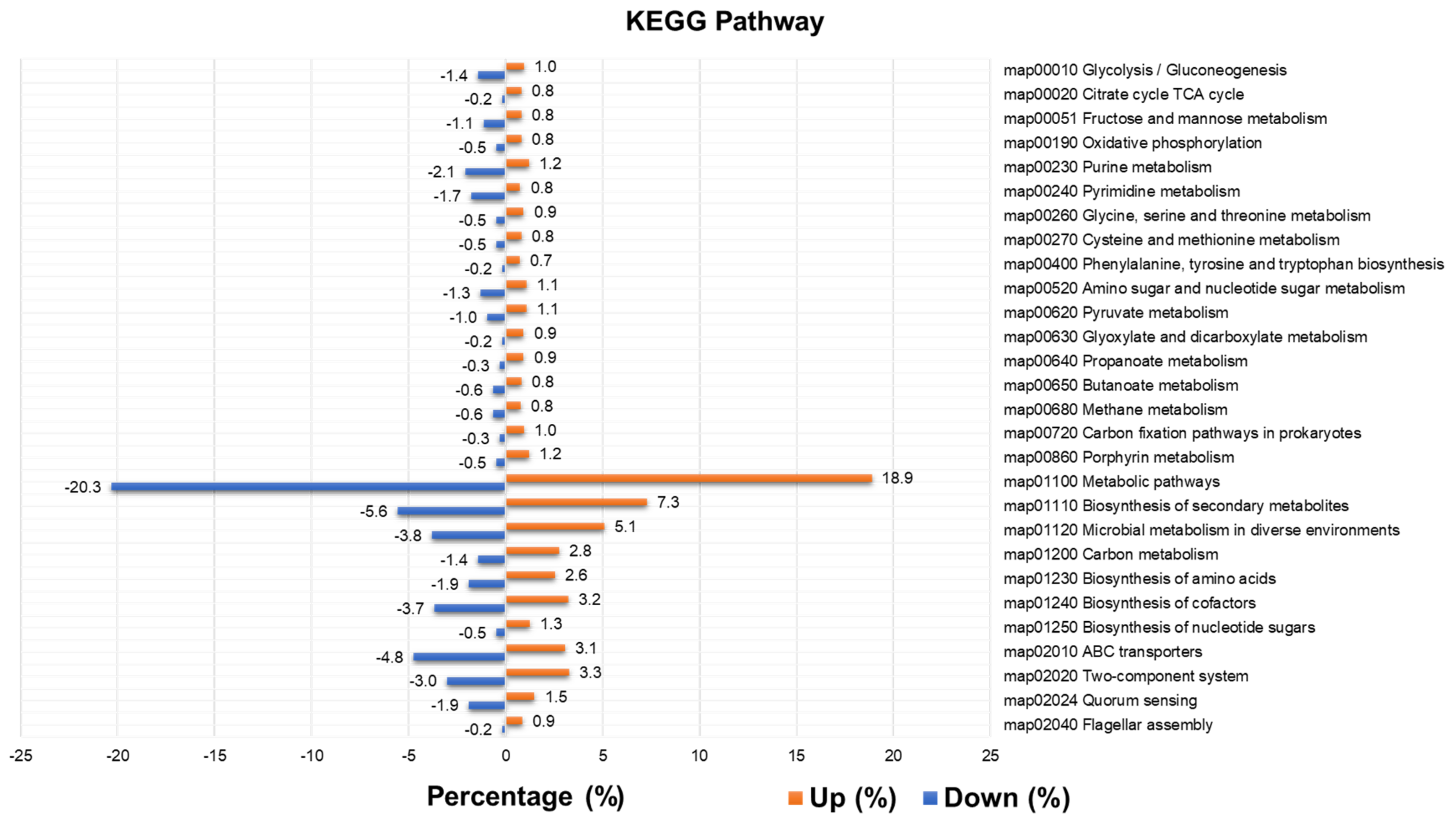
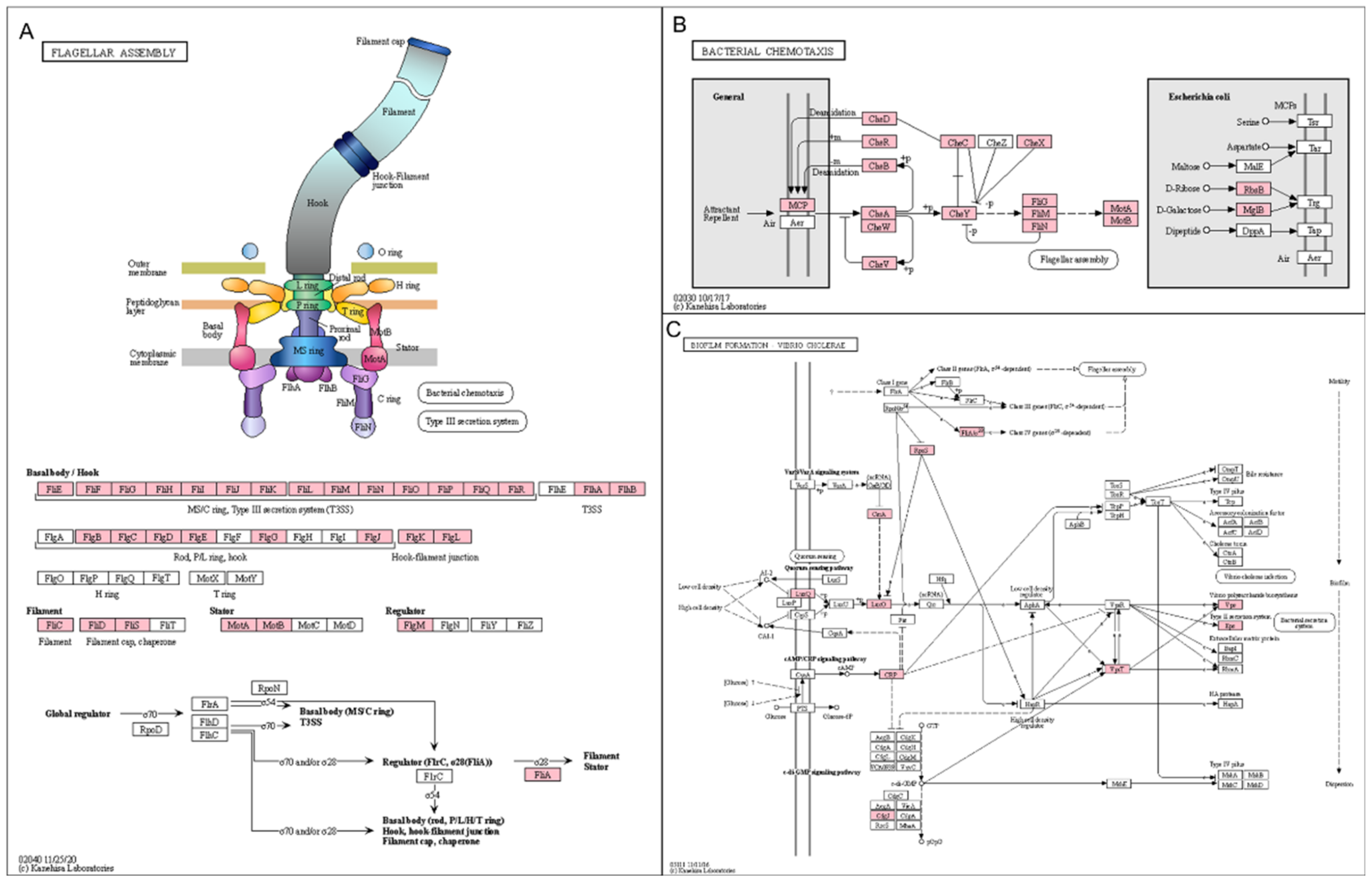
2.9. Functional Annotation of Identified Contigs According to Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathways and Identification of Differentially Expressed Enzymes
2.10. Binning of Metatranscriptome Data and Functional Classification of Identified Bins
2.11. Comparison of Three Different Approaches for Identification of Microorganisms from Vagina Samples
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Trimming, De Novo Transcriptome Assembly, and Deletion of Human Sequences
5.2. Identification and Classification of rRNA and tRNA
5.3. Prediction of ORFs, Taxonomical Classification, and Functional Annotation
5.4. Mapping of Reads and Normalization of Mapped Read Number
5.5. Identification of Differentially Expressed COGs and Enzymes
5.6. Identification of Biomarkers at Species Level Using Logistic Regression and Random Forest Algorithms
5.7. Reconstruction of Bacterial Genomes from Vaginal Metatranscriptome Dataset
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Berg, G.; Rybakova, D.; Fischer, D.; Cernava, T.; Vergès, M.-C.C.; Charles, T.; Chen, X.; Cocolin, L.; Eversole, K.; Corral, G.H. Microbiome definition re-visited: Old concepts and new challenges. Microbiome 2020, 8, 103. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, J.R.; Ravel, J. The vocabulary of microbiome research: A proposal. Microbiome 2015, 3, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Normandin, S.; Wolfe, C.T. Vitalism and the Scientific Image in Post-Enlightenment Life Science, 1800–2010; Springer: Berlin/Heidelberg, Germany, 2013; Volume 2. [Google Scholar]
- Gilbert, J.A.; Jansson, J.K.; Knight, R. Earth microbiome project and global systems biology. MSystems 2018, 3, e00217-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ursell, L.K.; Metcalf, J.L.; Parfrey, L.W.; Knight, R. Defining the human microbiome. Nutr. Rev. 2012, 70, S38–S44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnhart, K.T.; Izquierdo, A.; Pretorius, E.S.; Shera, D.M.; Shabbout, M.; Shaunik, A. Baseline dimensions of the human vagina. Hum. Reprod. 2006, 21, 1618–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravel, J.; Gajer, P.; Abdo, Z.; Schneider, G.M.; Koenig, S.S.; McCulle, S.L.; Karlebach, S.; Gorle, R.; Russell, J.; Tacket, C.O. Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. USA 2011, 108, 4680–4687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.H.; Marrazzo, J.M. The vaginal microbiome: Current understanding and future directions. J. Infect. Dis. 2016, 214, S36–S41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, B.; Forney, L.J.; Ravel, J. Vaginal microbiome: Rethinking health and disease. Annu. Rev. Microbiol. 2012, 66, 371–389. [Google Scholar] [CrossRef] [Green Version]
- Petrova, M.I.; Reid, G.; Vaneechoutte, M.; Lebeer, S. Lactobacillus iners: Friend or foe? Trends Microbiol. 2017, 25, 182–191. [Google Scholar] [CrossRef]
- Miller, E.A.; Beasley, D.E.; Dunn, R.R.; Archie, E.A. Lactobacilli dominance and vaginal pH: Why is the human vaginal microbiome unique? Front. Microbiol. 2016, 7, 1936. [Google Scholar] [CrossRef]
- Forsum, U.; Holst, E.; Larsson, P.-G.; Vasquez, A.; Jakobsson, T.; Mattsby-Baltzer, I. Bacterial vaginosis—A microbiological and immunological enigma. APMIS 2005, 113, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Lu, Y.; Chen, T.; Li, R. The female vaginal microbiome in health and bacterial vaginosis. Front. Cell. Infect. Microbiol. 2021, 11, 271. [Google Scholar] [CrossRef] [PubMed]
- Redelinghuys, M.J.; Geldenhuys, J.; Jung, H.; Kock, M.M. Bacterial vaginosis: Current diagnostic avenues and future opportunities. Front. Cell. Infect. Microbiol. 2020, 10, 354. [Google Scholar] [CrossRef] [PubMed]
- Purahong, W.; Wubet, T.; Lentendu, G.; Schloter, M.; Pecyna, M.J.; Kapturska, D.; Hofrichter, M.; Krüger, D.; Buscot, F. Life in leaf litter: Novel insights into community dynamics of bacteria and fungi during litter decomposition. Mol. Ecol. 2016, 25, 4059–4074. [Google Scholar] [CrossRef]
- Maritz, J.M.; Rogers, K.H.; Rock, T.M.; Liu, N.; Joseph, S.; Land, K.M.; Carlton, J.M. An 18S rRNA workflow for characterizing protists in sewage, with a focus on zoonotic trichomonads. Microb. Ecol. 2017, 74, 923–936. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.S.; Spakowicz, D.J.; Hong, B.-Y.; Petersen, L.M.; Demkowicz, P.; Chen, L.; Leopold, S.R.; Hanson, B.M.; Agresta, H.O.; Gerstein, M. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 2019, 10, 5029. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Hui, P.C.; Hui, M.; Yeoh, Y.K.; Wong, P.Y.; Chan, M.C.; Wong, M.C.; Ng, S.C.; Chan, F.K.; Chan, P.K. Impact of preservation method and 16S rRNA hypervariable region on gut microbiota profiling. Msystems 2019, 4, e00271-18. [Google Scholar] [CrossRef] [Green Version]
- Peterson, D.; Bonham, K.S.; Rowland, S.; Pattanayak, C.W.; Klepac-Ceraj, V.; Consortium, R. Comparative analysis of 16S rRNA gene and metagenome sequencing in pediatric gut microbiomes. Front. Microbiol. 2021, 12, 670336. [Google Scholar] [CrossRef]
- Hyman, R.W.; Fukushima, M.; Jiang, H.; Fung, E.; Rand, L.; Johnson, B.; Vo, K.C.; Caughey, A.B.; Hilton, J.F.; Davis, R.W. Diversity of the vaginal microbiome correlates with preterm birth. Reprod. Sci. 2014, 21, 32–40. [Google Scholar] [CrossRef] [Green Version]
- Fettweis, J.M.; Serrano, M.G.; Brooks, J.P.; Edwards, D.J.; Girerd, P.H.; Parikh, H.I.; Huang, B.; Arodz, T.J.; Edupuganti, L.; Glascock, A.L. The vaginal microbiome and preterm birth. Nat. Med. 2019, 25, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Freitas, A.C.; Chaban, B.; Bocking, A.; Rocco, M.; Yang, S.; Hill, J.E.; Money, D.M. The vaginal microbiome of pregnant women is less rich and diverse, with lower prevalence of Mollicutes, compared to non-pregnant women. Sci. Rep. 2017, 7, 9212. [Google Scholar] [CrossRef]
- Shen, J.; Song, N.; Williams, C.J.; Brown, C.J.; Yan, Z.; Xu, C.; Forney, L.J. Effects of low dose estrogen therapy on the vaginal microbiomes of women with atrophic vaginitis. Sci. Rep. 2016, 6, 24380. [Google Scholar] [CrossRef] [Green Version]
- Gottschick, C.; Deng, Z.L.; Vital, M.; Masur, C.; Abels, C.; Pieper, D.H.; Rohde, M.; Mendling, W.; Wagner-Dobler, I. Treatment of biofilms in bacterial vaginosis by an amphoteric tenside pessary-clinical study and microbiota analysis. Microbiome 2017, 5, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feehily, C.; Crosby, D.; Walsh, C.J.; Lawton, E.M.; Higgins, S.; McAuliffe, F.M.; Cotter, P.D. Shotgun sequencing of the vaginal microbiome reveals both a species and functional potential signature of preterm birth. NPJ Biofilms Microbiomes 2020, 6, 50. [Google Scholar] [CrossRef]
- Deng, Z.L.; Gottschick, C.; Bhuju, S.; Masur, C.; Abels, C.; Wagner-Dobler, I. Metatranscriptome Analysis of the Vaginal Microbiota Reveals Potential Mechanisms for Protection against Metronidazole in Bacterial Vaginosis. Msphere 2018, 3, e00262-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macklaim, J.M.; Fernandes, A.D.; Di Bella, J.M.; Hammond, J.-A.; Reid, G.; Gloor, G.B. Comparative meta-RNA-seq of the vaginal microbiota and differential expression by Lactobacillus iners in health and dysbiosis. Microbiome 2013, 1, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shakya, M.; Lo, C.-C.; Chain, P.S. Advances and challenges in metatranscriptomic analysis. Front. Genet. 2019, 10, 904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freitas, T.A.K.; Li, P.-E.; Scholz, M.B.; Chain, P.S. Accurate read-based metagenome characterization using a hierarchical suite of unique signatures. Nucleic Acids Res. 2015, 43, e69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [Green Version]
- Truong, D.T.; Franzosa, E.A.; Tickle, T.L.; Scholz, M.; Weingart, G.; Pasolli, E.; Tett, A.; Huttenhower, C.; Segata, N. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 2015, 12, 902–903. [Google Scholar] [CrossRef]
- Kim, D.; Song, L.; Breitwieser, F.P.; Salzberg, S.L. Centrifuge: Rapid and sensitive classification of metagenomic sequences. Genome Res. 2016, 26, 1721–1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Leung, H.C.; Yiu, S.-M.; Chin, F.Y. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [Green Version]
- Tamames, J.; Puente-Sanchez, F. SqueezeMeta, A Highly Portable, Fully Automatic Metagenomic Analysis Pipeline. Front. Microbiol. 2018, 9, 3349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayanasamy, S.; Jarosz, Y.; Muller, E.E.; Heintz-Buschart, A.; Herold, M.; Kaysen, A.; Laczny, C.C.; Pinel, N.; May, P.; Wilmes, P. IMP: A pipeline for reproducible reference-independent integrated metagenomic and metatranscriptomic analyses. Genome Biol. 2016, 17, 260. [Google Scholar] [CrossRef] [Green Version]
- Sequeira, J.C.; Rocha, M.; Alves, M.M.; Salvador, A.F. MOSCA: An automated pipeline for integrated metagenomics and metatranscriptomics data analysis. In Proceedings of the International Conference on Practical Applications of Computational Biology & Bioinformatics, Toledo, Spain, 20–22 May 2018; pp. 183–191. [Google Scholar]
- Kuczynski, J.; Lauber, C.L.; Walters, W.A.; Parfrey, L.W.; Clemente, J.C.; Gevers, D.; Knight, R. Experimental and analytical tools for studying the human microbiome. Nat. Rev. Genet. 2011, 13, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Guigo, R.; Agarwal, P.; Abril, J.F.; Burset, M.; Fickett, J.W. An assessment of gene prediction accuracy in large DNA sequences. Genome Res. 2000, 10, 1631–1642. [Google Scholar] [CrossRef] [Green Version]
- Ye, S.H.; Siddle, K.J.; Park, D.J.; Sabeti, P.C. Benchmarking Metagenomics Tools for Taxonomic Classification. Cell 2019, 178, 779–794. [Google Scholar] [CrossRef]
- Zou, Y.; Xue, W.; Luo, G.; Deng, Z.; Qin, P.; Guo, R.; Sun, H.; Xia, Y.; Liang, S.; Dai, Y.; et al. 1520 reference genomes from cultivated human gut bacteria enable functional microbiome analyses. Nat. Biotechnol. 2019, 37, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Nissen, J.N.; Johansen, J.; Allesøe, R.L.; Sønderby, C.K.; Armenteros, J.J.A.; Grønbech, C.H.; Jensen, L.J.; Nielsen, H.B.; Petersen, T.N.; Winther, O. Improved metagenome binning and assembly using deep variational autoencoders. Nat. Biotechnol. 2021, 39, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Rinke, C.; Chuvochina, M.; Chaumeil, P.-A.; Woodcroft, B.J.; Evans, P.N.; Hugenholtz, P.; Tyson, G.W. Recovery of nearly 8000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2017, 2, 1533–1542. [Google Scholar] [CrossRef] [PubMed]
- Holm, J.B.; France, M.T.; Ma, B.; McComb, E.; Robinson, C.K.; Mehta, A.; Tallon, L.J.; Brotman, R.M.; Ravel, J. Comparative Metagenome-Assembled Genome Analysis of “Candidatus Lachnocurva vaginae”, Formerly Known as Bacterial Vaginosis-Associated Bacterium−1 (BVAB1). Front. Cell. Infect. Microbiol. 2020, 10, 117. [Google Scholar] [CrossRef] [Green Version]
- van der Veer, C.; Hertzberger, R.Y.; Bruisten, S.M.; Tytgat, H.L.P.; Swanenburg, J.; de Kat Angelino-Bart, A.; Schuren, F.; Molenaar, D.; Reid, G.; de Vries, H.; et al. Comparative genomics of human Lactobacillus crispatus isolates reveals genes for glycosylation and glycogen degradation: Implications for in vivo dominance of the vaginal microbiota. Microbiome 2019, 7, 49. [Google Scholar] [CrossRef] [Green Version]
- Nilsen, T.; Swedek, I.; Lagenaur, L.A.; Parks, T.P. Novel Selective Inhibition of Lactobacillus iners by Lactobacillus-Derived Bacteriocins. Appl. Environ. Microbiol. 2020, 86, e01594–20. [Google Scholar] [CrossRef]
- Kort, R. Personalized therapy with probiotics from the host by TripleA. Trends Biotechnol. 2014, 32, 291–293. [Google Scholar] [CrossRef]
- Spiegel, C.A.; Roberts, M. Mobiluncus gen. nov., Mobiluncus curtisii subsp. curtisii sp. nov., Mobiluncus curtisii subsp. holmesii subsp. nov., and Mobiluncus mulieris sp. nov., curved rods from the human vagina. Int. J. Syst. Evol. 1984, 34, 177–184. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, C.A. Mobiluncus curtisii and Mobiluncus mulieris, curved motile bacteria from the human vagina. Clin. Microbiol. Newsl. 1984, 6, 163–165. [Google Scholar] [CrossRef]
- Webre, D.J.; Wolanin, P.M.; Stock, J.B. Bacterial chemotaxis. Curr. Biol. 2003, 13, R47–R49. [Google Scholar] [CrossRef] [Green Version]
- Valderrama, J.A.; Durante-Rodríguez, G.; Blázquez, B.; García, J.L.; Carmona, M.; Díaz, E. Bacterial degradation of benzoate: Cross-regulation between aerobic and anaerobic pathways. J. Biol. Chem. 2012, 287, 10494–10508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, J.; Machado, D.; Cerca, N. Unveiling the role of Gardnerella vaginalis in polymicrobial bacterial vaginosis biofilms: The impact of other vaginal pathogens living as neighbors. ISME J. 2019, 13, 1306–1317. [Google Scholar] [CrossRef] [PubMed]
- van Teijlingen, N.H.; Helgers, L.C.; Zijlstra-Willems, E.M.; van Hamme, J.L.; Ribeiro, C.M.; Strijbis, K.; Geijtenbeek, T.B. Vaginal dysbiosis associated-bacteria Megasphaera elsdenii and Prevotella timonensis induce immune activation via dendritic cells. J. Reprod. Immunol. 2020, 138, 103085. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Reuter, K.; Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.-J.; Tappu, R. MEGAN community edition-interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [Green Version]
- Hyatt, D.; Chen, G.L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2016, 44, D67–D72. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. A new generation of homology search tools based on probabilistic inference. In Genome Informatics 2009: Genome Informatics Series Vol. 23; World Scientific: Singapore, 2009; pp. 205–211. [Google Scholar]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Doak, T.G. A parsimony approach to biological pathway reconstruction/inference for genomes and metagenomes. PLoS Comput. Biol. 2009, 5, e1000465. [Google Scholar] [CrossRef] [Green Version]
- Caspi, R.; Billington, R.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Midford, P.E.; Ong, Q.; Ong, W.K. The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 2018, 46, D633–D639. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Federico, A.; Faits, T.; Manimaran, S.; Segrè, D.; Monti, S.; Johnson, W.E. Animalcules: Interactive microbiome analytics and visualization in R. Microbiome 2021, 9, 76. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.-W.; Simmons, B.A.; Singer, S.W. MaxBin 2.0: An automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 2016, 32, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef]
- Sieber, C.M.; Probst, A.J.; Sharrar, A.; Thomas, B.C.; Hess, M.; Tringe, S.G.; Banfield, J.F. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 2018, 3, 836–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laczny, C.C.; Sternal, T.; Plugaru, V.; Gawron, P.; Atashpendar, A.; Margossian, H.H.; Coronado, S.; Van der Maaten, L.; Vlassis, N.; Wilmes, P. VizBin-an application for reference-independent visualization and human-augmented binning of metagenomic data. Microbiome 2015, 3, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxonomy | Number of Contigs | Proportion | No. of Taxonomies |
|---|---|---|---|
| Super kingdoms | 276,994 | 96.80% | 4 |
| Phyla | 261,853 | 91.50% | 25 |
| Classes | 252,721 | 88.30% | 32 |
| Orders | 242,180 | 84.60% | 58 |
| Families | 218,460 | 76.30% | 99 |
| Genera | 192,420 | 67.20% | 209 |
| Species | 122,133 | 42.70% | 339 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, W.K.; Jo, Y.; Jeong, S. De Novo Assembly and Annotation of the Vaginal Metatranscriptome Associated with Bacterial Vaginosis. Int. J. Mol. Sci. 2022, 23, 1621. https://doi.org/10.3390/ijms23031621
Cho WK, Jo Y, Jeong S. De Novo Assembly and Annotation of the Vaginal Metatranscriptome Associated with Bacterial Vaginosis. International Journal of Molecular Sciences. 2022; 23(3):1621. https://doi.org/10.3390/ijms23031621
Chicago/Turabian StyleCho, Won Kyong, Yeonhwa Jo, and Seri Jeong. 2022. "De Novo Assembly and Annotation of the Vaginal Metatranscriptome Associated with Bacterial Vaginosis" International Journal of Molecular Sciences 23, no. 3: 1621. https://doi.org/10.3390/ijms23031621
APA StyleCho, W. K., Jo, Y., & Jeong, S. (2022). De Novo Assembly and Annotation of the Vaginal Metatranscriptome Associated with Bacterial Vaginosis. International Journal of Molecular Sciences, 23(3), 1621. https://doi.org/10.3390/ijms23031621