
Update on Novel Targeted Therapy for Pleural Organization and Fibrosis


Abstract
1. Introduction
2. Organizing Pleuritis and Its Place as an Important Problem in Pleural Medicine
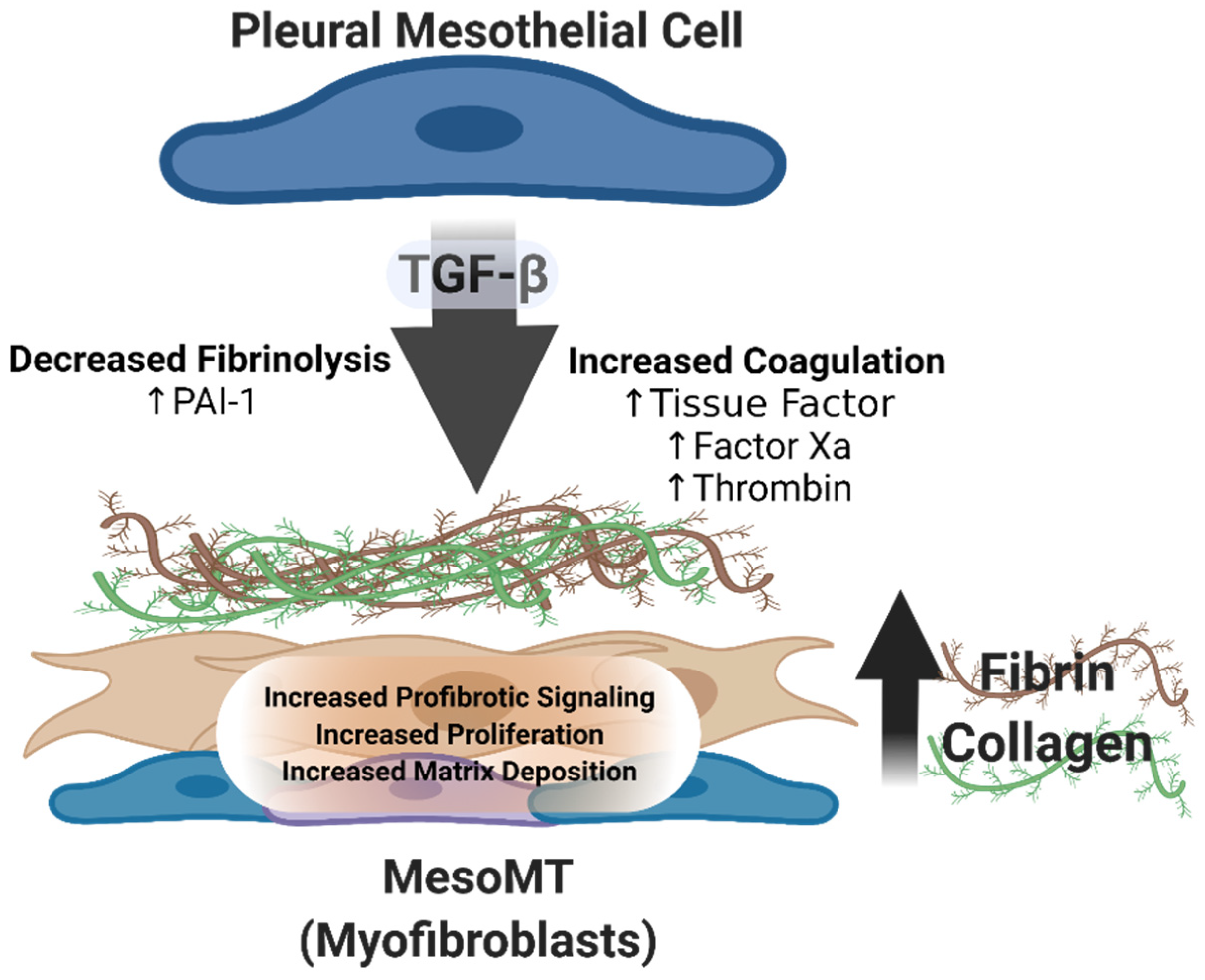
3. The Pathogenesis of Pleural Organization and the Role of Aberrant Fibrin Turnover and Mesomesenchymal Transition (MesoMT)
4. PAI-1
5. Mesothelial Cells
6. MesoMT
7. Novel Targets for Interventional Therapy

{kind=link}
| Novel Targets | Description |
|---|---|
| GSK-3β; Glycogen Synthase Kinase 3β | A serine/threonine kinase reported to regulate the function of glycogen synthase. Found to be activated by TGF-β and other MesoMT mediators by phosphoyratlion of tyrosine 216. Inhibition of GSK-3β with 9-ING-41 reversed pleural fibrosis [31,43]. |
| DOCK2; dedicator of cytokinesis 2 | Rac1 activating protein previously shown to regulate cellular phenotype. Recently found to be upregulated in pleural injury. DOCK2 deficiency protects against S. pneumoniae mediated pleural fibrosis [28]. |
| Myocardin | Smooth and cardiac muscle cell specific transcriptional coactivator of serum response factor. Found to be upregulated in pleural fibrosis and contributes to disease progression [29]. |
| EPCR; endothelial protein C receptor | An important regulator of Protein C whose expression was thought to be limited to the endothelium. Recently found to be important in the progression of pleural fibrosis [30]. |
| NOX1; NADPH oxidase 1 | A member of the NOX family whose expression is enhanced by factor Xa and thrombin in PMCs. Recently reported to be important for the progression of pleural fibrosis in vivo [27]. |
| uPAR; urokinase plasminogen activator receptor | A cell surface glycoprotein responsible for binding and localizing uPA to the surface of PMCs. Reported to be critical for the induction of MesoMT and the progression of PF [25,26,59]. |
| αβ crystallin | A small heat shock protein known to be enhanced by TGF-β. Reported to promote the progression of pulmonary and pleural fibrosis [73,74]. |
| CSP7; caveolin spanning peptide seven amino acid deletion fragment | A fragment of the caveolin spanning peptide. Reported to attenuate the progression of pulmonary fibrosis in murine models [75,76]. |
| mTOR; mechanistic target of rapamycin | A highly conserved serine/threonine kinase which regulates diverse cellular activities. Therapeutic targeting of the mTORC1 pathway was shown to block collagen synthesis in lung fibroblasts [81]. |
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Komissarov, A.A.; Rahman, N.; Lee, Y.C.G.; Florova, G.; Shetty, S.; Idell, R.; Ikebe, M.; Das, K.; Tucker, T.A.; Idell, S. Fibrin turnover and pleural organization: Bench to bedside. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L757–L768. [Google Scholar] [CrossRef]
- Popowicz, N.; Idell, S.; Lee, Y.C.G. Pathogenesis of pleural infection: A complex warfare. Respirology 2018, 23, 8–9. [Google Scholar] [CrossRef]
- Balbir-Gurman, A.; Yigla, M.; Nahir, A.M.; Braun-Moscovici, Y. Rheumatoid pleural effusion. Semin. Arthritis Rheum 2006, 35, 368–378. [Google Scholar] [CrossRef]
- Colice, G.L.; Curtis, A.; Deslauriers, J.; Heffner, J.; Light, R.; Littenberg, B.; Sahn, S.; Weinstein, R.A.; Yusen, R.D. Medical and surgical treatment of parapneumonic effusions: An evidence-based guideline. Chest 2000, 118, 1158–1171. [Google Scholar] [CrossRef]
- Light, R.W. Parapneumonic effusions and empyema. Proc. Am. Thorac. Soc. 2006, 3, 75–80. [Google Scholar] [CrossRef]
- Idell, S.; Rahman, N.M. Intrapleural Fibrinolytic Therapy for Empyema and Pleural Loculation: Knowns and Unknowns. Ann. Am. Thorac. Soc. 2018, 15, 515–517. [Google Scholar] [CrossRef]
- Beckert, L.; Brockway, B.; Simpson, G.; Southcott, A.M.; Lee, Y.C.G.; Rahman, N.; Light, R.W.; Shoemaker, S.; Gillies, J.; Komissarov, A.A.; et al. Phase 1 trial of intrapleural LTI-01; single chain urokinase in complicated parapneumonic effusions or empyema. JCI Insight 2019, 4, e127470. [Google Scholar] [CrossRef]
- Huggins, J.T.; Sahn, S.A. Causes and management of pleural fibrosis. Respirology 2004, 9, 441–447. [Google Scholar] [CrossRef]
- Antony, V.B.; Mohammed, K.A. Pathophysiology of pleural space infections. Semin. Respir. Infect. 1999, 14, 9–17. [Google Scholar]
- Antony, V.B.; Hott, J.W.; Kunkel, S.L.; Godbey, S.W.; Burdick, M.D.; Strieter, R.M. Pleural mesothelial cell expression of C-C (monocyte chemotactic peptide) and C-X-C (interleukin 8) chemokines. Am. J. Respir. Cell Mol. Biol. 1995, 12, 581–588. [Google Scholar] [CrossRef]
- Hsieh, C.Y.; Sheu, J.R.; Yang, C.H.; Chen, W.L.; Tsai, J.H.; Chung, C.L. Thrombin Upregulates PAI-1 and Mesothelial-Mesenchymal Transition Through PAR-1 and Contributes to Tuberculous Pleural Fibrosis. Int. J. Mol. Sci. 2019, 20, 5076. [Google Scholar] [CrossRef] [PubMed]
- Tucker, T.; Idell, S. Plasminogen-plasmin system in the pathogenesis and treatment of lung and pleural injury. Semin. Thromb. Hemost. 2013, 39, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Mutsaers, S.E.; Prele, C.M.; Brody, A.R.; Idell, S. Pathogenesis of pleural fibrosis. Respirology 2004, 9, 428–440. [Google Scholar] [CrossRef] [PubMed]
- Karandashova, S.; Florova, G.; Azghani, A.O.; Komissarov, A.A.; Koenig, K.; Tucker, T.A.; Allen, T.C.; Stewart, K.; Tvinnereim, A.; Idell, S. Intrapleural adenoviral delivery of human plasminogen activator inhibitor-1 exacerbates tetracycline-induced pleural injury in rabbits. Am. J. Respir. Cell Mol. Biol. 2013, 48, 44–52. [Google Scholar] [CrossRef]
- Tucker, T.A.; Jeffers, A.; Alvarez, A.; Owens, S.; Koenig, K.; Quaid, B.; Komissarov, A.A.; Florova, G.; Kothari, H.; Pendurthi, U.; et al. Plasminogen activator inhibitor-1 deficiency augments visceral mesothelial organization, intrapleural coagulation, and lung restriction in mice with carbon black/bleomycin-induced pleural injury. Am. J. Respir. Cell Mol. Biol. 2014, 50, 316–327. [Google Scholar] [CrossRef]
- Maskell, N.A.; Davies, C.W.; Nunn, A.J.; Hedley, E.L.; Gleeson, F.V.; Miller, R.; Gabe, R.; Rees, G.L.; Peto, T.E.; Woodhead, M.A.; et al. U.K. Controlled trial of intrapleural streptokinase for pleural infection. N. Engl. J. Med. 2005, 352, 865–874. [Google Scholar] [CrossRef]
- Rahman, N.M.; Maskell, N.A.; West, A.; Teoh, R.; Arnold, A.; Mackinlay, C.; Peckham, D.; Davies, C.W.; Ali, N.; Kinnear, W.; et al. Intrapleural use of tissue plasminogen activator and DNase in pleural infection. N. Engl. J. Med. 2011, 365, 518–526. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumors: Wounds That Do Not Heal-A Historical Perspective with a Focus on the Fundamental Roles of Increased Vascular Permeability and Clotting. Semin. Thromb. Hemost. 2019, 45, 576–592. [Google Scholar] [CrossRef]
- Mutsaers, S.E.; Kalomenidis, I.; Wilson, N.A.; Lee, Y.C. Growth factors in pleural fibrosis. Curr. Opin. Pulm. Med. 2006, 12, 251–258. [Google Scholar] [CrossRef]
- Maeda, J.; Ueki, N.; Ohkawa, T.; Iwahashi, N.; Nakano, T.; Hada, T.; Higashino, K. Local production and localization of transforming growth factor-beta in tuberculous pleurisy. Clin. Exp. Immunol. 1993, 92, 32–38. [Google Scholar] [CrossRef]
- Marie, C.; Losser, M.R.; Fitting, C.; Kermarrec, N.; Payen, D.; Cavaillon, J.M. Cytokines and soluble cytokine receptors in pleural effusions from septic and nonseptic patients. Am. J. Respir. Crit. Care Med. 1997, 156, 1515–1522. [Google Scholar] [CrossRef] [PubMed]
- Kalomenidis, I.; Guo, Y.; Lane, K.B.; Hawthorne, M.; Light, R.W. Transforming growth factor-beta3 induces pleurodesis in rabbits and collagen production of human mesothelial cells. Chest 2005, 127, 1335–1340. [Google Scholar] [PubMed]
- Idell, S. The pathogenesis of pleural space loculation and fibrosis. Curr. Opin. Pulm. Med. 2008, 14, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Tucker, T.A.; Idell, S. The Contribution of the Urokinase Plasminogen Activator and the Urokinase Receptor to Pleural and Parenchymal Lung Injury and Repair: A Narrative Review. Int. J. Mol. Sci. 2021, 22, 1437. [Google Scholar] [CrossRef] [PubMed]
- Logan, R.; Jeffers, A.; Qin, W.; Owens, S.; Chauhan, P.; Komatsu, S.; Ikebe, M.; Idell, S.; Tucker, T.A. TGF-beta regulation of the uPA/uPAR axis modulates mesothelial-mesenchymal transition (MesoMT). Sci. Rep. 2021, 11, 21210. [Google Scholar] [CrossRef] [PubMed]
- Tucker, T.A.; Williams, L.; Koenig, K.; Kothari, H.; Komissarov, A.A.; Florova, G.; Mazar, A.P.; Allen, T.C.; Bdeir, K.; Mohan Rao, L.V.; et al. Lipoprotein receptor-related protein 1 regulates collagen 1 expression, proteolysis, and migration in human pleural mesothelial cells. Am. J. Respir. Cell Mol. Biol. 2012, 46, 196–206. [Google Scholar] [CrossRef]
- Qin, W.; Jeffers, A.; Owens, S.; Chauhan, P.; Komatsu, S.; Qian, G.; Guo, X.; Ikebe, M.; Idell, S.; Tucker, T.A. NOX1 Promotes Mesothelial-Mesenchymal Transition through Modulation of Reactive Oxygen Species-mediated Signaling. Am. J. Respir. Cell Mol. Biol. 2021, 64, 492–503. [Google Scholar] [CrossRef]
- Qian, G.; Adeyanju, O.; Roy, S.; Sunil, C.; Jeffers, A.; Guo, X.; Ikebe, M.; Idell, S.; Tucker, T.A. DOCK2 Promotes Pleural Fibrosis by Modulating Mesothelial to Mesenchymal Transition. Am. J. Respir. Cell Mol. Biol. 2021. [Google Scholar] [CrossRef]
- Tucker, T.; Tsukasaki, Y.; Sakai, T.; Mitsuhashi, S.; Komatsu, S.; Jeffers, A.; Idell, S.; Ikebe, M. Myocardin Is Involved in Mesothelial-Mesenchymal Transition of Human Pleural Mesothelial Cells. Am. J. Respir. Cell Mol. Biol. 2019, 61, 86–96. [Google Scholar] [CrossRef]
- Keshava, S.; Magisetty, J.; Tucker, T.A.; Kujur, W.; Mulik, S.; Esmon, C.T.; Idell, S.; Rao, L.V.M.; Pendurthi, U.R. Endothelial Cell Protein C Receptor Deficiency Attenuates Streptococcus pneumoniae-induced Pleural Fibrosis. Am. J. Respir. Cell Mol. Biol. 2021, 64, 477–491. [Google Scholar] [CrossRef]
- Jeffers, A.; Qin, W.; Owens, S.; Koenig, K.B.; Komatsu, S.; Giles, F.J.; Schmitt, D.M.; Idell, S.; Tucker, T.A. Glycogen Synthase Kinase-3beta Inhibition with 9-ING-41 Attenuates the Progression of Pulmonary Fibrosis. Sci. Rep. 2019, 9, 18925. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Lagares, D. Evasion of apoptosis by myofibroblasts: A hallmark of fibrotic diseases. Nat. Rev. Rheumatol. 2020, 16, 11–31. [Google Scholar] [CrossRef] [PubMed]
- Hecker, L.; Logsdon, N.J.; Kurundkar, D.; Kurundkar, A.; Bernard, K.; Hock, T.; Meldrum, E.; Sanders, Y.Y.; Thannickal, V.J. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci. Transl. Med. 2014, 6, 231ra47. [Google Scholar] [CrossRef] [PubMed]
- Zolak, J.S.; Jagirdar, R.; Surolia, R.; Karki, S.; Oliva, O.; Hock, T.; Guroji, P.; Ding, Q.; Liu, R.M.; Bolisetty, S.; et al. Pleural mesothelial cell differentiation and invasion in fibrogenic lung injury. Am. J. Pathol. 2013, 182, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Mubarak, K.K.; Montes-Worboys, A.; Regev, D.; Nasreen, N.; Mohammed, K.A.; Faruqi, I.; Hensel, E.; Baz, M.A.; Akindipe, O.A.; Fernandez-Bussy, S.; et al. Parenchymal trafficking of pleural mesothelial cells in idiopathic pulmonary fibrosis. Eur. Respir. J. 2012, 39, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Nasreen, N.; Mohammed, K.A.; Mubarak, K.K.; Baz, M.A.; Akindipe, O.A.; Fernandez-Bussy, S.; Antony, V.B. Pleural mesothelial cell transformation into myofibroblasts and haptotactic migration in response to TGF-beta1 in vitro. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L115–L124. [Google Scholar] [CrossRef] [PubMed]
- Owens, S.; Jeffers, A.; Boren, J.; Tsukasaki, Y.; Koenig, K.; Ikebe, M.; Idell, S.; Tucker, T.A. Mesomesenchymal transition of pleural mesothelial cells is PI3K and NF-kappaB dependent. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L1265–L1273. [Google Scholar] [CrossRef] [PubMed]
- Gharaee-Kermani, M.; Gyetko, M.R.; Hu, B.; Phan, S.H. New insights into the pathogenesis and treatment of idiopathic pulmonary fibrosis: A potential role for stem cells in the lung parenchyma and implications for therapy. Pharm. Res. 2007, 24, 819–841. [Google Scholar] [CrossRef] [PubMed]
- Lama, V.N.; Phan, S.H. The extrapulmonary origin of fibroblasts: Stem/progenitor cells and beyond. Proc. Am. Thorac. Soc. 2006, 3, 373–376. [Google Scholar] [CrossRef]
- Nasreen, N.; Mohammed, K.A.; Antony, V.B. Silencing the receptor EphA2 suppresses the growth and haptotaxis of malignant mesothelioma cells. Cancer 2006, 107, 2425–2435. [Google Scholar] [CrossRef]
- Decologne, N.; Kolb, M.; Margetts, P.J.; Menetrier, F.; Artur, Y.; Garrido, C.; Gauldie, J.; Camus, P.; Bonniaud, P. TGF-beta1 induces progressive pleural scarring and subpleural fibrosis. J. Immunol. 2007, 179, 6043–6051. [Google Scholar] [CrossRef] [PubMed]
- Idell, S.; Florova, G.; Shetty, S.; Tucker, T.; Idell, R.; Koenig, K.; Azghani, A.; Rahman, N.M.; Komissarov, A. Precision-guided, Personalized Intrapleural Fibrinolytic Therapy for Empyema and Complicated Parapneumonic Pleural Effusions: The Case for the Fibrinolytic Potential. Clin. Pulm. Med. 2017, 24, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Boren, J.; Shryock, G.; Fergis, A.; Jeffers, A.; Owens, S.; Qin, W.; Koenig, K.B.; Tsukasaki, Y.; Komatsu, S.; Ikebe, M.; et al. Inhibition of Glycogen Synthase Kinase 3beta Blocks Mesomesenchymal Transition and Attenuates Streptococcus pneumonia-Mediated Pleural Injury in Mice. Am. J. Pathol. 2017, 187, 2461–2472. [Google Scholar] [CrossRef]
- Decologne, N.; Wettstein, G.; Kolb, M.; Margetts, P.; Garrido, C.; Camus, P.; Bonniaud, P. Bleomycin induces pleural and subpleural fibrosis in the presence of carbon particles. Eur. Respir. J. 2010, 35, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Tucker, T.A.; Jeffers, A.; Boren, J.; Quaid, B.; Owens, S.; Koenig, K.B.; Tsukasaki, Y.; Florova, G.; Komissarov, A.A.; Ikebe, M.; et al. Organizing empyema induced in mice by Streptococcus pneumoniae: Effects of plasminogen activator inhibitor-1 deficiency. Clin. Transl. Med. 2016, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Cuzzocrea, S.; McDonald, M.C.; Filipe, H.M.; Costantino, G.; Mazzon, E.; Santagati, S.; Caputi, A.P.; Thiemermann, C. Effects of tempol, a membrane-permeable radical scavenger, in a rodent model of carrageenan-induced pleurisy. Eur. J. Pharmacol. 2000, 390, 209–222. [Google Scholar] [CrossRef]
- Fusco, R.; Gugliandolo, E.; Biundo, F.; Campolo, M.; Di Paola, R.; Cuzzocrea, S. Inhibition of inflammasome activation improves lung acute injury induced by carrageenan in a mouse model of pleurisy. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2017, 31, 3497–3511. [Google Scholar] [CrossRef]
- Chung, J.Y.; Chan, M.K.; Li, J.S.; Chan, A.S.; Tang, P.C.; Leung, K.T.; To, K.F.; Lan, H.Y.; Tang, P.M. TGF-beta Signaling: From Tissue Fibrosis to Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 7575. [Google Scholar] [CrossRef]
- Ghatak, S.; Hascall, V.C.; Markwald, R.R.; Feghali-Bostwick, C.; Artlett, C.M.; Gooz, M.; Bogatkevich, G.S.; Atanelishvili, I.; Silver, R.M.; Wood, J.; et al. Transforming growth factor beta1 (TGFbeta1)-induced CD44V6-NOX4 signaling in pathogenesis of idiopathic pulmonary fibrosis. J. Biol. Chem. 2017, 292, 10490–10519. [Google Scholar] [CrossRef]
- Jiang, F.; Liu, G.S.; Dusting, G.J.; Chan, E.C. NADPH oxidase-dependent redox signaling in TGF-beta-mediated fibrotic responses. Redox Biol. 2014, 2, 267–272. [Google Scholar] [CrossRef]
- Barnes, J.L.; Gorin, Y. Myofibroblast differentiation during fibrosis: Role of NAD(P)H oxidases. Kidney Int. 2011, 79, 944–956. [Google Scholar] [CrossRef] [PubMed]
- Hecker, L.; Vittal, R.; Jones, T.; Jagirdar, R.; Luckhardt, T.R.; Horowitz, J.C.; Pennathur, S.; Martinez, F.J.; Thannickal, V.J. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 2009, 15, 1077–1081. [Google Scholar] [CrossRef] [PubMed]
- Cucoranu, I.; Clempus, R.; Dikalova, A.; Phelan, P.J.; Ariyan, S.; Dikalov, S.; Sorescu, D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 2005, 97, 900–907. [Google Scholar] [CrossRef]
- Rysenkova, K.D.; Klimovich, P.S.; Shmakova, A.A.; Karagyaur, M.N.; Ivanova, K.A.; Aleksandrushkina, N.A.; Tkachuk, V.A.; Rubina, K.A.; Semina, E.V. Urokinase receptor deficiency results in EGFR-mediated failure to transmit signals for cell survival and neurite formation in mouse neuroblastoma cells. Cell Signal 2020, 75, 109741. [Google Scholar] [CrossRef]
- Gilder, A.S.; Natali, L.; Van Dyk, D.M.; Zalfa, C.; Banki, M.A.; Pizzo, D.P.; Wang, H.; Klemke, R.L.; Mantuano, E.; Gonias, S.L. The Urokinase Receptor Induces a Mesenchymal Gene Expression Signature in Glioblastoma Cells and Promotes Tumor Cell Survival in Neurospheres. Sci. Rep. 2018, 8, 2982. [Google Scholar] [CrossRef] [PubMed]
- Magnussen, S.N.; Hadler-Olsen, E.; Costea, D.E.; Berg, E.; Jacobsen, C.C.; Mortensen, B.; Salo, T.; Martinez-Zubiaurre, I.; Winberg, J.O.; Uhlin-Hansen, L.; et al. Cleavage of the urokinase receptor (uPAR) on oral cancer cells: Regulation by transforming growth factor—beta1 (TGF-beta1) and potential effects on migration and invasion. BMC Cancer 2017, 17, 350. [Google Scholar] [CrossRef]
- van Veen, M.; Matas-Rico, E.; van de Wetering, K.; Leyton-Puig, D.; Kedziora, K.M.; De Lorenzi, V.; Stijf-Bultsma, Y.; van den Broek, B.; Jalink, K.; Sidenius, N.; et al. Negative regulation of urokinase receptor activity by a GPI-specific phospholipase C in breast cancer cells. Elife 2017, 6, e23649. [Google Scholar] [CrossRef]
- Raghu, H.; Lakka, S.S.; Gondi, C.S.; Mohanam, S.; Dinh, D.H.; Gujrati, M.; Rao, J.S. Suppression of uPA and uPAR attenuates angiogenin mediated angiogenesis in endothelial and glioblastoma cell lines. PLoS ONE 2010, 5, e12458. [Google Scholar] [CrossRef]
- Tucker, T.A.; Dean, C.; Komissarov, A.A.; Koenig, K.; Mazar, A.P.; Pendurthi, U.; Allen, T.; Idell, S. The urokinase receptor supports tumorigenesis of human malignant pleural mesothelioma cells. Am. J. Respir. Cell Mol. Biol. 2010, 42, 685–696. [Google Scholar] [CrossRef]
- Doble, B.W.; Woodgett, J.R. GSK-3: Tricks of the trade for a multi-tasking kinase. J. Cell Sci. 2003, 116, 1175–1186. [Google Scholar] [CrossRef]
- Bijur, G.N.; Jope, R.S. Glycogen synthase kinase-3 beta is highly activated in nuclei and mitochondria. Neuroreport 2003, 14, 2415–2419. [Google Scholar] [CrossRef] [PubMed]
- Ugolkov, A.; Qiang, W.; Bondarenko, G.; Procissi, D.; Gaisina, I.; James, C.D.; Chandler, J.; Kozikowski, A.; Gunosewoyo, H.; O’Halloran, T.; et al. Combination Treatment with the GSK-3 Inhibitor 9-ING-41 and CCNU Cures Orthotopic Chemoresistant Glioblastoma in Patient-Derived Xenograft Models. Transl. Oncol. 2017, 10, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Frame, S.; Cohen, P.; Biondi, R.M. A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol. Cell 2001, 7, 1321–1327. [Google Scholar] [CrossRef]
- Bhat, R.V.; Shanley, J.; Correll, M.P.; Fieles, W.E.; Keith, R.A.; Scott, C.W.; Lee, C.M. Regulation and localization of tyrosine216 phosphorylation of glycogen synthase kinase-3beta in cellular and animal models of neuronal degeneration. Proc. Natl. Acad. Sci. USA 2000, 97, 11074–11079. [Google Scholar] [CrossRef] [PubMed]
- Baarsma, H.A.; Engelbertink, L.H.; van Hees, L.J.; Menzen, M.H.; Meurs, H.; Timens, W.; Postma, D.S.; Kerstjens, H.A.; Gosens, R. Glycogen synthase kinase-3 (GSK-3) regulates TGF-beta(1)-induced differentiation of pulmonary fibroblasts. Br. J. Pharmacol. 2013, 169, 590–603. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yang, S.; Yang, Z.; Ma, L.; Jiang, D.; Mao, J.; Jiao, B.; Cai, Z. Inhibition of GSK-3beta decreases NF-kappaB-dependent gene expression and impairs the rat liver regeneration. J. Cell. Biochem. 2007, 102, 1281–1289. [Google Scholar] [CrossRef]
- Parmacek, M.S. Myocardin-related transcription factors: Critical coactivators regulating cardiovascular development and adaptation. Circ. Res. 2007, 100, 633–644. [Google Scholar] [CrossRef]
- Wang, D.; Chang, P.S.; Wang, Z.; Sutherland, L.; Richardson, J.A.; Small, E.; Krieg, P.A.; Olson, E.N. Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell 2001, 105, 851–862. [Google Scholar] [CrossRef]
- Sisson, T.H.; Ajayi, I.O.; Subbotina, N.; Dodi, A.E.; Rodansky, E.S.; Chibucos, L.N.; Kim, K.K.; Keshamouni, V.G.; White, E.S.; Zhou, Y.; et al. Inhibition of myocardin-related transcription factor/serum response factor signaling decreases lung fibrosis and promotes mesenchymal cell apoptosis. Am. J. Pathol. 2015, 185, 969–986. [Google Scholar] [CrossRef]
- Luchsinger, L.L.; Patenaude, C.A.; Smith, B.D.; Layne, M.D. Myocardin-related transcription factor-A complexes activate type I collagen expression in lung fibroblasts. J. Biol. Chem. 2011, 286, 44116–44125. [Google Scholar] [CrossRef]
- Guo, X.; Chen, S.Y. Dedicator of Cytokinesis 2 in Cell Signaling Regulation and Disease Development. J. Cell. Physiol. 2017, 232, 1931–1940. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, V.S.; Nelson, D.A.; Marriott, I.; Bost, K.L. Alpha beta-crystallin expression and presentation following infection with murine gammaherpesvirus 68. Autoimmunity 2013, 46, 399–408. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bellaye, P.S.; Wettstein, G.; Burgy, O.; Besnard, V.; Joannes, A.; Colas, J.; Causse, S.; Marchal-Somme, J.; Fabre, A.; Crestani, B.; et al. The small heat-shock protein alphaB-crystallin is essential for the nuclear localization of Smad4: Impact on pulmonary fibrosis. J. Pathol. 2014, 232, 458–472. [Google Scholar] [CrossRef] [PubMed]
- Bellaye, P.S.; Burgy, O.; Colas, J.; Fabre, A.; Marchal-Somme, J.; Crestani, B.; Kolb, M.; Camus, P.; Garrido, C.; Bonniaud, P. Antifibrotic role of alphaB-crystallin inhibition in pleural and subpleural fibrosis. Am. J. Respir. Cell Mol. Biol. 2015, 52, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Marudamuthu, A.S.; Bhandary, Y.P.; Fan, L.; Radhakrishnan, V.; MacKenzie, B.; Maier, E.; Shetty, S.K.; Nagaraja, M.R.; Gopu, V.; Tiwari, N.; et al. Caveolin-1-derived peptide limits development of pulmonary fibrosis. Sci. Transl. Med. 2019, 11, eaat2848. [Google Scholar] [CrossRef] [PubMed]
- Nagaraja, M.R.; Tiwari, N.; Shetty, S.K.; Marudamuthu, A.S.; Fan, L.; Ostrom, R.S.; Fu, J.; Gopu, V.; Radhakrishnan, V.; Idell, S.; et al. p53 Expression in Lung Fibroblasts Is Linked to Mitigation of Fibrotic Lung Remodeling. Am. J. Pathol. 2018, 188, 2207–2222. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Li, X.; Zhang, J. mTOR Signaling in Cancer and mTOR Inhibitors in Solid Tumor Targeting Therapy. Int. J. Mol. Sci. 2019, 20, 755. [Google Scholar] [CrossRef]
- Lawrence, J.; Nho, R. The Role of the Mammalian Target of Rapamycin (mTOR) in Pulmonary Fibrosis. Int. J. Mol. Sci. 2018, 19, 778. [Google Scholar] [CrossRef]
- Mercer, P.F.; Woodcock, H.V.; Eley, J.D.; Plate, M.; Sulikowski, M.G.; Durrenberger, P.F.; Franklin, L.; Nanthakumar, C.B.; Man, Y.; Genovese, F.; et al. Exploration of a potent PI3 kinase/mTOR inhibitor as a novel anti-fibrotic agent in IPF. Thorax 2016, 71, 701–711. [Google Scholar] [CrossRef]
- Hall, M.N. mTOR-what does it do? Transpl. Proc. 2008, 40, S5–S8. [Google Scholar] [CrossRef]
- Woodcock, H.V.; Eley, J.D.; Guillotin, D.; Plate, M.; Nanthakumar, C.B.; Martufi, M.; Peace, S.; Joberty, G.; Poeckel, D.; Good, R.B.; et al. The mTORC1/4E-BP1 axis represents a critical signaling node during fibrogenesis. Nat. Commun. 2019, 10, 6. [Google Scholar] [CrossRef] [PubMed]

| Fibrinolysin | ||
|---|---|---|
| tPA (Alteplase) | ||
| tPA/DNAse | ||
| scuPA * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tucker, T.A.; Idell, S. Update on Novel Targeted Therapy for Pleural Organization and Fibrosis. Int. J. Mol. Sci. 2022, 23, 1587. https://doi.org/10.3390/ijms23031587
Tucker TA, Idell S. Update on Novel Targeted Therapy for Pleural Organization and Fibrosis. International Journal of Molecular Sciences. 2022; 23(3):1587. https://doi.org/10.3390/ijms23031587
Chicago/Turabian StyleTucker, Torry A., and Steven Idell. 2022. "Update on Novel Targeted Therapy for Pleural Organization and Fibrosis" International Journal of Molecular Sciences 23, no. 3: 1587. https://doi.org/10.3390/ijms23031587
APA StyleTucker, T. A., & Idell, S. (2022). Update on Novel Targeted Therapy for Pleural Organization and Fibrosis. International Journal of Molecular Sciences, 23(3), 1587. https://doi.org/10.3390/ijms23031587