
Structural Insights into the Substrate Transport Mechanisms in GTR Transporters through Ensemble Docking

 , , , ,
, , , , 
Abstract
:1. Introduction
2. Results
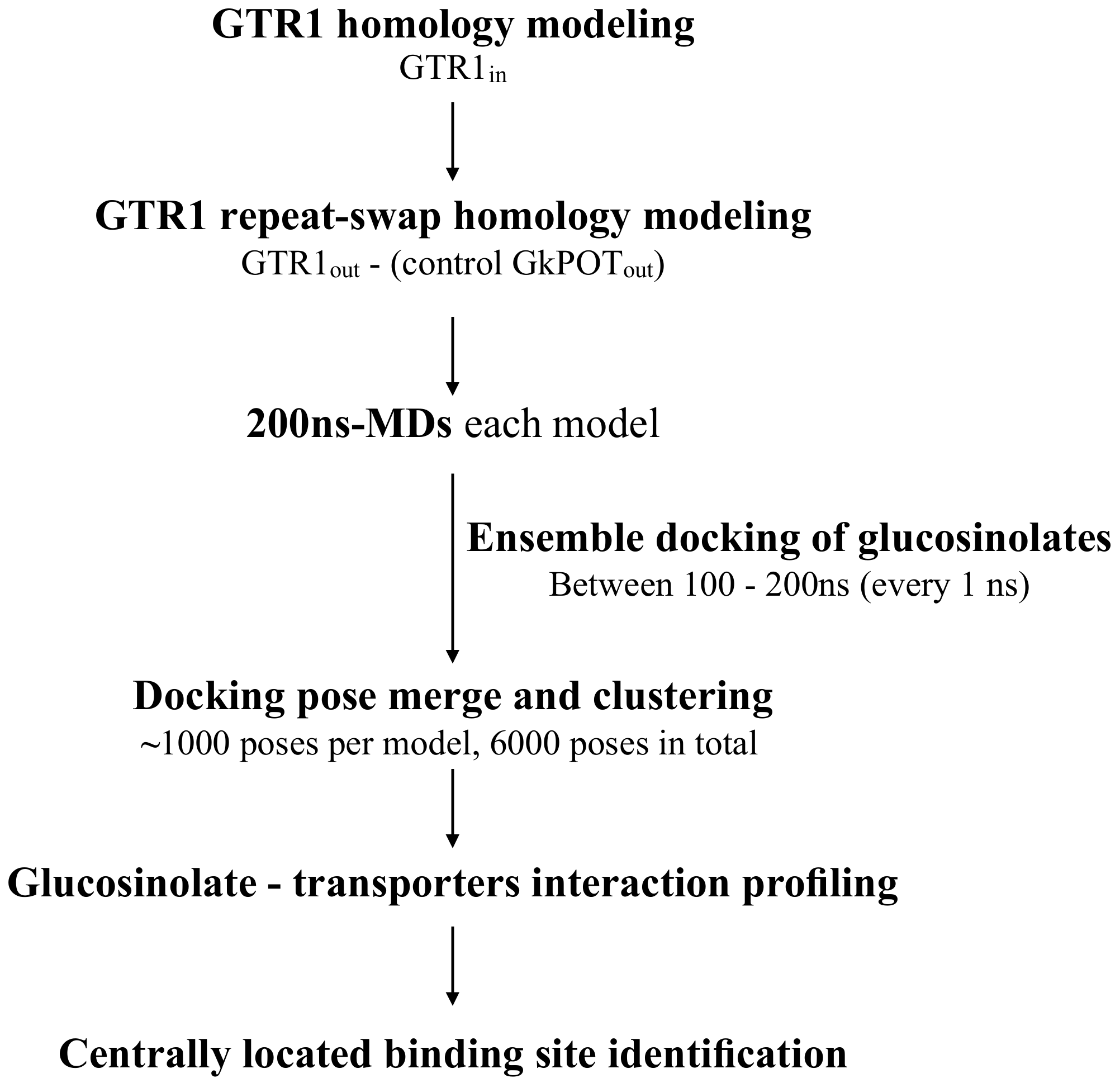
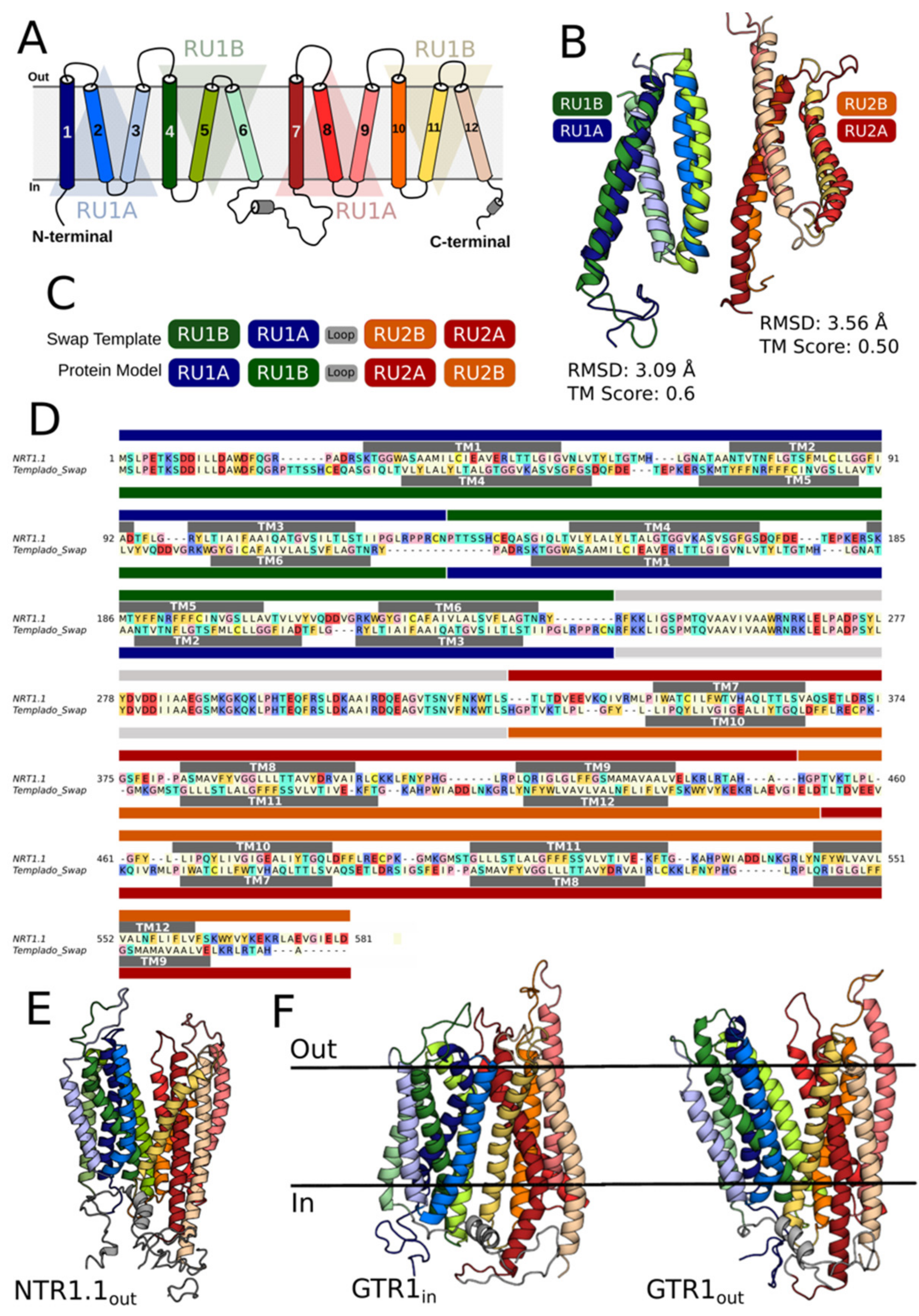
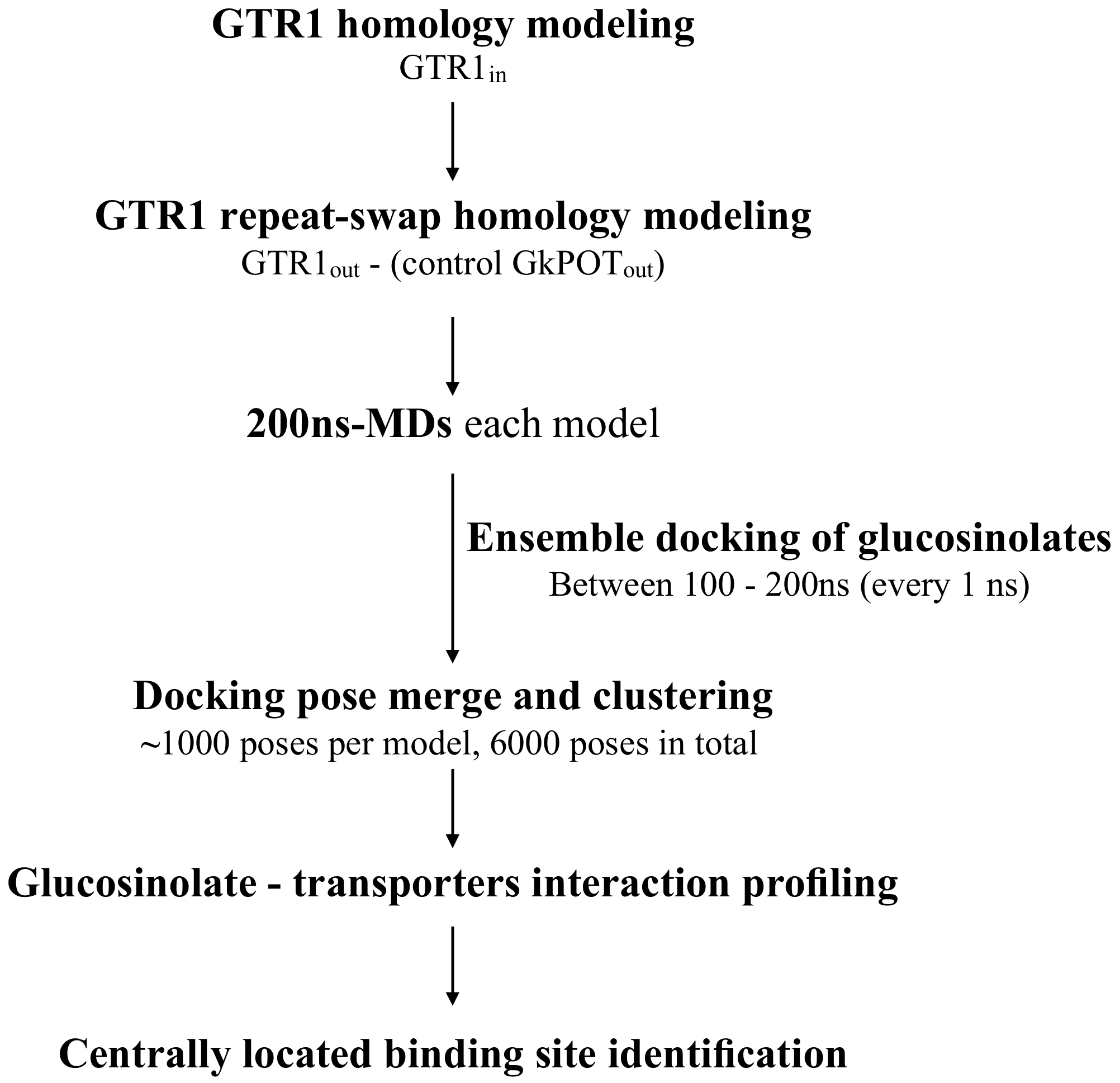
2.1. Molecular Modeling of GTR1 in Both Inward- and Outward-Facing Conformations
2.2. Generation of GTR1 Ensemble Conformations
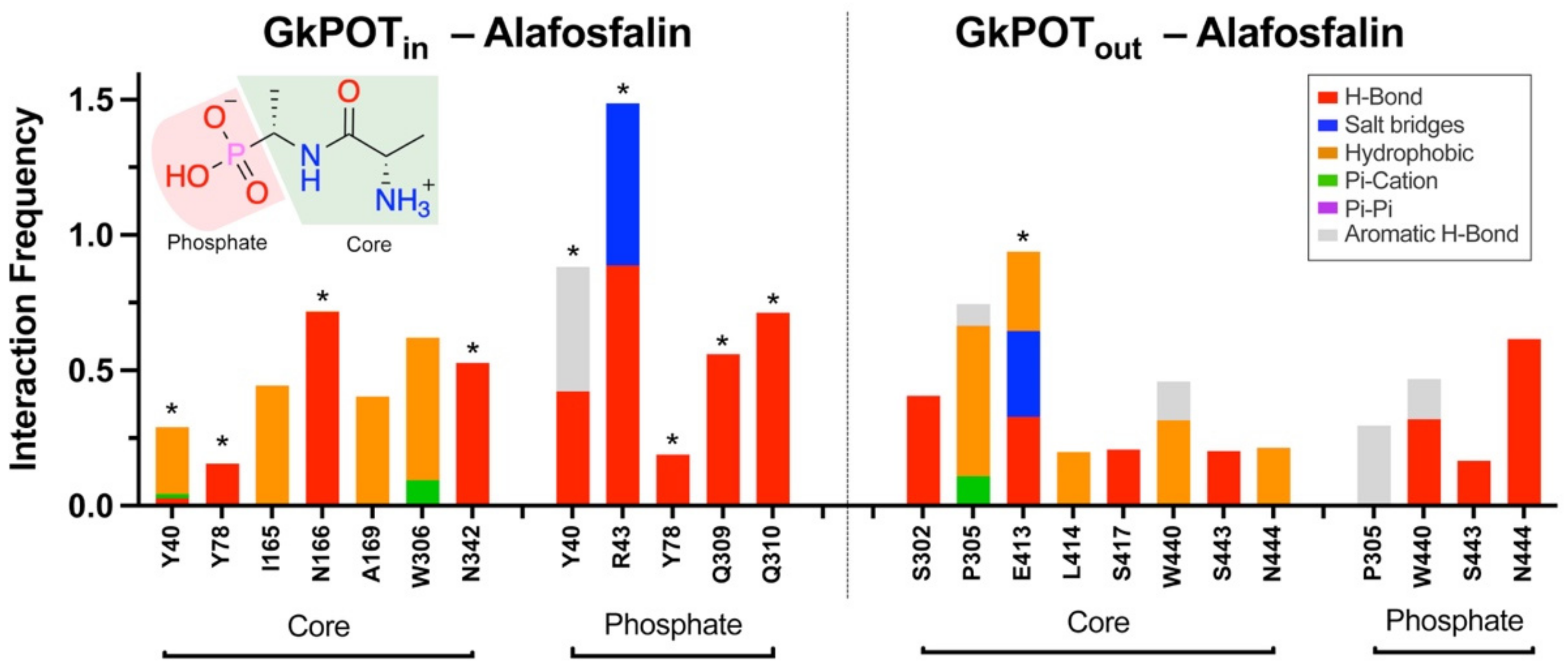
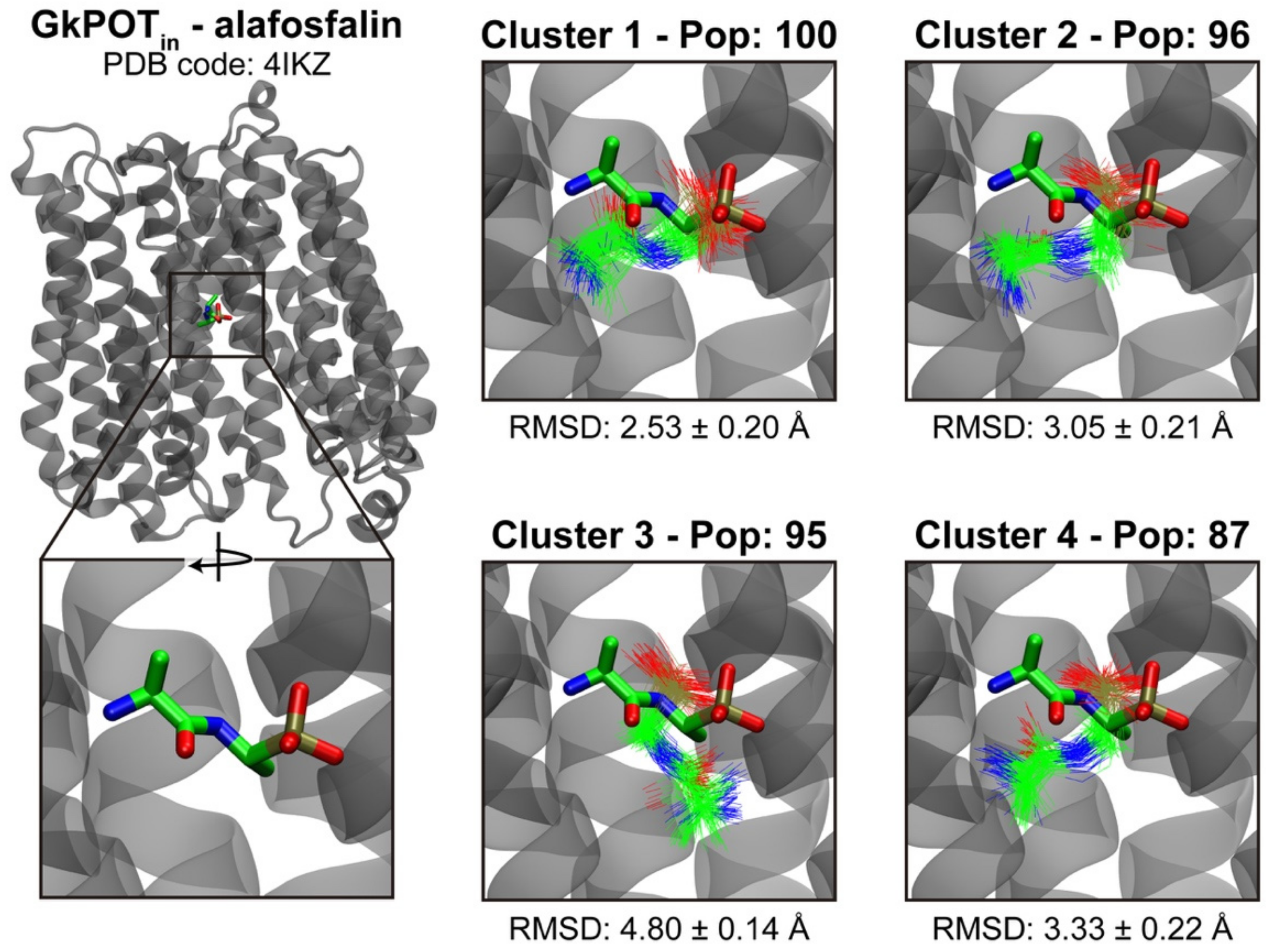
2.3. Testing the Ensemble Docking Pipeline
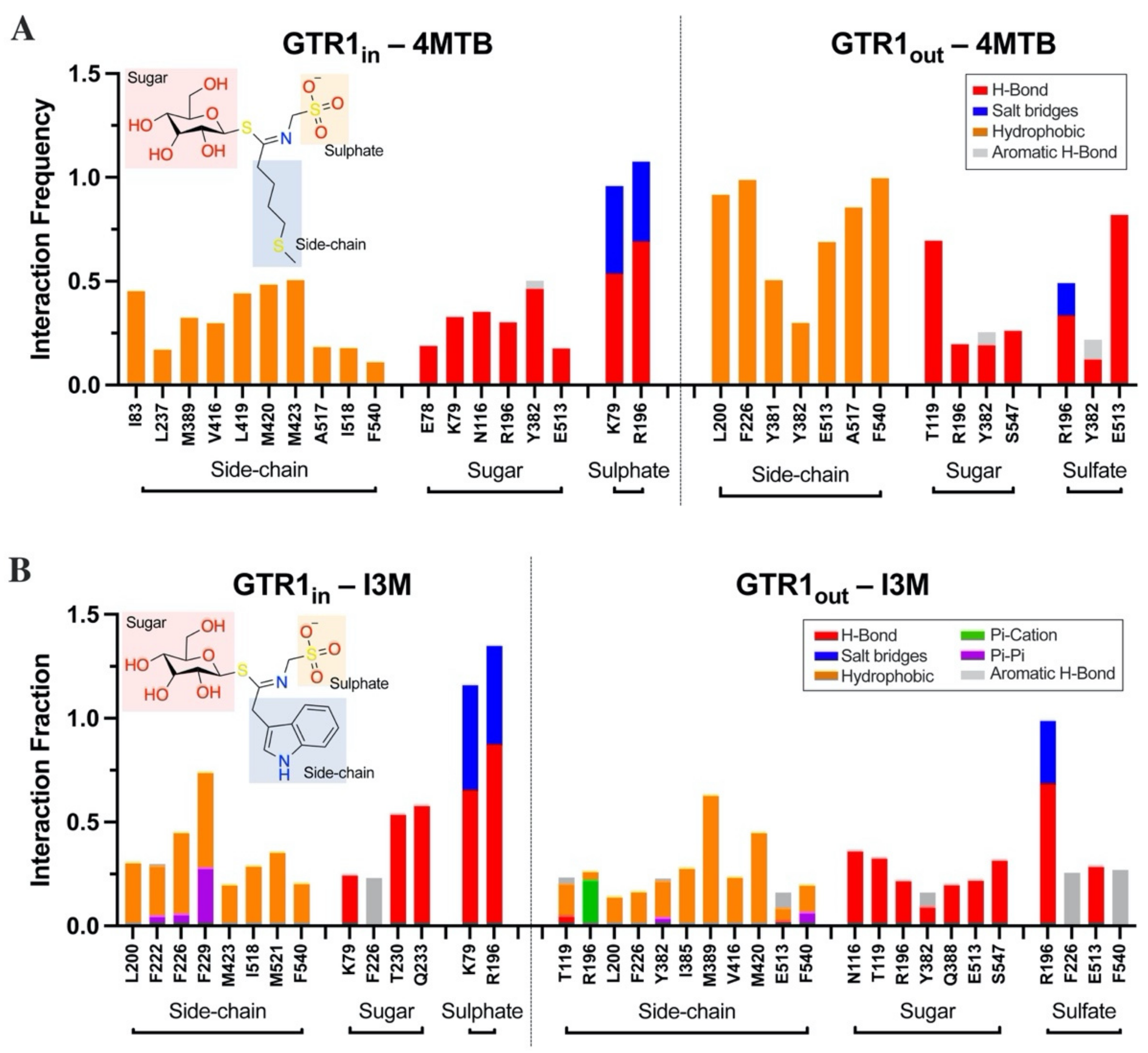
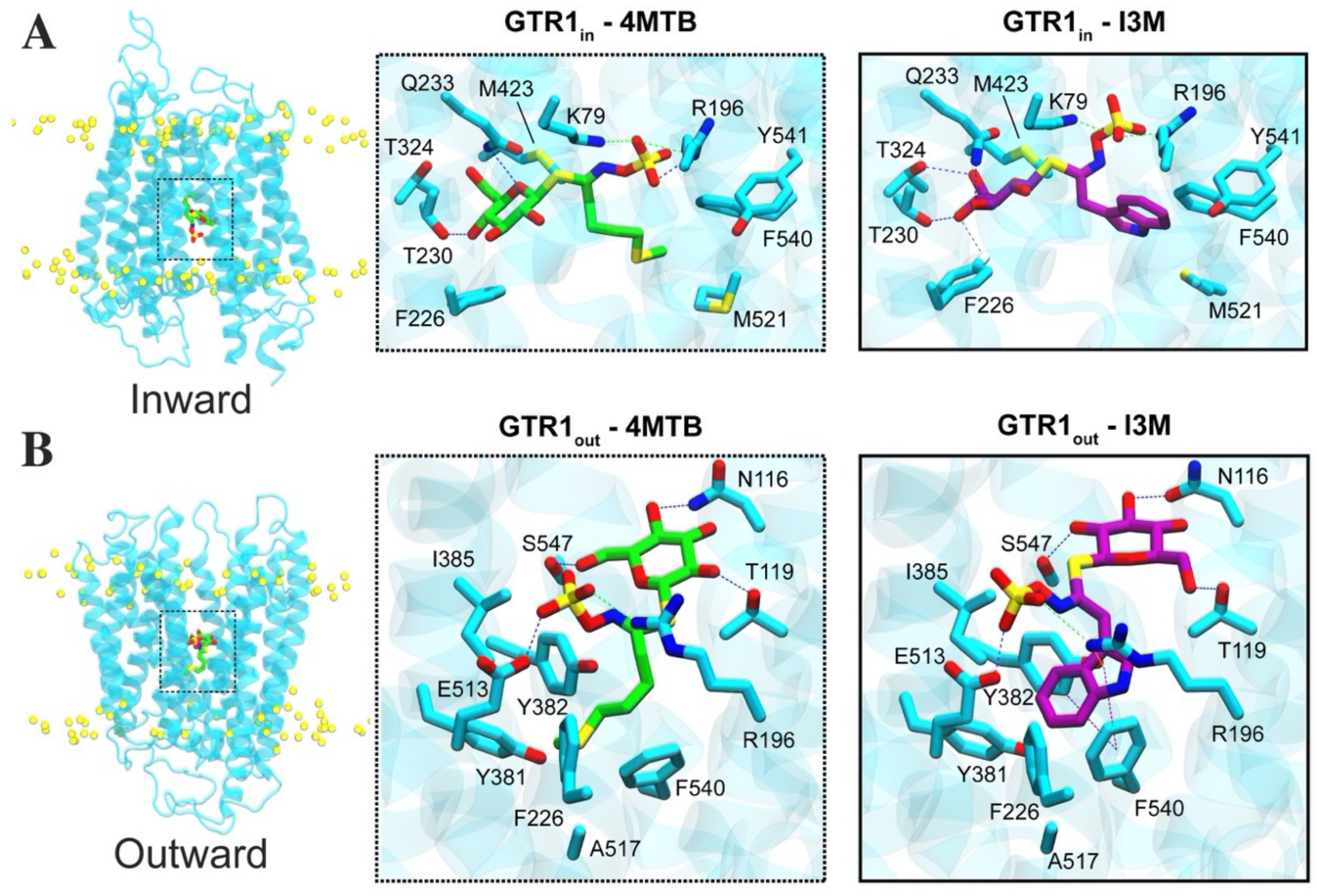
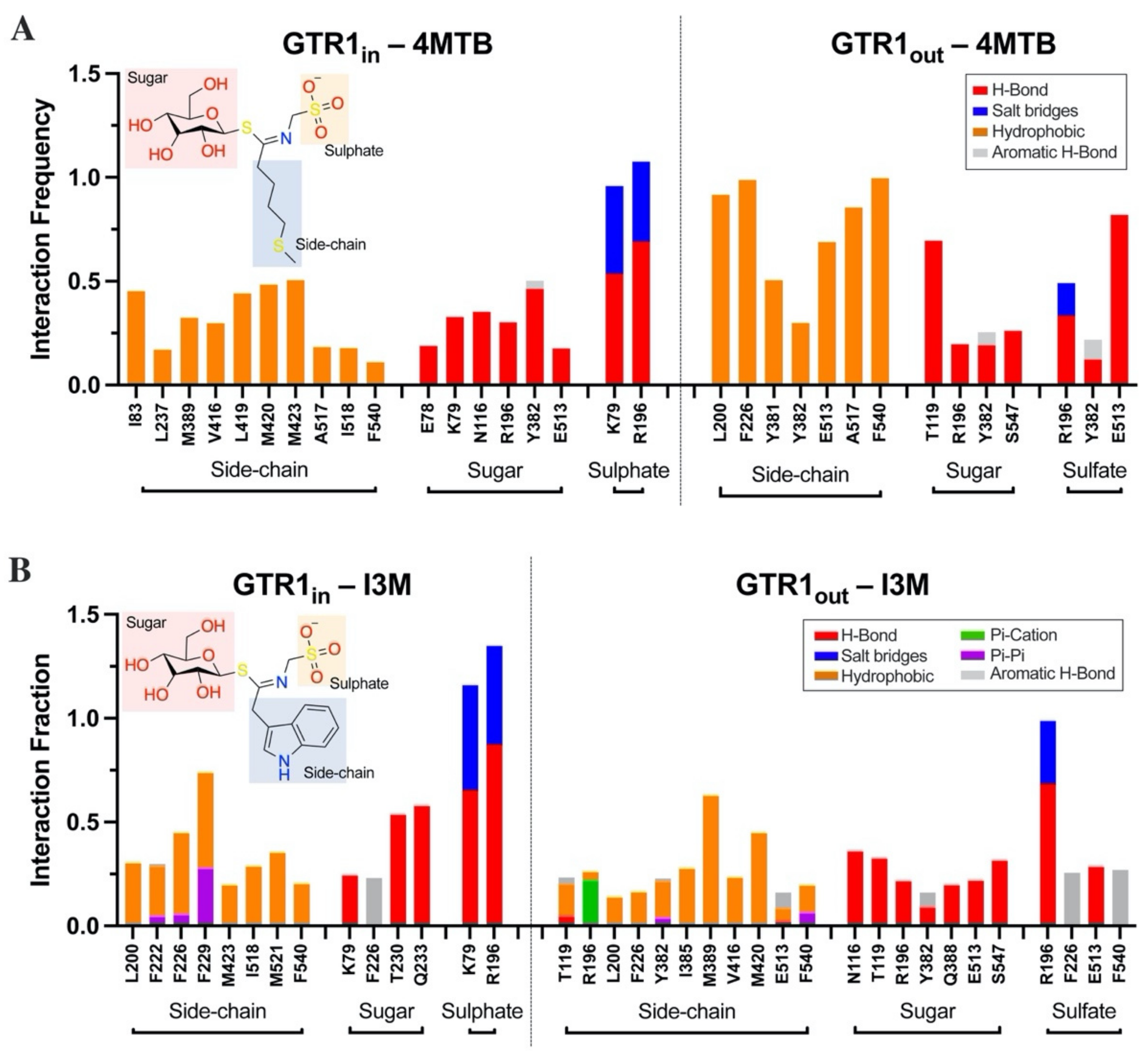
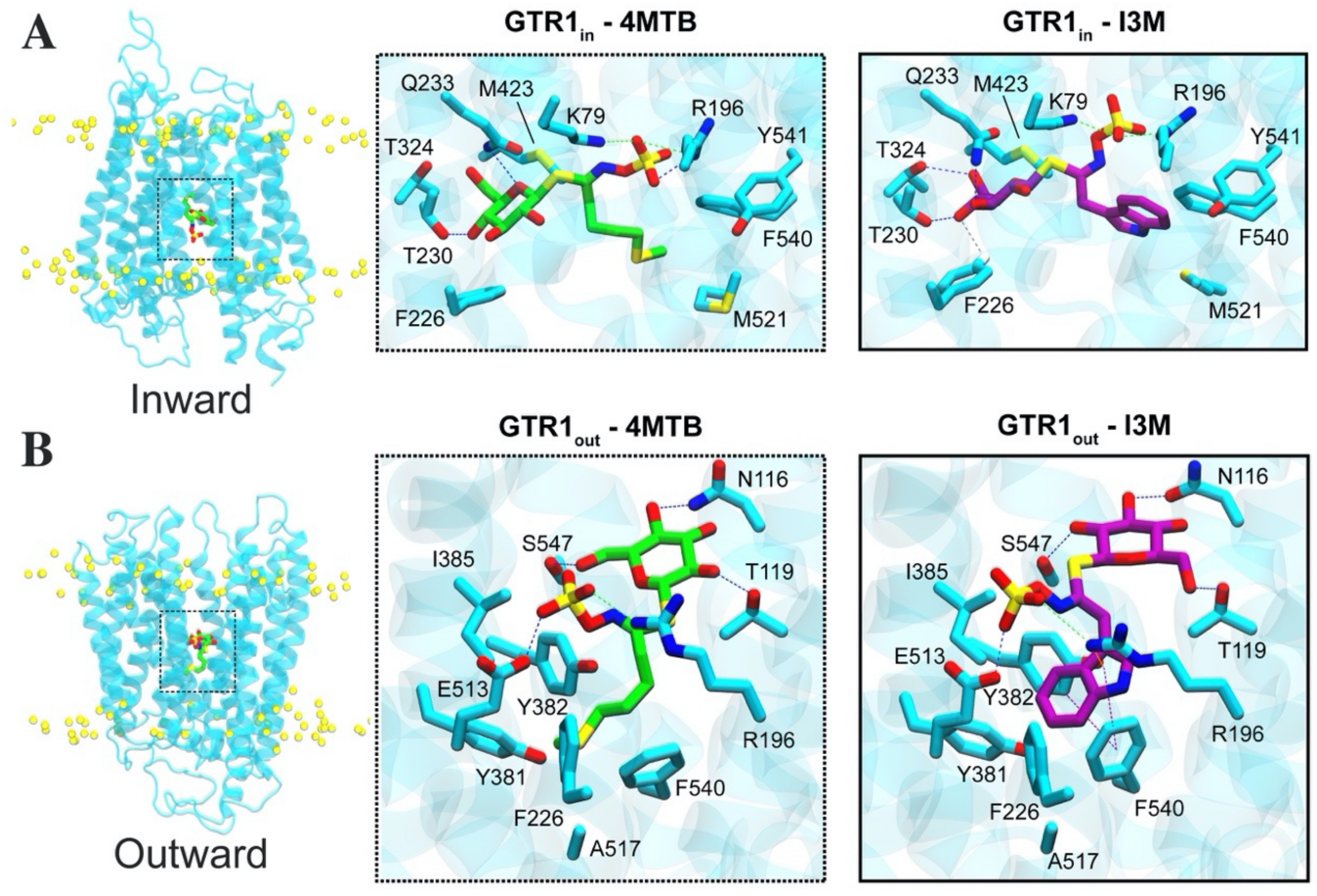
2.4. Glucosinolate Centrally Located Binding Site Revealed by Ensemble Docking
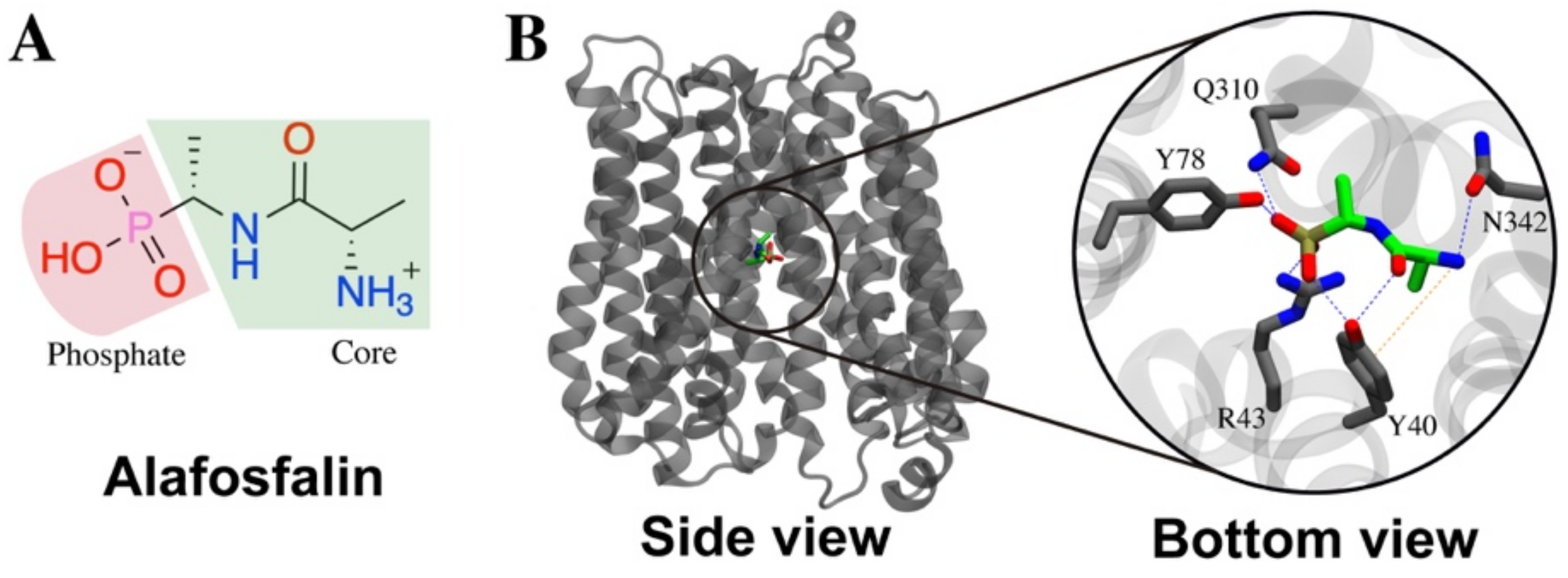
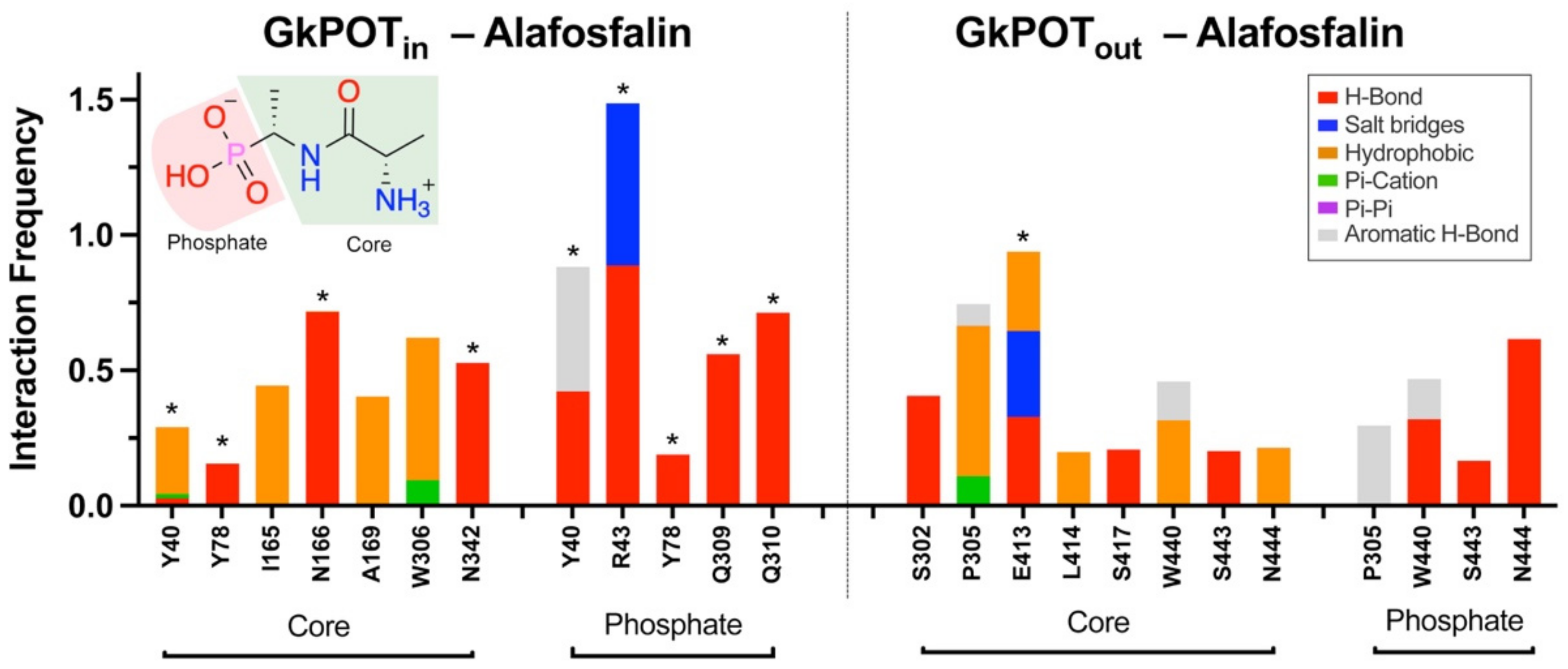
2.5. Contacts of Glucosinolates with Key Residues of the Centrally Located Binding Site and the Nature of the Chemical Interactions
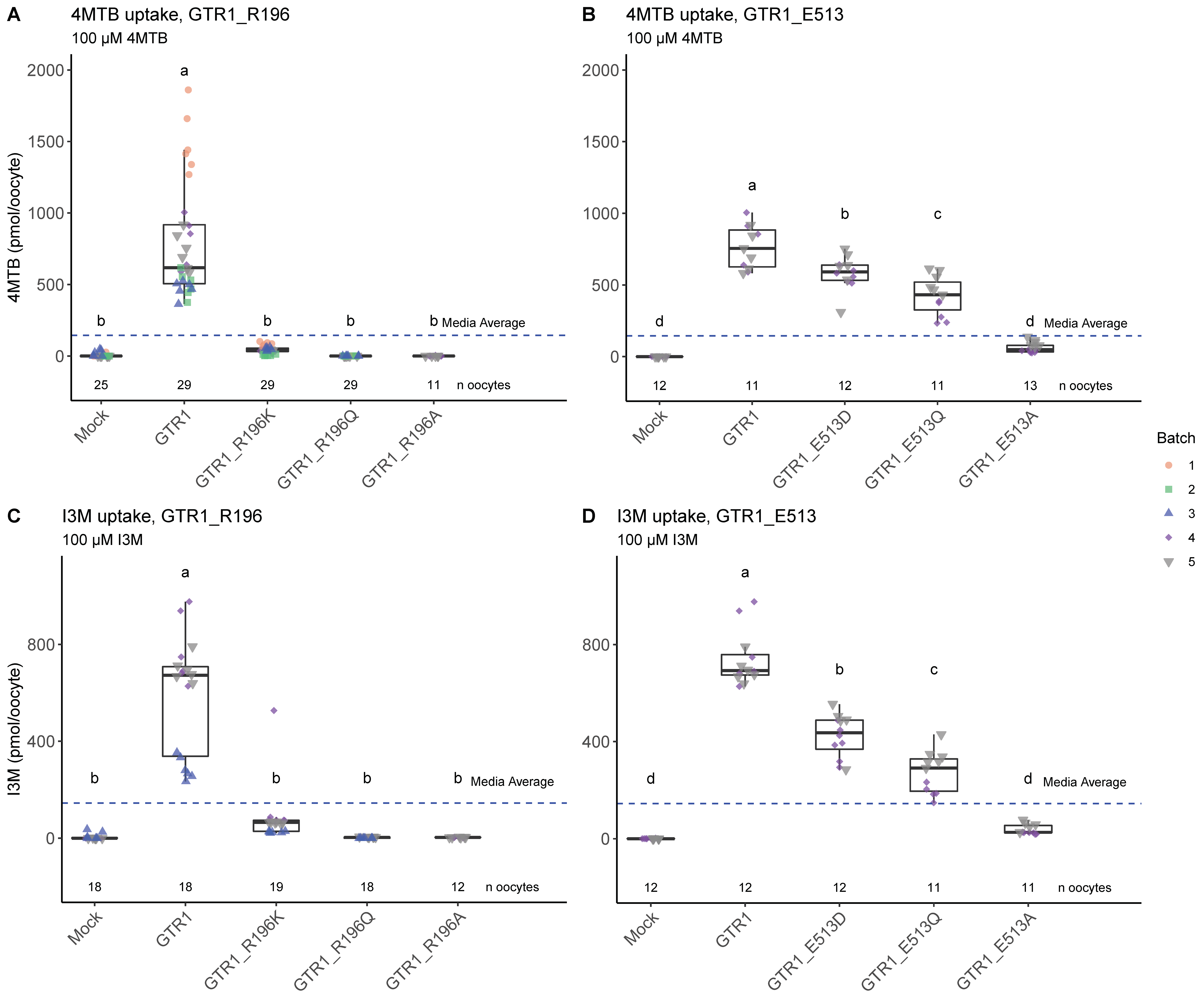
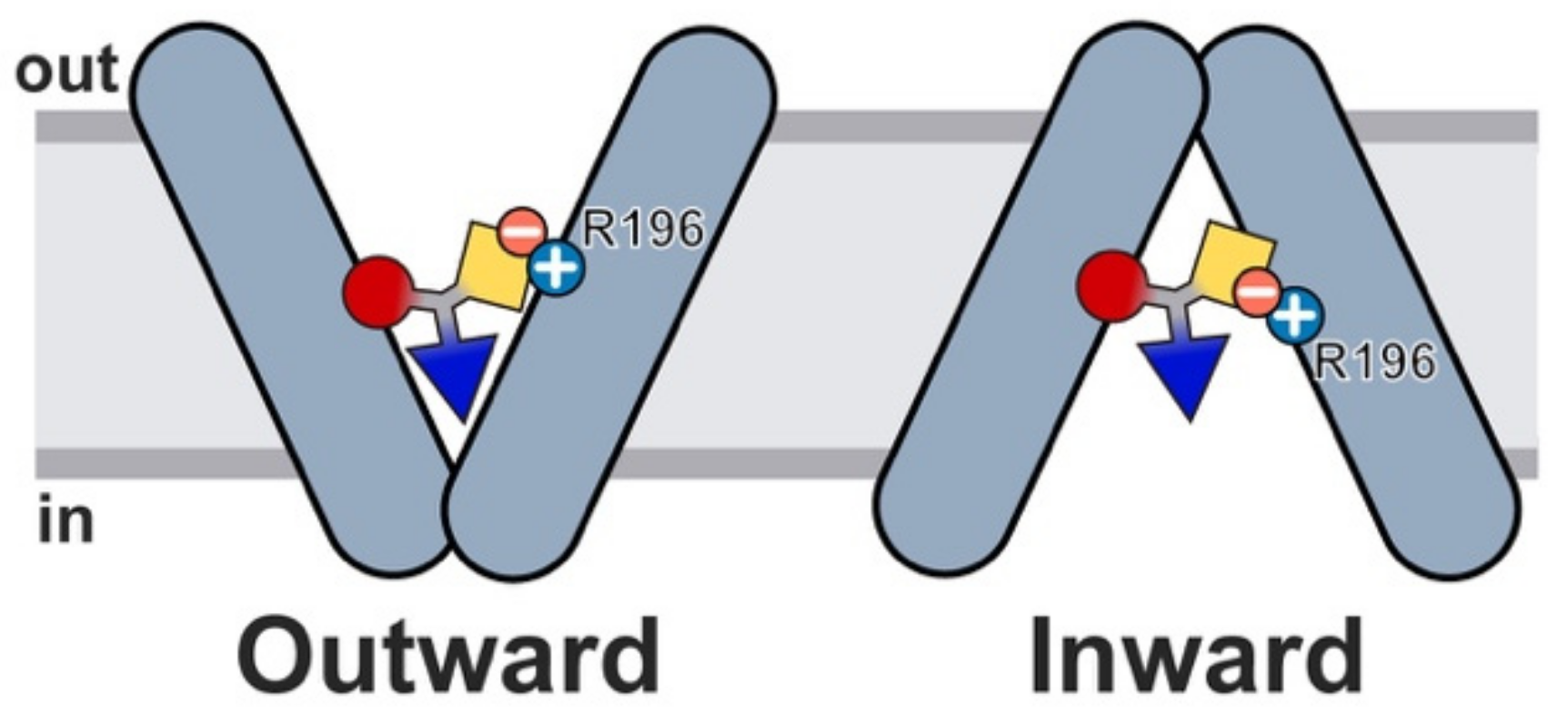
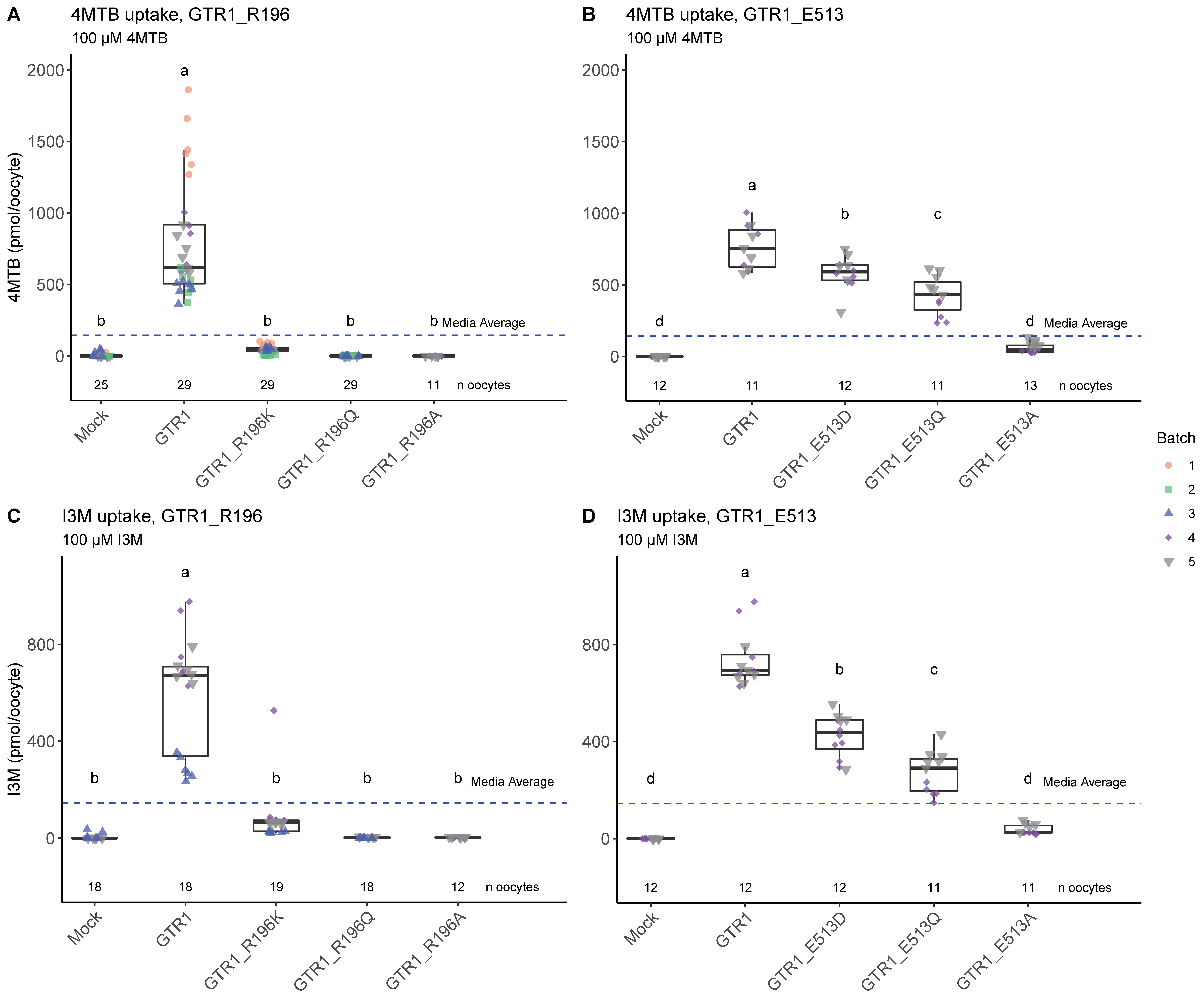
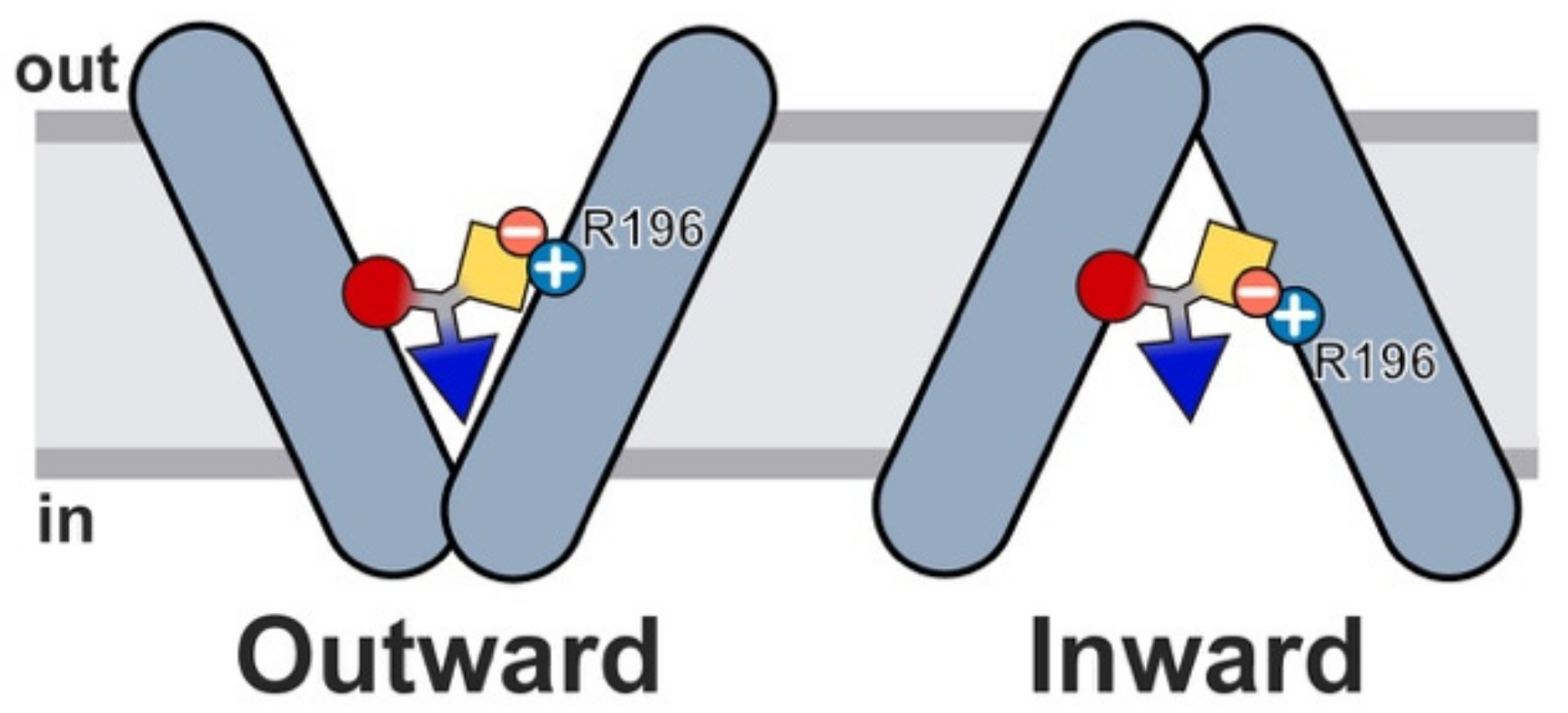
2.6. Glucosinolate Transport Is Governed by Ionic Interactions
3. Discussion
4. Materials and Methods
4.1. Molecular Modeling of GTR1
4.2. Molecular Modeling of GkPOT
4.3. Molecular Dynamics Simulations (MDs)
4.4. Ensemble Docking of 4MTB and I3M into GTR1 Transporter
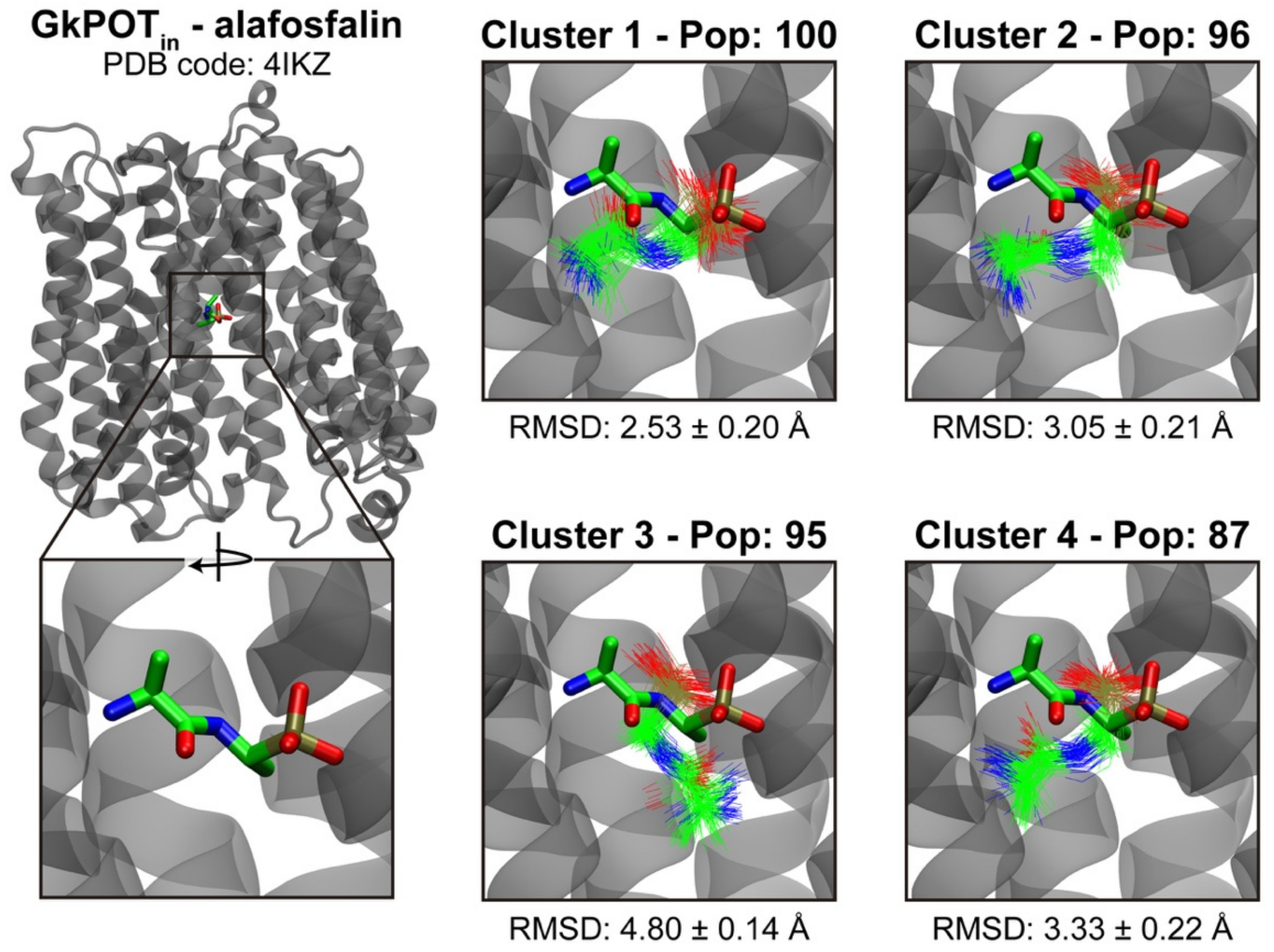
4.5. Clustering and Docking Postprocessing
4.6. Cloning of Mutants for Xenopus Oocyte Expression and cRNA Generation
4.7. Xenopus Oocytes Transport Assay
4.8. Glucosinolate Analysis by LC-MS/MS
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Corratgé-Faillie, C.; Lacombe, B. Substrate (un)specificity of Arabidopsis NRT1/PTR FAMILY (NPF) proteins. J. Exp. Bot. 2017, 68, 3107–3113. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.; Newstead, S. Molecular basis of nitrate uptake by the plant nitrate transporter NRT1.1. Nature 2014, 507, 68–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Bankston, J.R.; Payandeh, J.; Hinds, T.R.; Zagotta, W.N.; Zheng, N. Crystal structure of the plant dual-affinity nitrate transporter NRT1.1. Nature 2014, 507, 73–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.E.; Clémençon, B.; Hediger, M.A. Proton-coupled oligopeptide transporter family SLC15: Physiological, pharmacological and pathological implications. Mol. Asp. Med. 2013, 34, 323–336. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Mao, G.; Liu, M.; Zhang, L.; Wang, X.; Zhang, X.C. Crystal structure of the E. coli peptide transporter YbgH. Structure 2014, 22, 1152–1160. [Google Scholar] [CrossRef] [Green Version]
- Doki, S.; Kato, H.E.; Solcan, N.; Iwaki, M.; Koyama, M.; Hattori, M.; Iwase, N.; Tsukazaki, T.; Sugita, Y.; Kandori, H.; et al. Structural basis for dynamic mechanism of proton-coupled symport by the peptide transporter POT. Proc. Natl. Acad. Sci. USA 2013, 110, 11343–11348. [Google Scholar] [CrossRef] [Green Version]
- Newstead, S.; Drew, D.; Cameron, A.D.; Postis, V.L.G.; Xia, X.; Fowler, P.W.; Ingram, J.C.; Carpenter, E.P.; Sansom, M.; McPherson, M.J.; et al. Crystal structure of a prokaryotic homologue of the mammalian oligopeptide—Proton symporters, PepT1 and PepT2. EMBO J. 2011, 30, 417–426. [Google Scholar] [CrossRef] [Green Version]
- Guettou, F.; Quistgaard, E.M.; Trésaugues, L.; Moberg, P.; Jegerschöld, C.; Zhu, L.; Jong, A.J.O.; Nordlund, P.; Löw, C. Structural insights into substrate recognition in proton-dependent oligopeptide transporters. EMBO Rep. 2013, 14, 804–810. [Google Scholar] [CrossRef] [Green Version]
- Guettou, F.; Quistgaard, E.M.; Raba, M.; Moberg, P.; Löw, C.; Nordlund, P. Selectivity mechanism of a bacterial homolog of the human drug-peptide transporters PepT1 and PepT2. Nat. Struct. Mol. Biol. 2014, 21, 728. [Google Scholar] [CrossRef] [Green Version]
- Yan, N. Structural Biology of the Major Facilitator Superfamily Transporters. Annu. Rev. Biophys. 2015, 44, 257–283. [Google Scholar] [CrossRef]
- Zhang, X.C.; Zhao, Y.; Heng, J.; Jiang, D. Energy coupling mechanisms of MFS transporters. Protein Sci. 2015, 24, 1560–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniel, H.; Spanier, B.; Kottra, G.; Weitz, D. From bacteria to man: Archaic proton-dependent peptide transporters at work. Physiology 2006, 21, 93–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newstead, S. Molecular insights into proton coupled peptide transport in the PTR family of oligopeptide transporters. Biochim. Biophys. Acta BBA-Gen. Subj. 2015, 1850, 488–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aduri, N.G.; Prabhala, B.K.; Ernst, H.A.; Jørgensen, F.S.; Olsen, L.; Mirza, O. Salt-bridge Swapping in the EXXERFXYY Motif of Proton Coupled Oligopeptide Transporters. J. Biol. Chem. 2015, 290, 29931–29940. [Google Scholar] [CrossRef] [Green Version]
- Jørgensen, M.E.; Olsen, C.E.; Geiger, D.; Mirza, O.; Halkier, B.A.; Nour-Eldin, H.H. A Functional EXXEK Motif is Essential for Proton Coupling and Active Glucosinolate Transport by NPF2.11. Plant Cell Physiol. 2015, 56, 2340–2350. [Google Scholar] [CrossRef] [Green Version]
- Redovnikovic, I.R.; Glivetic, T.; Delonga, K.; Vorkapic-Furac, J. Glucosinolates and their potential role in plant. Period. Biol. 2008, 110, 297–309. [Google Scholar]
- Agerbirk, N.; Olsen, C.E. Glucosinolate structures in evolution. Phytochemistry 2012, 77, 16–45. [Google Scholar] [CrossRef]
- Nour-Eldin, H.; Andersen, T.G.; Burow, M.; Madsen, S.R.; Jørgensen, M.E.; Olsen, C.E.; Dreyer, I.; Hedrich, R.; Geiger, D.; Halkier, B.A. NRT/PTR transporters are essential for translocation of glucosinolate defence compounds to seeds. Nature 2012, 488, 531–534. [Google Scholar] [CrossRef]
- Jørgensen, M.; Xu, D.; Crocoll, C.; Ramírez, D.; Motawia, M.; Olsen, C.; Nour-Eldin, H.; Halkie, B. Origin and evolution of a transporter substrate specificity. eLife 2017, 6, e19466. [Google Scholar] [CrossRef]
- Lyons, J.A.; Parker, J.L.; Solcan, N.; Brinth, A.; Li, D.; Shah, S.T.A.; Caffrey, M.; Newstead, S. Structural basis for polyspecificity in the POT family of proton-coupled oligopeptide transporters. EMBO Rep. 2014, 15, 886–893. [Google Scholar] [CrossRef]
- Solcan, N.; Kwok, J.; Fowler, P.W.; Cameron, A.D.; Drew, D.; Iwata, S.; Newstead, S. Alternating access mechanism in the POT family of oligopeptide transporters. EMBO J. 2012, 31, 3411–3421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaro, R.E.; Baudry, J.; Chodera, J.; Demir, Ö.; McCammon, J.A.; Miao, Y.; Smith, J.C. Ensemble docking in drug discovery. Biophys. J. 2018, 114, 2271–2278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.-Y.; Zou, X. Ensemble docking of multiple protein structures: Considering protein structural variations in molecular docking. Proteins Struct. Funct. Bioinform. 2007, 66, 399–421. [Google Scholar] [CrossRef] [PubMed]
- Caballero, J. The latest automated docking technologies for novel drug discovery. Expert Opin. Drug Discov. 2021, 16, 625–645. [Google Scholar] [CrossRef]
- Novoa, E.M.; de Pouplana, L.R.; Barril, X.; Orozco, M. Ensemble docking from homology models. J. Chem. Theory Comput. 2010, 6, 2547–2557. [Google Scholar] [CrossRef]
- Ellingson, S.R.; Miao, Y.; Baudry, J.; Smith, J.C. Multi-conformer ensemble docking to difficult protein targets. J. Phys. Chem. B 2015, 119, 1026–1034. [Google Scholar] [CrossRef]
- Tam, H.-K.; Malviya, V.N.; Foong, W.-E.; Herrmann, A.; Malloci, G.; Ruggerone, P.; Vargiu, A.V.; Pos, K.M. Binding and transport of carboxylated drugs by the multidrug transporter AcrB. J. Mol. Biol. 2020, 432, 861–877. [Google Scholar] [CrossRef]
- Malvacio, I.; Buonfiglio, R.; D’Atanasio, N.; Serra, G.; Bosin, A.; Di Giorgio, F.P.; Ruggerone, P.; Ombrato, R.; Vargiu, A.V. Molecular basis for the different interactions of congeneric substrates with the polyspecific transporter AcrB. Biochim. Biophys. Acta BBA-Biomembr. 2019, 1861, 1397–1408. [Google Scholar] [CrossRef]
- Kaufmann, K.W.; Dawson, E.S.; Henry, L.K.; Field, J.R.; Blakely, R.D.; Meiler, J. Structural determinants of species-selective substrate recognition in human and Drosophila serotonin transporters revealed through computational docking studies. Proteins Struct. Funct. Bioinform. 2009, 74, 630–642. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Mandell, D.J.; Coutsias, E.A.; Kortemme, T. Sub-angstrom accuracy in protein loop reconstruction by robotics-inspired conformational sampling. Nat. Methods 2009, 6, 551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohl, C.A.; Strauss, C.E.M.; Misura, K.M.S.; Baker, D. Protein Structure Prediction Using Rosetta. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2004; Volume 383, pp. 66–93. [Google Scholar]
- Stamm, M.; Staritzbichler, R.; Khafizov, K.; Forrest, L.R. AlignMe—A membrane protein sequence alignment web server. Nucleic Acids Res. 2014, 42, W246–W251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.S.; Eramian, D.; Shen, M.; Pieper, U.; Sali, A. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinform. 2006, 15, 5–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchan, D.W.A.; Minneci, F.; Nugent, T.C.O.; Bryson, K.; Jones, D.T. Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 2013, 41, W349–W357. [Google Scholar] [CrossRef]
- Tsirigos, K.D.; Peters, C.; Shu, N.; Käll, L.; Elofsson, A. The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Res. 2015, 43, W401–W407. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Ray, A.; Lindahl, E.; Wallner, B. Model quality assessment for membrane proteins. Bioinformatics 2010, 26, 3067–3074. [Google Scholar] [CrossRef] [Green Version]
- Forrest, L.R.; Zhang, Y.-W.; Jacobs, M.T.; Gesmonde, J.; Xie, L.; Honig, B.H.; Rudnick, G. Mechanism for alternating access in neurotransmitter transporters. Proc. Natl. Acad. Sci. USA 2008, 105, 10338–10343. [Google Scholar] [CrossRef] [Green Version]
- Vergara-Jaque, A.; Fenollar-Ferrer, C.; Kaufmann, D.; Forrest, L.R. Repeat-swap homology modeling of secondary active transporters: Updated protocol and prediction of elevator-type mechanisms. Front. Pharmacol. 2015, 6, 183. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Skolnick, J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK a prediction and protonation state generation for drug-like molecules. J. Comput. Aided. Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical pK a predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Merz, K.M. Application of the Nosé-Hoover chain algorithm to the study of protein dynamics. J. Phys. Chem. 1996, 100, 1927–1937. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; IEEE: Manhattan, NY, USA, 2006; p. 43. [Google Scholar]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L. OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- DeLano, W.L. PyMOL; DeLano Sci: San Carlos, CA, USA, 2002; Volume 700. [Google Scholar]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Jiménez, A.; Peña-Varas, C.; Borrego-Muñoz, P.; Arrue, L.; Alegría-Arcos, M.; Nour-Eldin, H.; Dreyer, I.; Nuñez-Vivanco, G.; Ramírez, D. Psc-db: A structured and searchable 3d-database for plant secondary compounds. Molecules 2021, 26, 1124. [Google Scholar] [CrossRef]
- Schrödinger LLC. Schrödinger Release 2020-3 LigPrep; Schrödinger LLC: New York, NY, USA, 2020. [Google Scholar]
- Totrov, M.; Abagyan, R. Flexible ligand docking to multiple receptor conformations: A practical alternative. Curr. Opin. Struct. Biol. 2008, 18, 178–184. [Google Scholar] [CrossRef] [Green Version]
- Feixas, F.; Lindert, S.; Sinko, W.; McCammon, A. Exploring the Role of Receptor Flexibility in Structure-Based Drug Discovery. Biophys. Chem. 2015, 186, 31–45. [Google Scholar] [CrossRef] [Green Version]
- Lorenzen, S.; Zhang, Y. Identification of near-native structures by clustering protein docking conformations. Proteins Struct. Funct. Bioinform. 2007, 68, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Shenkin, P.S.; Mcdonald, D.Q. Cluster Analysis of Molecular Conformations. J. Comput. Chem. 1994, 15, 899–916. [Google Scholar] [CrossRef]
- Bottegoni, G.; Cavalli, A.; Recanatini, M. A comparative study on the application of hierarchical-agglomerative clustering approaches to organize outputs of reiterated docking runs. J. Chem. Inf. Model. 2006, 46, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, D.; Arévalo, B.; Martínez, G.; Rinné, S.; Sepúlveda, F.V.; Decher, N.; González, W. Side fenestrations provide “anchor” for a stable binding of A1899 to the pore of TASK-1 potassium channels. Mol. Pharm. 2017, 14, 2197–2208. [Google Scholar] [CrossRef]
- Peña-Varas, C.; Ramírez, D. Docking Post-Processing: Interaction Frequency among Multiple-Cluster Conformers; Zenodo: 2021. Available online: https://zenodo.org/record/5498074#.YfU_evgRU2w.
- Nour-Eldin, H.H.; Hansen, B.G.; Nørholm, M.H.H.; Jensen, J.K.; Halkier, B.A. Advancing uracil-excision based cloning towards an ideal technique for cloning PCR fragments. Nucleic Acids Res. 2006, 34, e122. [Google Scholar] [CrossRef] [Green Version]
- Geu-Flores, F.; Nour-Eldin, H.H.; Nielsen, M.T.; Halkier, B.A. USER fusion: A rapid and efficient method for simultaneous fusion and cloning of multiple PCR products. Nucleic Acids Res. 2007, 35, e55. [Google Scholar] [CrossRef] [Green Version]
- Nørholm, M.H.H.; Nour-Eldin, H.H.; Brodersen, P.; Mundy, J.; Halkier, B.A. Expression of the Arabidopsis high-affinity hexose transporter STP13 correlates with programmed cell death. FEBS Lett. 2006, 580, 2381–2387. [Google Scholar] [CrossRef] [Green Version]
- Crocoll, C.; Halkier, B.A.; Burow, M. Analysis and quantification of glucosinolates. Curr. Protoc. Plant Biol. 2016, 1, 385–409. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystallographic Structures (Inward-Facing Conformation) | |||
| NTR1.1in (PDB code: 4OH3) | GkPOTin (PDB code: 4IKZ) | ||
| Model in an inward-facing conformation | |||
| GTR1in | |||
| Models in an outward-facing conformation | |||
| NTR1.1out | GkPOTout | GTR1out | |
| Cluster Nº | GkPOTin | GkPOTout |
|---|---|---|
| 1 | 100 | 142 |
| 2 | 96 | 142 |
| 3 | 95 | 91 |
| 4 | 87 | 86 |
| GTR1in-4MTB | GTR1in-I3M | GTR1out-4MTB | GTR1out-I3M | |
|---|---|---|---|---|
| 98 | 186 | 510 | 184 | |
| 81 | 173 | 96 | ||
| 73 | 88 | |||
| 70 | ||||
| 53 | ||||
| 50 | ||||
| 49 | ||||
| # Significant cluster | 7 | 2 | 1 | 3 |
| Total population | 986 | 965 | 979 | 993 |
| Total population Significant clusters | 474 | 359 | 510 | 368 |
| % conformers in significant clusters | 48.07% | 37.20% | 52.09% | 37.06% |
| GTR1 | GkPOT (PDB: 4IKZ) [6] | PepTSt (PDB: 4D2C) [20] | PepTSt (PDB: 4D2D) [20] | Function [14] |
|---|---|---|---|---|
| E75 | E32 | E22 | E22 | ExxE[K/R] motif [6,14,15] |
| E78 * | E35 | E25 | E25 | ExxE[K/R] motif [6] |
| K79 * | R36 | R26 * | R26 | ExxE[K/R] motif [6,15] |
| I82 | Y39 | Y29 | Y29 | Peptide specificity [21] |
| I83 | Y40 * | Y30 * | Y30 | Peptide specificity [6] |
| L86 | R43 * | R33 | R33 | Peptide specificity [6] |
| N116 * | Y78 * | Y68 | Y68 | Peptide binding [6] |
| T119 * | G81 | G71 | G71 | ------ |
| L200 | S140 | S130 | S130 | ------ |
| R196 * | K136 | K126 * | K126 | ExxE[K/R]-interactor [6] |
| F222 | F158 | F148 | F148 | ------ |
| F226 | Y162 | V152 | V152 | ------ |
| F229 | I165 | I155 | I155 | ------ |
| T230 | N166 | N156 | N156 | Peptide binding [6] |
| Q233 | A169 | A159 | A159 | ------ |
| Y382 | W306 | W296 | W296 | ------ |
| I385 | Q309 | E299 | E299 | Peptide binding/protonation [2,3] |
| T386 | E310Q * | E300 | E300 | ------ |
| M389 | S313 | S303 | S303 * | ------ |
| L419 | N342 * | N328 * | N328 | Peptide binding [6] |
| M420 | P343 | P329 | P329 | ------ |
| E513 | E413 | E400 | E400 * | Proton translocation [6] |
| A517 | S417 | S404 | S404 | ------ |
| F540 | W440 | W427 | W427 | ------ |
| Y541 | F441 | F428 | F428 | ------ |
| A544 | N444 | S431 | S431 | ------ |
| S547 * | A447 | G434 | G434 | ------ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peña-Varas, C.; Kanstrup, C.; Vergara-Jaque, A.; González-Avendaño, M.; Crocoll, C.; Mirza, O.; Dreyer, I.; Nour-Eldin, H.; Ramírez, D. Structural Insights into the Substrate Transport Mechanisms in GTR Transporters through Ensemble Docking. Int. J. Mol. Sci. 2022, 23, 1595. https://doi.org/10.3390/ijms23031595
Peña-Varas C, Kanstrup C, Vergara-Jaque A, González-Avendaño M, Crocoll C, Mirza O, Dreyer I, Nour-Eldin H, Ramírez D. Structural Insights into the Substrate Transport Mechanisms in GTR Transporters through Ensemble Docking. International Journal of Molecular Sciences. 2022; 23(3):1595. https://doi.org/10.3390/ijms23031595
Chicago/Turabian StylePeña-Varas, Carlos, Christa Kanstrup, Ariela Vergara-Jaque, Mariela González-Avendaño, Christoph Crocoll, Osman Mirza, Ingo Dreyer, Hussam Nour-Eldin, and David Ramírez. 2022. "Structural Insights into the Substrate Transport Mechanisms in GTR Transporters through Ensemble Docking" International Journal of Molecular Sciences 23, no. 3: 1595. https://doi.org/10.3390/ijms23031595
APA StylePeña-Varas, C., Kanstrup, C., Vergara-Jaque, A., González-Avendaño, M., Crocoll, C., Mirza, O., Dreyer, I., Nour-Eldin, H., & Ramírez, D. (2022). Structural Insights into the Substrate Transport Mechanisms in GTR Transporters through Ensemble Docking. International Journal of Molecular Sciences, 23(3), 1595. https://doi.org/10.3390/ijms23031595