
Virtual Combinatorial Chemistry and Pharmacological Screening: A Short Guide to Drug Design

 ,
,  ,
,  ,
,  and
and 
Abstract
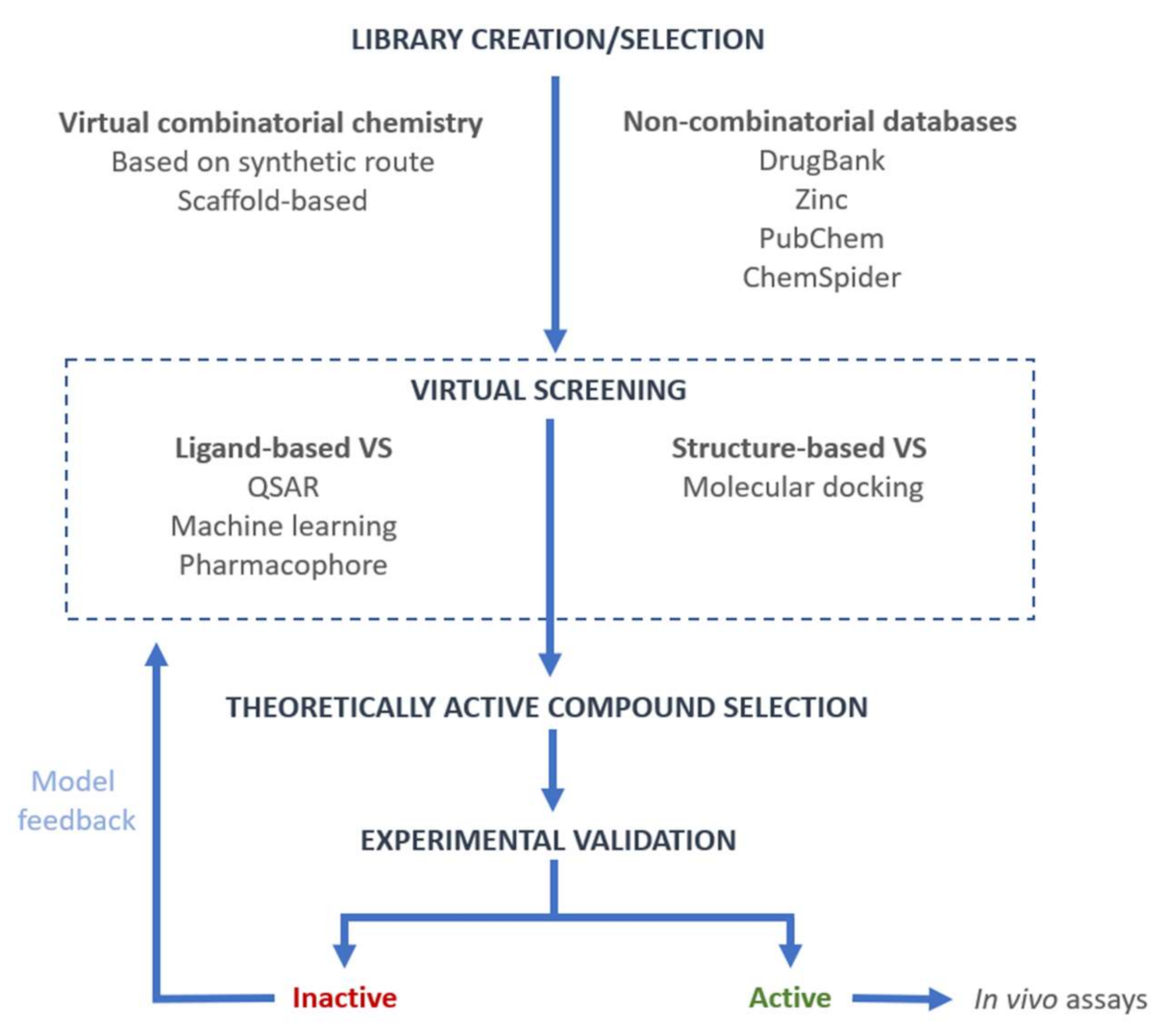
:1. Introduction
2. Virtual Combinatorial Library Creation
2.1. Types of Combinatorial Libraries
2.2. Generation of Combinatorial Libraries
3. Virtual Screening
3.1. Methods Used in Virtual Screening
3.1.1. Ligand-Based Virtual Screening (LBVS)
3.1.2. Structure-Based Virtual Screening (SBVS)
4. Applications and Current Trends
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Garrett, M.D.; Workman, P. Discovering novel chemotherapeutic drugs for the third millennium. Eur. J. Cancer 1999, 35, 2010–2030. [Google Scholar] [CrossRef]
- Guido, R.V.C.; Oliva, G.; Andricopulo, A.D. Modern drug discovery technologies: Opportunities and challenges in lead discovery. Comb. Chem. High Throughput Screen. 2011, 14, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.J.; Zheng, W.; Tropsha, A. Rational Combinatorial Library Design. 2. Rational Design of Targeted Combinatorial Peptide Libraries Using Chemical Similarity Probe and the Inverse QSAR Approaches. J. Chem. Inf. Comput. Sci. 1998, 38, 259–268. [Google Scholar] [CrossRef] [PubMed]
- De Julian-Ortiz, J.V. Virtual darwinian drug design: QSAR inverse problem, virtual combinatorial chemistry, and computational screening. Comb. Chem. High Throughput Screen. 2001, 4, 295–310. [Google Scholar] [CrossRef]
- López-Vallejo, F.; Caulfield, T.; Martinez-Mayorga, K.; Giulianotti, M.A.; Nefzi, A.; Houghten, R.A.; Medina-Franco, J.L. Integrating Virtual Screening and Combinatorial Chemistry for Accelerated Drug Discovery. Comb. Chem. High Throughput Screen. 2011, 14, 475–487. [Google Scholar] [CrossRef]
- Bajorath, J. Integration of virtual and high-throughput screening. Nat. Rev. Drug Discov. 2002, 1, 882–894. [Google Scholar] [CrossRef]
- Lill, M. Virtual Screening in Drug Design. Methods Mol. Biol. 2013, 993, 1–12. [Google Scholar] [CrossRef]
- Jahn, A.; Hinselmann, G.; Fechner, N.; Zell, A. Optimal assignment methods for ligand-based virtual screening. J. Cheminformatics 2009, 1, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Maia, E.H.B.; Assis, L.C.; De Oliveira, T.A.; Da Silva, A.M.; Taranto, A.G. Structure-Based Virtual Screening: From Classical to Artificial Intelligence. Front. Chem. 2020, 8, 343. [Google Scholar] [CrossRef]
- Bauer, J.; Spanton, S.; Henry, R.; Quick, J.; Dziki, W.; Porter, W.; Morris, J. Ritonavir: An Extraordinary Example of Conformational Polymorphism. Pharm. Res. 2001, 18, 859–866. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, J.; Xiao, Y.; Wang, T.; Huang, X. The Effects of Polymorphism on Physicochemical Properties and Pharmacodynamics of Solid Drugs. Curr. Pharm. Des. 2018, 24, 2375–2382. [Google Scholar] [CrossRef] [PubMed]
- Drebushchak, V.A.; McGregor, L.; Rychkov, D.A. Cooling rate “window” in the crystallization of metacetamol form II. J. Therm. Anal. Calorim. 2017, 127, 1807–1814. [Google Scholar] [CrossRef]
- Mazurek, A.H.; Szeleszczuk, Ł.; Pisklak, D.M. Periodic DFT Calculations—Review of Applications in the Pharmaceutical Sciences. Pharmaceutics 2020, 12, 415. [Google Scholar] [CrossRef] [PubMed]
- Vainio, M.J.; Kogej, T.; Raubacher, F. Automated Recycling of Chemistry for Virtual Screening and Library Design. J. Chem. Inf. Model. 2012, 52, 1777–1786. [Google Scholar] [CrossRef] [PubMed]
- Schneider, G. Trends in virtual combinatorial library design. Curr. Med. Chem. 2002, 9, 2095–2101. [Google Scholar] [CrossRef]
- Nikolay, K.; Svetlana, A.; Nina, J. Combinatorial generation of molecules by virtual software reactor. Sci. Work Union Sci. Bulg. Plovdiv 2017, 11, 214–219. [Google Scholar]
- Lessel, U.; Wellenzohn, B.; Lilienthal, M.; Claussen, H. Searching Fragment Spaces with Feature Trees. J. Chem. Inf. Model. 2009, 49, 270–279. [Google Scholar] [CrossRef]
- Nicolaou, C.A.; Watson, I.A.; Hu, H.; Wang, J.-B. The Proximal Lilly Collection: Mapping, Exploring and Exploiting Feasible Chemical Space. J. Chem. Inf. Model. 2016, 56, 1253–1266. [Google Scholar] [CrossRef]
- Hu, Q.; Peng, Z.; Sutton, S.C.; Na, J.; Kostrowicki, J.; Yang, B.; Thacher, T.; Kong, X.; Mattaparti, S.; Zhou, J.Z.; et al. Pfizer Global Virtual Library (PGVL): A Chemistry Design Tool Powered by Experimentally Validated Parallel Synthesis Information. ACS Comb. Sci. 2012, 14, 579–589. [Google Scholar] [CrossRef]
- Humbeck, L.; Weigang, S.; Schäfer, T.; Mutzel, P.; Koch, O. CHI PMUNK: A Virtual Synthesizable Small-Molecule Library for Medicinal Chemistry, Exploitable for Protein-Protein Interaction Modulators. ChemMedChem 2018, 13, 532–539. [Google Scholar] [CrossRef] [Green Version]
- Massarotti, A. Investigation of the Click-Chemical Space for Drug Design Using ZINClick. Methods Mol. Biol. 2021, 2266, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Saldívar-González, F.I.; Lenci, E.; Calugi, L.; Medina-Franco, J.L.; Trabocchi, A. Computational-aided design of a library of lactams through a diversity-oriented synthesis strategy. Bioorganic Med. Chem. 2020, 28, 115539. [Google Scholar] [CrossRef] [PubMed]
- Karthikeyan, M.; Pandit, D.; Vyas, R. ChemScreener: A Distributed Computing Tool for Scaffold based Virtual Screening. Comb. Chem. High Throughput Screen. 2015, 18, 544–561. [Google Scholar] [CrossRef] [PubMed]
- Krier, M.; de Araújo-Júnior, J.X.; Schmitt, M.; Duranton, J.; Justiano-Basaran, H.; Lugnier, C.; Bourguignon, J.-J.; Rognan, D. Design of Small-Sized Libraries by Combinatorial Assembly of Linkers and Functional Groups to a Given Scaffold: Application to the Structure-Based Optimization of a Phosphodiesterase 4 Inhibitor. J. Med. Chem. 2005, 48, 3816–3822. [Google Scholar] [CrossRef]
- Bueso-Bordils, J.I.; Perez-Gracia, M.T.; Suay-Garcia, B.; Duart, M.J.; Algarra, R.V.M.; Zamora, L.L.; Anton-Fos, G.M.; Lopez, P.A.A. Topological pattern for the search of new active drugs against methicillin resistant Staphylococcus aureus. Eur. J. Med. Chem. 2017, 138, 807–815. [Google Scholar] [CrossRef]
- Kouman, K.C.; Keita, M.; N’Guessan, R.K.; Owono, L.C.O.; Megnassan, E.; Frecer, V.; Miertus, S. Structure-Based Design and in Silico Screening of Virtual Combinatorial Library of Benzamides Inhibiting 2-trans Enoyl-Acyl Carrier Protein Reductase of Mycobacterium tuberculosis with Favorable Predicted Pharmacokinetic Profiles. Int. J. Mol. Sci. 2019, 20, 4730. [Google Scholar] [CrossRef] [Green Version]
- Lauro, G.; Terracciano, S.; Cantone, V.; Ruggiero, D.; Fischer, K.; Pace, S.; Werz, O.; Bruno, I.; Bifulco, G. A Combinatorial Virtual Screening Approach Driving the Synthesis of 2,4-Thiazolidinedione-Based Molecules as New Dual mPGES-1/5-LO Inhibitors. ChemMedChem 2020, 15, 481–489. [Google Scholar] [CrossRef]
- Saldívar-González, F.I.; Huerta-García, C.S.; Medina-Franco, J.L. Chemoinformatics-based enumeration of chemical libraries: A tutorial. J. Cheminformatics 2020, 12, 1–25. [Google Scholar] [CrossRef]
- Fang, G.; Xue, M.; Su, M.; Hu, D.; Li, Y.; Xiong, B.; Ma, L.; Meng, T.; Chen, Y.; Li, J.; et al. CCLab—a multi-objective genetic algorithm based combinatorial library design software and an application for histone deacetylase inhibitor design. Bioorganic Med. Chem. Lett. 2012, 22, 4540–4545. [Google Scholar] [CrossRef]
- Gillet, V.J.; Khatib, W.; Willett, P.; Fleming, P.J.; Green, D.V.S. Combinatorial Library Design Using a Multiobjective Genetic Algorithm. J. Chem. Inf. Comput. Sci. 2002, 42, 375–385. [Google Scholar] [CrossRef]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Thiel, K.; Wiswedel, B. KNIME—The Konstanz information miner: Version 2.0 and beyond. ACM SIGKDD Explor. Newsl. 2009, 11, 26–31. [Google Scholar] [CrossRef] [Green Version]
- Landrum, G. RDKit. Available online: https://www.rdkit.org/ (accessed on 28 October 2021).
- Sander, T.; Freyss, J.; Von Korff, M.; Rufener, C. DataWarrior: An Open-Source Program For Chemistry Aware Data Visualization and Analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Reactor|ChemAxon. Available online: https://chemaxon.com/products/reactor (accessed on 28 October 2021).
- Library synthesizer—Tripod Development. Available online: https://tripod.nih.gov/?p=370 (accessed on 28 October 2021).
- Schüller, A.; Hähnke, V.; Schneider, G. SmiLib v2.0: A Java-Based Tool for Rapid Combinatorial Library Enumeration. QSAR Comb. Sci. 2007, 26, 407–410. [Google Scholar] [CrossRef]
- Chemical Computing Group (CCG)|Computer-Aided Molecular Design. Available online: https://www.chemcomp.com/ (accessed on 28 October 2021).
- Schrödinger. Available online: https://www.schrodinger.com/ (accessed on 28 October 2021).
- Optibrium. Available online: https://www.optibtium.com/startdrop/startdrop-nova.php (accessed on 28 October 2021).
- ChemDraw. Available online: https://perkinelmerinformatics.com/products/research/chemdraw/ (accessed on 4 November 2021).
- GLARE. Available online: https://glare.sourcefoge.net/ (accessed on 28 October 2021).
- Shoichet, B.K. Virtual screening of chemical libraries. Nature 2004, 432, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A.; Di Giovanni, C. Virtual Screening Strategies in Drug Discovery: A Critical Review. Curr. Med. Chem. 2013, 20, 2839–2860. [Google Scholar] [CrossRef] [PubMed]
- Tanrikulu, Y.; Krüger, B.; Proschak, E. The holistic integration of virtual screening in drug discovery. Drug Discov. Today 2013, 18, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Ripphausen, P.; Nisius, B.; Bajorath, J. State-of-the-art in ligand-based virtual screening. Drug Discov. Today 2011, 16, 372–376. [Google Scholar] [CrossRef]
- Spiegel, J.; Senderowitz, H. Evaluation of QSAR Equations for Virtual Screening. Int. J. Mol. Sci. 2020, 21, 7828. [Google Scholar] [CrossRef]
- Tropsha, A.; Golbraikh, A. Predictive QSAR Modeling Workflow, Model Applicability Domains, and Virtual Screening. Curr. Pharm. Des. 2007, 13, 3494–3504. [Google Scholar] [CrossRef]
- Suay-Garcia, B.; Bueso-Bordils, J.I.; Falcó, A.; Pérez-Gracia, M.T.; Antón-Fos, G.; Alemán-López, P. Quantitative structure–activity relationship methods in the discovery and development of antibacterials. WIREs Comput. Mol. Sci. 2020, 10, e1472. [Google Scholar] [CrossRef]
- Gini, G. QSAR: What Else? Methods Mol. Biol. 2018, 1800, 79–105. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.U. Descriptors and their selection methods in QSAR analysis: Paradigm for drug design. Drug Discov. Today 2016, 21, 1291–1302. [Google Scholar] [CrossRef] [PubMed]
- Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors; Wiley-VCH: Weinheim, Germany, 2000. [Google Scholar] [CrossRef]
- LaPointe, S.M.; Weaver, D.F. A Review of Density Functional Theory Quantum Mechanics as Applied to Pharmaceutically Relevant Systems. Curr. Comput. Aided-Drug Des. 2007, 3, 290–296. [Google Scholar] [CrossRef]
- Perkins, R.; Fang, H.; Tong, W.; Welsh, W.J. Quantitative structure-activity relationship methods: Perspectives on drug discovery and toxicology. Environ. Toxicol. Chem. 2003, 22, 1666–1679. [Google Scholar] [CrossRef]
- Liu, P.; Long, W. Current Mathematical Methods Used in QSAR/QSPR Studies. Int. J. Mol. Sci. 2009, 10, 1978–1998. [Google Scholar] [CrossRef]
- Li, S.; Zhang, S.; Chen, D.; Jiang, X.; Liu, B.; Zhang, H.; Rachakunta, M.; Zuo, Z. Identification of Novel TRPC5 Inhibitors by Pharmacophore-Based and Structure-Based Approaches. Comput. Biol. Chem. 2020, 87, 107302. [Google Scholar] [CrossRef]
- Wolber, G. 3D pharmacophore elucidation and virtual screening. Drug Discov. Today Technol. 2011, 7, e203–e204. [Google Scholar] [CrossRef]
- Hessler, G.; Baringhaus, K.-H. The scaffold hopping potential of pharmacophores. Drug Discov. Today Technol. 2011, 7, e263–e269. [Google Scholar] [CrossRef]
- Liu, S.; Alnammi, M.; Ericksen, S.S.; Voter, A.F.; Ananiev, G.E.; Keck, J.L.; Hoffmann, F.M.; Wildman, S.A.; Gitter, A. Practical Model Selection for Prospective Virtual Screening. J. Chem. Inf. Model. 2018, 59, 282–293. [Google Scholar] [CrossRef] [Green Version]
- Kuntz, I.D.; Blaney, J.M.; Oatley, S.J.; Langridge, R.; Ferrin, T.E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982, 161, 269–288. [Google Scholar] [CrossRef]
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-Based Virtual Screening for Drug Discovery: Principles, Applications and Recent Advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.S.; Pati, S.P.; Kumar, P.P.; Pradeep, H.N.; Sastry, G.N. Virtual Screening in Drug Discovery—A Computational Perspective. Curr. Protein Pept. Sci. 2007, 8, 329–351. [Google Scholar] [CrossRef]
- Sun, H. Pharmacophore-Based Virtual Screening. Curr. Med. Chem. 2008, 15, 1018–1024. [Google Scholar] [CrossRef]
- Eldridge, M.D.; Murray, C.W.; Auton, T.R.; Paolini, G.V.; Mee, R.P. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput. Aided Mol. Des. 1997, 11, 425–445. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, Z.; Li, J.; Han, L.; Zhao, Z.; Wang, R. Comparative Assessment of Scoring Functions on an Updated Benchmark: 1. Compilation of the Test Set. J. Chem. Inf. Model. 2014, 54, 1700–1716. [Google Scholar] [CrossRef] [PubMed]
- Gohlkea, H.; Hendlicha, M.; Klebea, G. Knowledge-based scoring function to predict protein-ligand interactions. J. Mol. Biol. 2000, 295, 337–356. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Zheng, Z.; Wang, T.; Li, P.; Merz, K.M., Jr. KECSA-Movable Type Implicit Solvation Model (KMTISM). J. Chem. Theory Comput. 2014, 11, 667–682. [Google Scholar] [CrossRef] [Green Version]
- Raha, K.; Peters, M.B.; Wang, B.; Yu, N.; Wollacott, A.M.; Westerhoff, L.; Merz, K.M. The role of quantum mechanics in structure-based drug design. Drug Discov. Today 2007, 12, 725–731. [Google Scholar] [CrossRef]
- Chen, Z.; Li, H.-L.; Zhang, Q.-J.; Bao, X.-G.; Yu, K.-Q.; Luo, X.-M.; Zhu, W.-L.; Jiang, H.-L. Pharmacophore-based virtual screening versus docking-based virtual screening: A benchmark comparison against eight targets. Acta Pharmacol. Sin. 2009, 30, 1694–1708. [Google Scholar] [CrossRef] [Green Version]
- Tanoli, Z.; Seemab, U.; Scherer, A.; Wennerberg, K.; Tang, J.; Vähä-Koskela, M. Exploration of databases and methods supporting drug repurposing: A comprehensive survey. Briefings Bioinform. 2021, 22, 1656–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S. Getting the most out of PubChem for virtual screening. Expert Opin. Drug Discov. 2016, 11, 843–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ZINC15. Available online: http://zinc15.docking.org/ (accessed on 17 November 2021).
- ChemSpider–Chemical Database. Royal Society of Chemistry, Cambridge, UK. Available online: http://www.chemspider.com/ (accessed on 15 December 2021).
- DrugBank. Available online: https://go.drugbank.com/ (accessed on 8 December 2021).
- Suay-Garcia, B.; Falcó, A.; Bueso-Bordils, J.I.; Anton-Fos, G.M.; Pérez-Gracia, M.T.; Alemán-López, P.A. Tree-Based QSAR Model for Drug Repurposing in the Discovery of New Antibacterial Compounds Against Escherichia coli. Pharmaceuticals 2020, 13, 431. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Wang, X.S.; Roth, B.L.; Golbraikh, A.; Tropsha, A. Application of Quantitative Structure–Activity Relationship Models of 5-HT1A Receptor Binding to Virtual Screening Identifies Novel and Potent 5-HT1A Ligands. J. Chem. Inf. Model. 2014, 54, 634–647. [Google Scholar] [CrossRef] [PubMed]
- Guasch, L.; Zakharov, A.V.; Tarasova, O.A.; Poroikov, V.V.; Liao, C.; Nicklaus, M.C. Novel HIV-1 Integrase Inhibitor Development by Virtual Screening Based on QSAR Models. Curr. Top. Med. Chem. 2016, 16, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Zaki, M.E.A.; Al-Hussain, S.A.; Masand, V.H.; Akasapu, S.; Bajaj, S.O.; El-Sayed, N.N.E.; Ghosh, A.; Lewaa, I. Identification of Anti-SARS-CoV-2 Compounds from Food Using QSAR-Based Virtual Screening, Molecular Docking, and Molecular Dynamics Simulation Analysis. Pharmaceuticals 2021, 14, 357. [Google Scholar] [CrossRef]
- Alamri, M.A.; Alamri, M.A. Pharmacophore and docking-based sequential virtual screening for the identification of novel Sigma 1 receptor ligands. Bioinformation 2019, 15, 586–595. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Yin, J.; Yao, J.; Xu, Z.; Tao, Y.; Zhang, H. Pharmacophore-Based Virtual Screening Toward the Discovery of Novel Anti-echinococcal Compounds. Front. Cell. Infect. Microbiol. 2020, 10, 118. [Google Scholar] [CrossRef]
- Poli, G.; Dimmito, M.P.; Mollica, A.; Zengin, G.; Benyhe, S.; Zador, F.; Stefanucci, A. Discovery of Novel µ-Opioid Receptor Inverse Agonist from a Combinatorial Library of Tetrapeptides through Structure-Based Virtual Screening. Molecules 2019, 24, 3872. [Google Scholar] [CrossRef] [Green Version]
- Shah, B.M.; Modi, P.; Trivedi, P. Pharmacophore- based virtual screening, 3D- QSAR, molecular docking approach for identification of potential dipeptidyl peptidase IV inhibitors. J. Biomol. Struct. Dyn. 2021, 39, 2021–2043. [Google Scholar] [CrossRef]
- Bommu, U.D.; Konidala, K.K.; Pabbaraju, N.; Yeguvapalli, S. QSAR modeling, pharmacophore-based virtual screening, and ensemble docking insights into predicting potential epigallocatechin gallate (EGCG) analogs against epidermal growth factor receptor. J. Recept. Signal Transduct. 2019, 39, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Vora, J.; Patel, S.; Sinha, S.; Sharma, S.; Srivastava, A.; Chhabria, M.; Shrivastava, N. Structure based virtual screening, 3D-QSAR, molecular dynamics and ADMET studies for selection of natural inhibitors against structural and non-structural targets of Chikungunya. J. Biomol. Struct. Dyn. 2018, 37, 3150–3161. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Tool/Software | Main Features | Ref. |
|---|---|---|
| CCLab | Based on a multi-objective genetic algorithm, including synthesis cost and drug-likeness. | [29] |
| MoSELECT | Based on a multi-objective genetic algorithm, including diversity and “drug-like” physicochemical properties, and a fitness function. | [30] |
| KNIME | Based on generic reactions. | [31] |
| RDKit | Based on generic reactions. | [32] |
| DataWarrior | Molecules are designed following a given generic reaction and a list of real reactant structures. | [33] |
| Library synthesizer | Creates libraries through specification of a central scaffold with connection points and a list of R groups. | [35] |
| SimLib v2.0 | Libraries are built using SMILES and a scaffold-based approach. | [36] |
| GLARE | Allows one to optimize reagent lists for the design of combinatorial libraries. | [41] |
| Reactor (ChemAxon) | Library generated using generic reactions and considering reaction rules that yield chemically feasible products. | [34] |
| Molecular Operating Environment (MOE) | Scaffold-based. New chemical compounds are generated by attaching R groups to a common skeleton with marked points. | [37] |
| Schrödinger | Creates library by substituting attachments on a core structure with fragments from reagent compounds. | [38] |
| Nova | Uses central scaffolds and a list of R groups. | [39] |
| ChemDraw | Uses central scaffolds and a list of R groups. | [40] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suay-García, B.; Bueso-Bordils, J.I.; Falcó, A.; Antón-Fos, G.M.; Alemán-López, P.A. Virtual Combinatorial Chemistry and Pharmacological Screening: A Short Guide to Drug Design. Int. J. Mol. Sci. 2022, 23, 1620. https://doi.org/10.3390/ijms23031620
Suay-García B, Bueso-Bordils JI, Falcó A, Antón-Fos GM, Alemán-López PA. Virtual Combinatorial Chemistry and Pharmacological Screening: A Short Guide to Drug Design. International Journal of Molecular Sciences. 2022; 23(3):1620. https://doi.org/10.3390/ijms23031620
Chicago/Turabian StyleSuay-García, Beatriz, Jose I. Bueso-Bordils, Antonio Falcó, Gerardo M. Antón-Fos, and Pedro A. Alemán-López. 2022. "Virtual Combinatorial Chemistry and Pharmacological Screening: A Short Guide to Drug Design" International Journal of Molecular Sciences 23, no. 3: 1620. https://doi.org/10.3390/ijms23031620
APA StyleSuay-García, B., Bueso-Bordils, J. I., Falcó, A., Antón-Fos, G. M., & Alemán-López, P. A. (2022). Virtual Combinatorial Chemistry and Pharmacological Screening: A Short Guide to Drug Design. International Journal of Molecular Sciences, 23(3), 1620. https://doi.org/10.3390/ijms23031620