
Deciphering Genetic Alterations of Taiwanese Patients with Pancreatic Adenocarcinoma through Targeted Sequencing

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Results
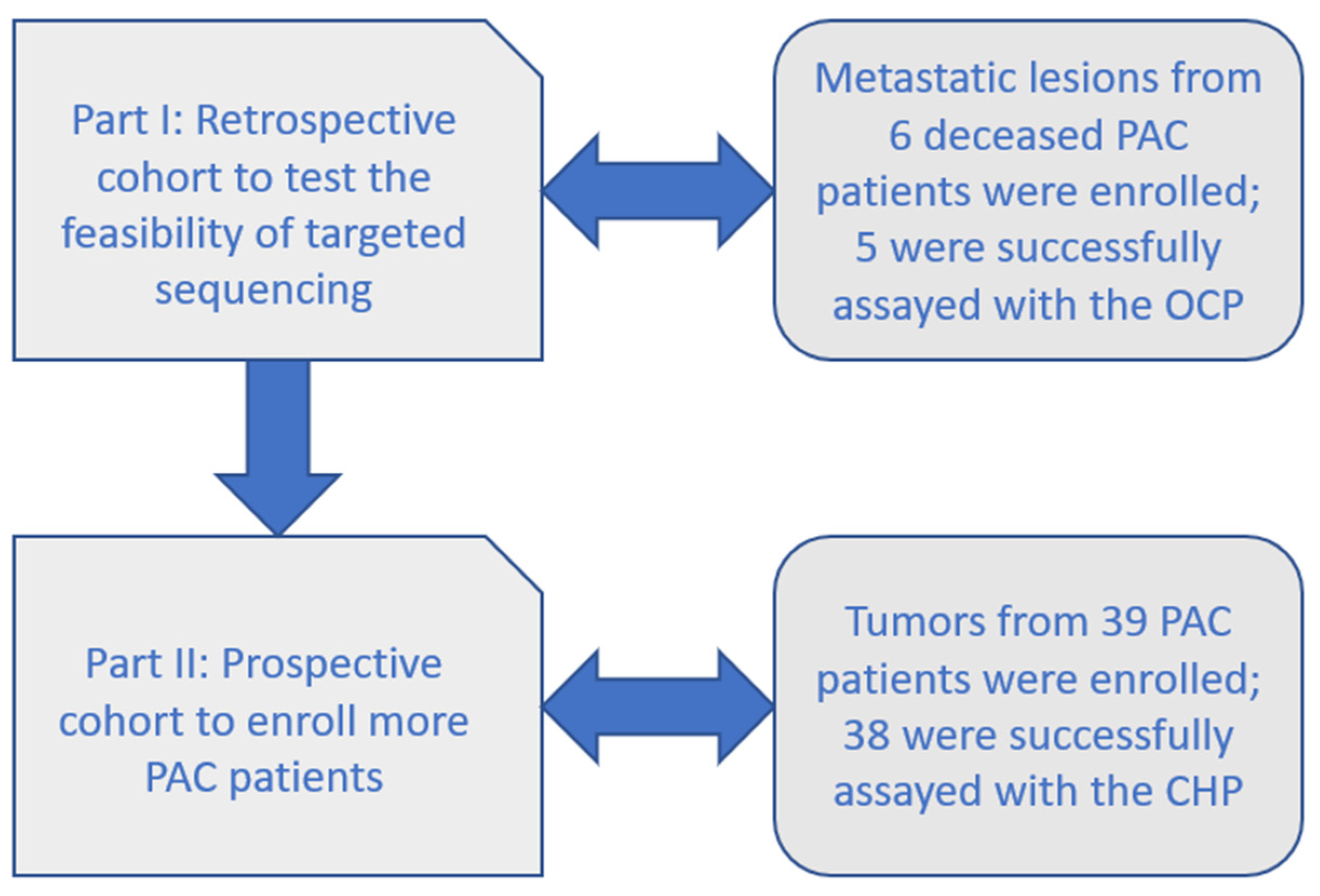
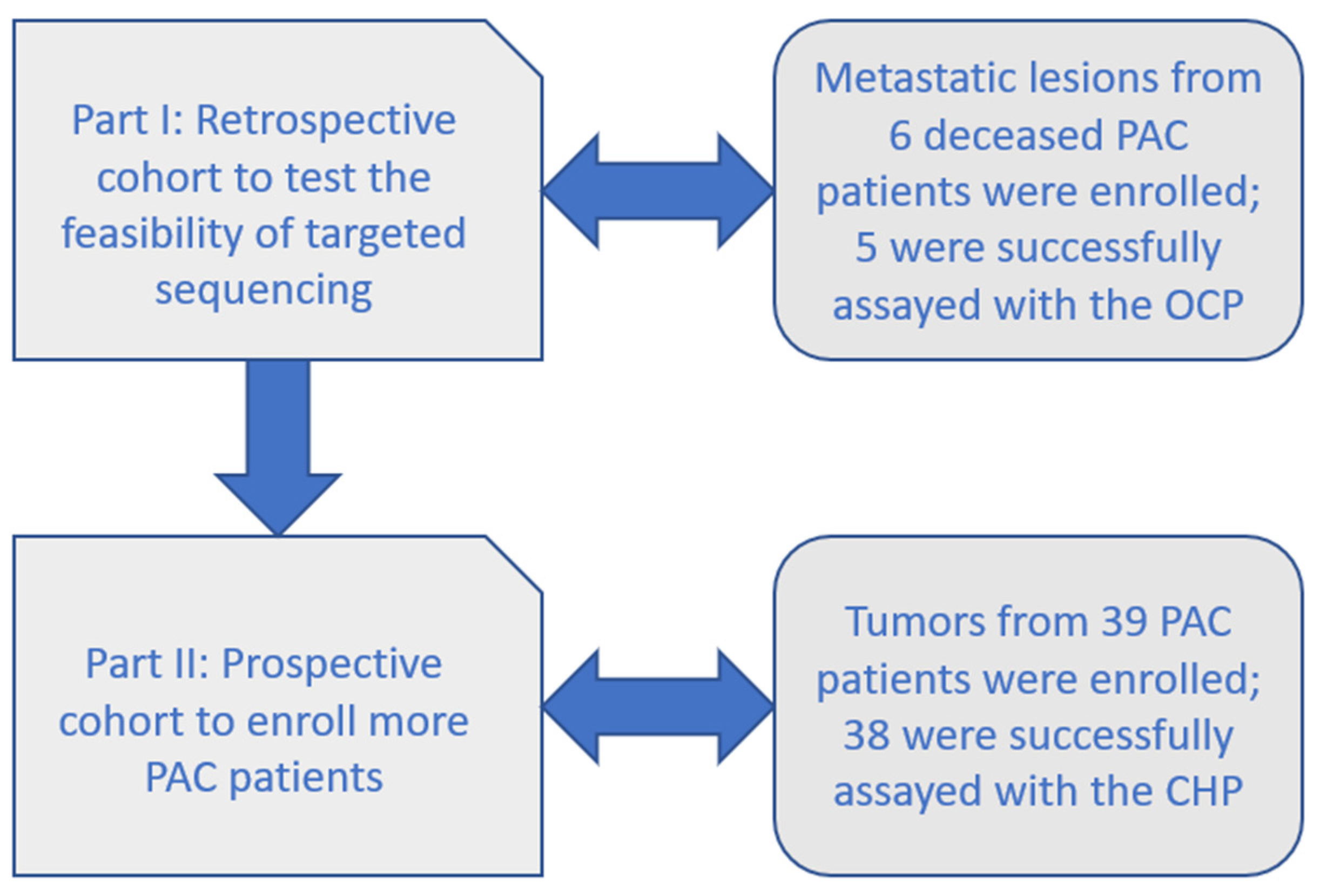
2.1. Part I: Retrospective Cohort with the OCP
2.2. Part II: Prospective Cohort with the CHP
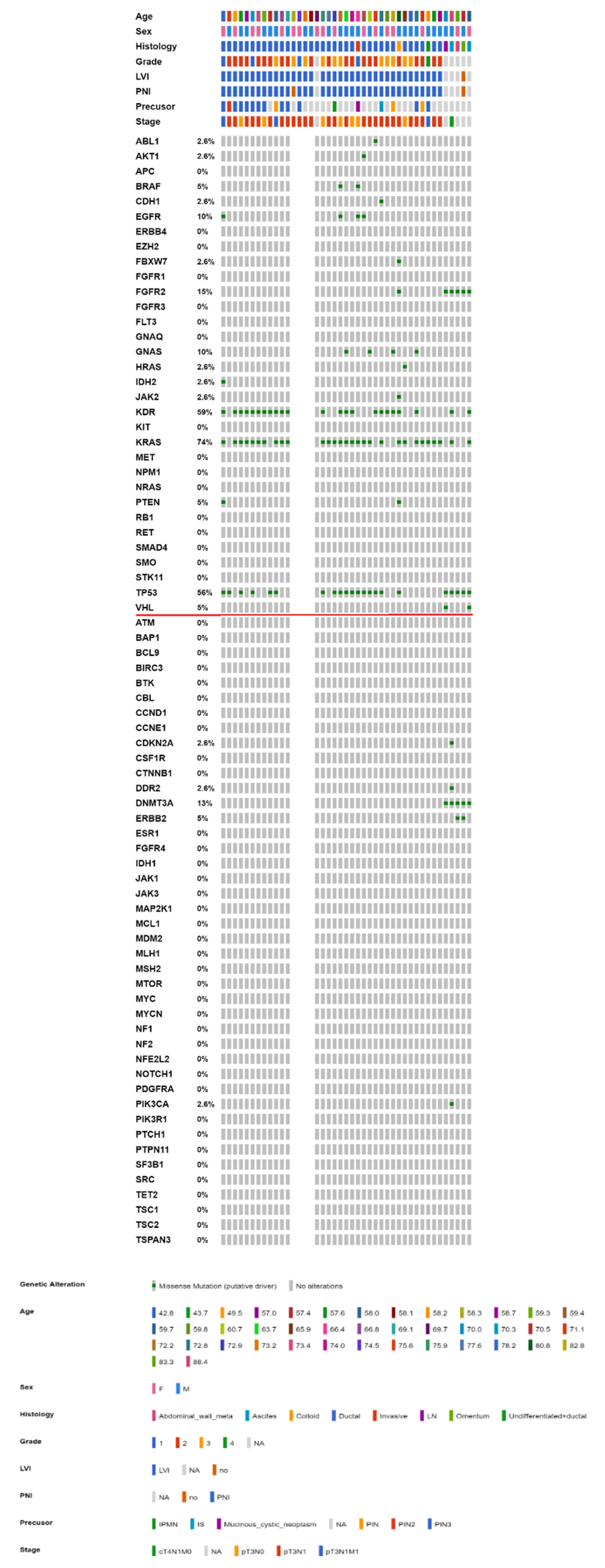
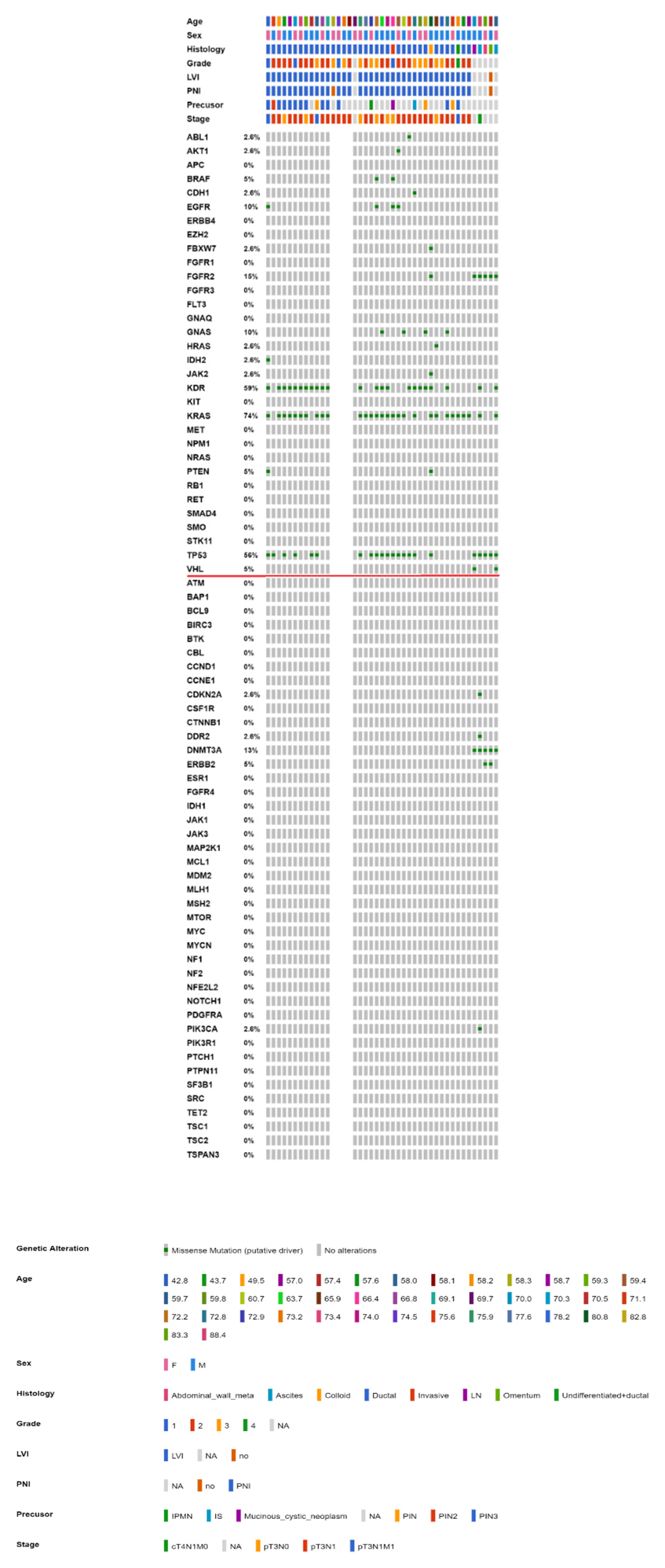
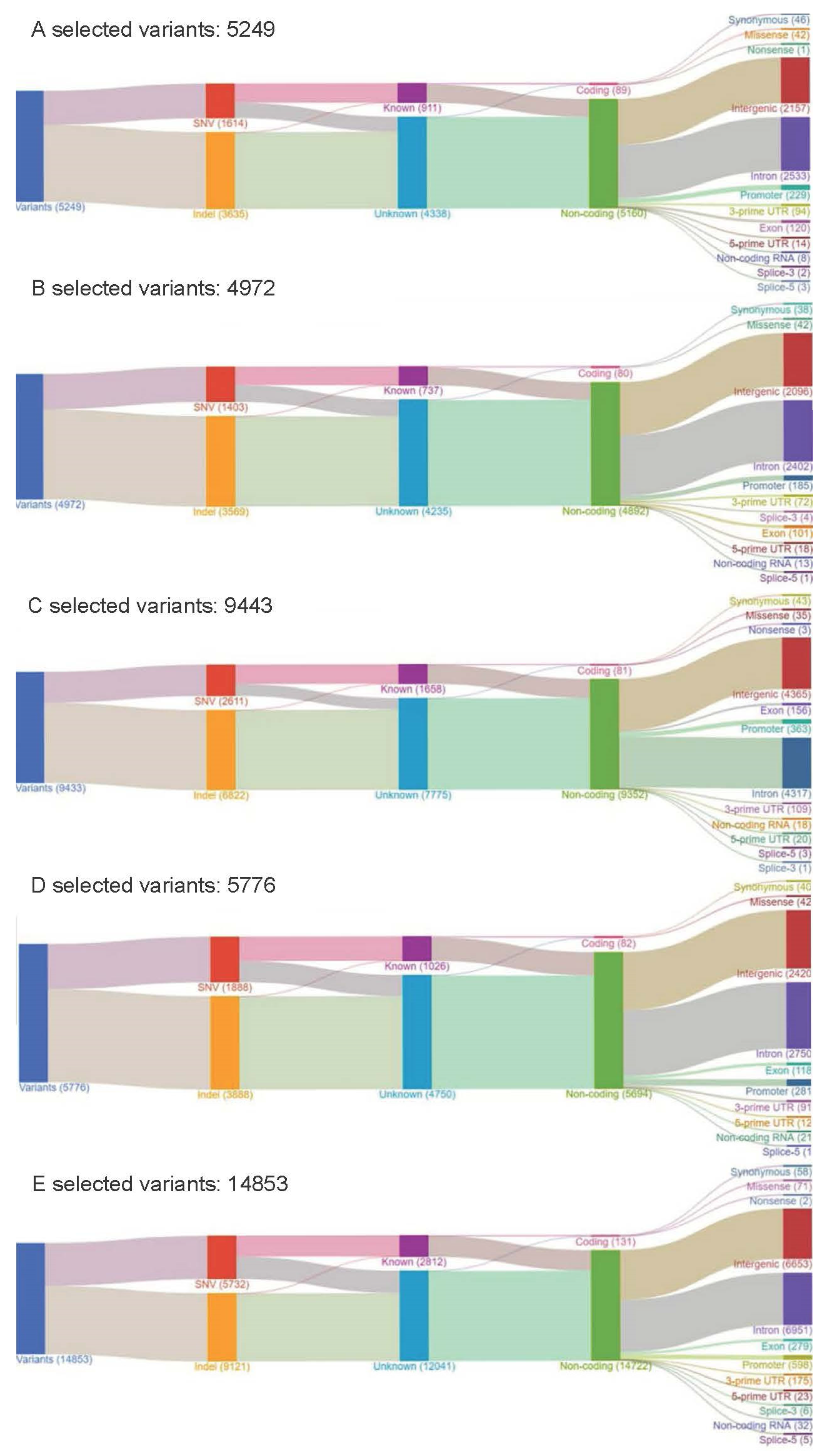
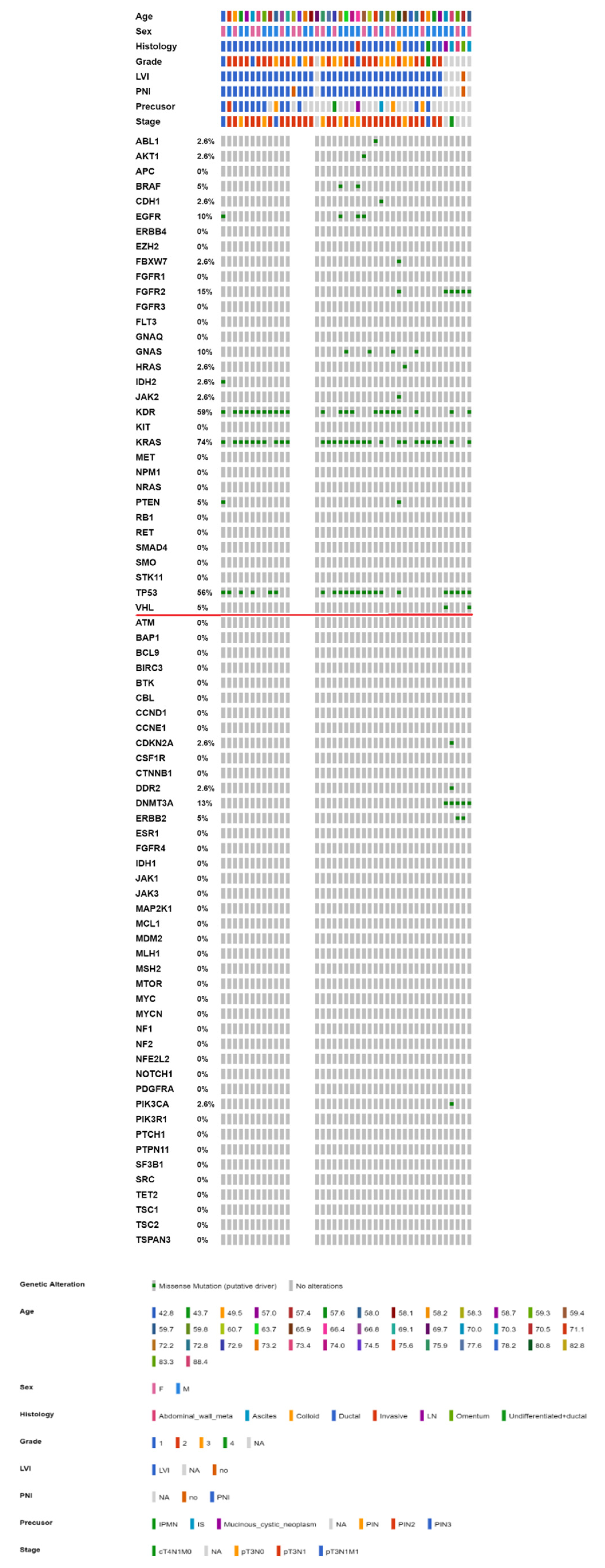
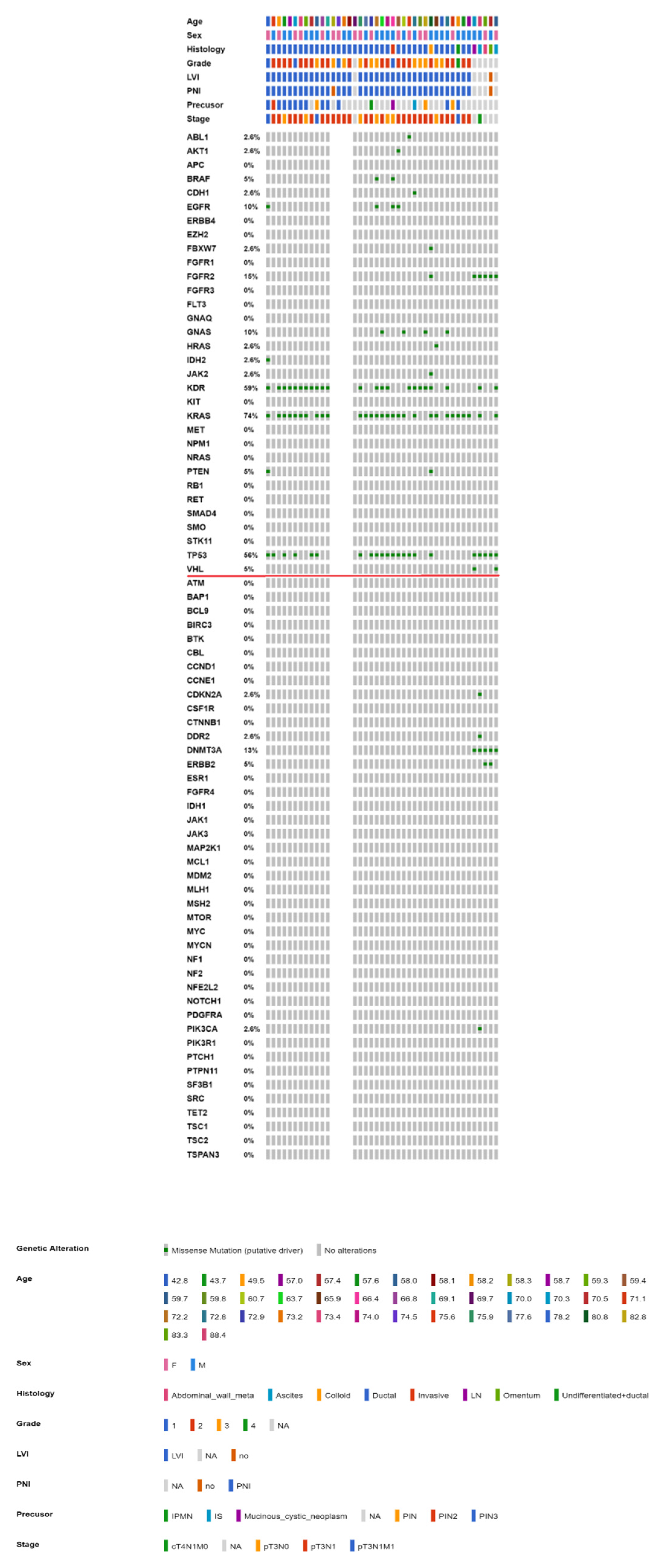
2.3. Mutational Landscape of Taiwanese Patients with PAC
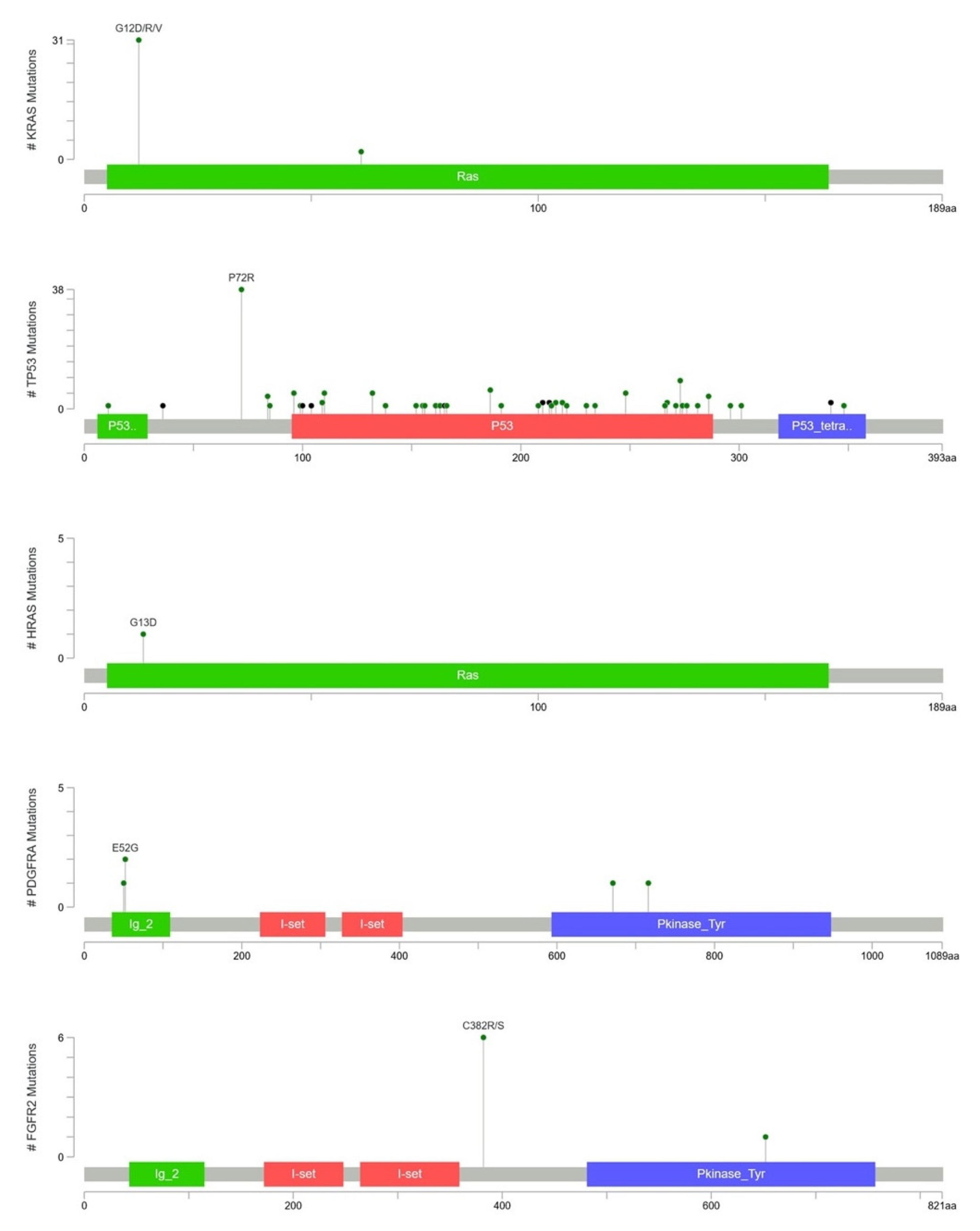
2.4. KRAS Mutations Were Most Prevalent in Taiwanese Patients with PAC
3. Discussion
4. Materials and Methods
4.1. Study Population
4.2. Nucleic Acid Extraction
4.3. Targeted Sequencing Panel
4.4. NGS Experiments
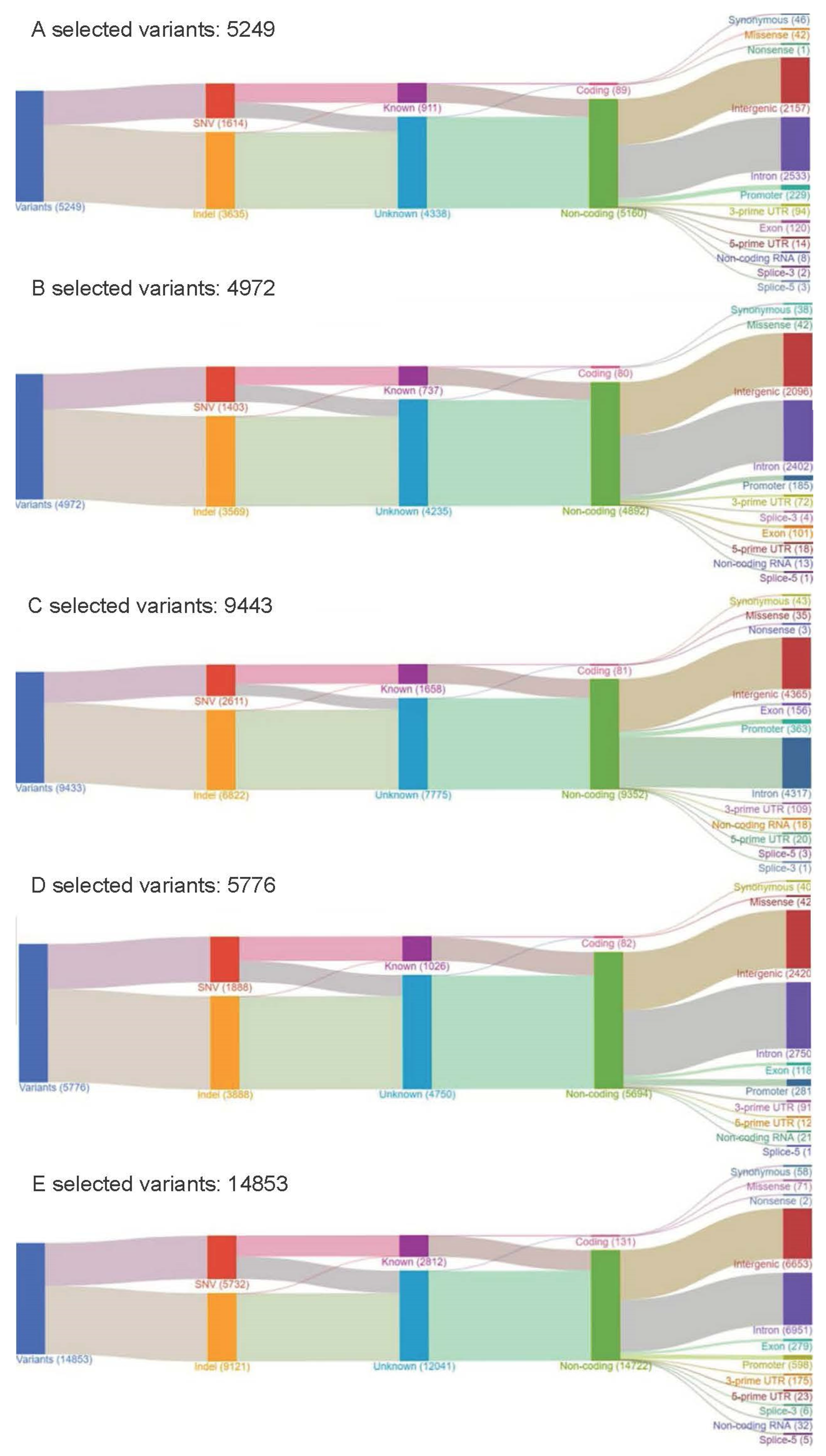
4.5. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ministry of Health and Welfare, Taiwan (R.O.C.). 2018 Statistics of Causes of Death. 2018. Available online: http://www.mohw.gov.tw/cp-16-48057-1.html (accessed on 19 March 2020).
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef] [PubMed]
- Grant, R.C.; Selander, I.; Connor, A.A.; Selvarajah, S.; Borgida, A.; Briollais, L.; Petersen, G.M.; Lerner-Ellis, J.; Holter, S.; Gallinger, S. Prevalence of germline mutations in cancer predisposition genes in patients with pancreatic cancer. Gastroenterology 2015, 148, 556–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, N.J.; Klein, A.P. Genome-wide sequencing to identify the cause of hereditary cancer syndromes: With examples from familial pancreatic cancer. Cancer Lett. 2013, 340, 227–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotterell, J. Exome sequencing reveals a potential mutational trajectory and treatments for a specific pancreatic cancer patient. Onco Targets Ther. 2014, 7, 655–662. [Google Scholar] [CrossRef] [Green Version]
- Cowley, M.J.; Chang, D.K.; Pajic, M.; Johns, A.L.; Waddell, N.; Grimmond, S.M.; Biankin, A.V. Understanding pancreatic cancer genomes. J. Hepatobiliary Pancreat. Sci. 2013, 20, 549–556. [Google Scholar] [CrossRef]
- Liang, W.S.; Craig, D.W.; Carpten, J.; Borad, M.J.; Demeure, M.J.; Weiss, G.J.; Izatt, T.; Sinari, S.; Christoforides, A.; Aldrich, J.; et al. Genome-wide characterization of pancreatic adenocarcinoma patients using next generation sequencing. PLoS ONE 2012, 7, e43192. [Google Scholar] [CrossRef] [Green Version]
- Brosens, L.A.; Hackeng, W.M.; Offerhaus, G.J.; Hruban, R.H.; Wood, L.D. Pancreatic adenocarcinoma pathology: Changing “landscape”. J. Gastrointest. Oncol. 2015, 6, 358–374. [Google Scholar]
- Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Ng, S.; Leiserson, M.D.M.; Niu, B.; McLellan, M.D.; Uzunangelov, V.; et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 2014, 158, 929–944. [Google Scholar] [CrossRef] [Green Version]
- Samuel, N.; Hudson, T.J. The molecular and cellular heterogeneity of pancreatic ductal adenocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 9, 77–87. [Google Scholar] [CrossRef]
- Lin, P.H.; Kuo, W.H.; Huang, A.C.; Lu, Y.S.; Lin, C.H.; Kuo, S.H.; Wang, M.Y.; Liu, C.Y.; Cheng, F.T.; Yeh, M.H.; et al. Multiple gene sequencing for risk assessment in patients with early-onset or familial breast cancer. Oncotarget 2016, 7, 8310–8320. [Google Scholar] [CrossRef] [Green Version]
- Liao, W.C.; Chien, K.L.; Lin, Y.L.; Wu, M.S.; Lin, J.T.; Wang, H.P.; Tu, Y.K. Adjuvant treatments for resected pancreatic adenocarcinoma: A systematic review and network meta-analysis. Lancet Oncol. 2013, 14, 1095–1103. [Google Scholar] [CrossRef]
- Visani, M.; de Biase, D.; Baccarini, P.; Fabbri, C.; Polifemo, A.M.; Zanini, N.; Pession, A.; Tallini, G. Multiple KRAS mutations in pancreatic adenocarcinoma: Molecular features of neoplastic clones indicate the selection of divergent populations of tumor cells. Int. J. Surg. Pathol. 2013, 21, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, l1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Mardis, E.R. Applying next-generation sequencing to pancreatic cancer treatment. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 477–486. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Pao, W.; Wang, T.Y.; Riely, G.J.; Miller, V.A.; Pan, Q.; Ladanyi, M.; Zakowski, M.F.; Heelan, R.T.; Kris, M.G.; Varmus, H.E. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005, 2, e17. [Google Scholar] [CrossRef] [Green Version]
- van Zandwijk, N.; Mathy, A.; Boerrigter, L.; Ruijter, H.; Tielen, I.; de Jong, D.; Baas, P.; Burgers, S.; Nederlof, P. EGFR and KRAS mutations as criteria for treatment with tyrosine kinase inhibitors: Retro- and prospective observations in non-small-cell lung cancer. Ann Oncol. 2007, 18, 99–103. [Google Scholar] [CrossRef]
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371, 1039–1049. [Google Scholar] [CrossRef]
- Singh, A.; Greninger, P.; Rhodes, D.; Koopman, L.; Violette, S.; Bardeesy, N.; Settleman, J. A gene expression signature associated with“K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell 2009, 15, 489–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.R.; O’Reilly, E.M. New Treatment Strategies for Metastatic Pancreatic Ductal Adenocarcinoma. Drugs 2020, 80, 647–669. [Google Scholar] [CrossRef] [PubMed]
- Guan, Z.; Lan, H.; Sun, D.; Wang, X.; Jin, K. A potential novel therapy for FGFR1-amplified pancreatic cancer with bone metastasis, screened by next-generation sequencing and a patient-derived xenograft model. Oncol. Lett. 2019, 17, 2303–2307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chae, Y.K.; Ranganath, K.; Hammerman, P.S.; Vaklavas, C.; Mohindra, N.; Kalyan, A.; Matsangou, M.; Costa, R.; Carneiro, B.; Villaflor, V.M.; et al. Inhibition of the fibroblast growth factor receptor (FGFR) pathway: The current landscape and barriers to clinical application. Oncotarget 2017, 8, 16052–16074. [Google Scholar] [CrossRef] [Green Version]
- Powers, M.P. The ever-changing world of gene fusions in cancer: A secondary gene fusion and progression. Oncogene 2019, 38, 7197–7199. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Fujimoto, H.; Gabazza, E.C. Efficacy of crizotinib in ALK fusion variants. J. Thorac. Dis. 2016, 8, E1381–E1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaherty, K.T.; Gray, R.; Chen, A.; Li, S.; Patton, D.; Hamilton, S.R.; Williams, P.M.; Mitchell, E.P.; Iafrate, A.J.; Sklar, J.; et al. NCI-MATCH Team. The Molecular Analysis for Therapy Choice (NCI-MATCH) Trial: Lessons for Genomic Trial Design. J. Natl. Cancer Inst. 2020, 112, 1021–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singhi, A.D.; George, B.; Greenbowe, J.R.; Chung, J.; Suh, J.; Maitra, A.; Klempner, S.J.; Hendifar, A.; Milind, J.M.; Golan, T.; et al. Real-Time Targeted Genome Profile Analysis of Pancreatic Ductal Adenocarcinomas Identifies Genetic Alterations That Might Be Targeted with Existing Drugs or Used as Biomarkers. Gastroenterology 2019, 156, 2242–2253.e4. [Google Scholar] [CrossRef] [Green Version]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Rainone, M.; Singh, I.; Salo-Mullen, E.E.; Stadler, Z.K.; O’Reilly, E.M. An Emerging Paradigm for Germline Testing in Pancreatic Ductal Adenocarcinoma and Immediate Implications for Clinical Practice: A Review. JAMA Oncol. 2020, 6, 764–771. [Google Scholar] [CrossRef]
- Roberts, N.J.; Jiao, Y.; Yu, J.; Kopelovich, L.; Petersen, G.M.; Bondy, M.L.; Gallinger, S.; Schwartz, A.G.; Syngal, S.; Cote, M.L.; et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov. 2012, 2, 41–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waddell, N.; Pajic, M.; Patch, A.-M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Young, G.; Wang, K.; He, J.; Otto, G.; Hawryluk, M.; Zwirco, Z.; Brennan, T.; Nahas, M.; Donahue, A.; Yelensky, R.; et al. Clinical next-generation sequencing was successfully applied to fine-needle aspirations of pulmonary and pancreatic neoplasms. Cancer Cytopathol. 2013, 121, 688–694. [Google Scholar] [CrossRef]
- Park, G.; Park, J.K.; Son, D.S.; Shin, S.H.; Kim, Y.J.; Jeon, H.J.; Lee, J.; Park, W.Y.; Lee, K.H.; Park, D. Utility of targeted deep sequencing for detecting circulating tumor DNA in pancreatic cancer patients. Sci. Rep. 2018, 8, 11631. [Google Scholar] [CrossRef] [PubMed]
- Hovelson, D.H.; McDaniel, A.S.; Cani, A.K.; Johnson, B.; Rhodes, K.; Williams, P.D.; Bandla, S.; Bien, G.; Choppa, P.; Hyland, F.; et al. Development and validation of a scalable next-generation sequencing system for assessing relevant somatic variants in solid tumors. Neoplasia 2015, 17, 385–399. [Google Scholar] [CrossRef] [Green Version]
- Boland, G.M.; Piha-Paul, S.A.; Subbiah, V.; Routbort, M.; Herbrich, S.M.; Baggerly, K.; Patel, K.P.; Brusco, L.; Horombe, C.; Naing, A.; et al. Clinical next generation sequencing to identify actionable aberrations in a phase I program. Oncotarget 2015, 6, 20099–20110. [Google Scholar] [CrossRef] [Green Version]
- Tsongalis, G.J.; Peterson, J.D.; de Abreu, F.B.; Tunkey, C.D.; Gallagher, T.L.; Strausbaugh, L.D.; Wells, W.A.; Amos, C.I. Routine use of the Ion Torrent AmpliSeq™ Cancer Hotspot Panel for identification of clinically actionable somatic mutations. Clin. Chem. Lab. Med. 2014, 52, 707–714. [Google Scholar] [CrossRef]
- Lennon, N.J.; Adalsteinsson, V.A.; Gabriel, S.B. Technological considerations for genome-guided diagnosis and management of cancer. Genome Med. 2016, 8, 112. [Google Scholar] [CrossRef] [Green Version]
- Chakravarty, D.; Gao, J.; Phillips, S.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A precision oncology knowledge base. JCO Precis. Oncol. 2017, 2017, PO.17.00011. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID/Tumor Source | No. of SNVs/MNVs | No. of INDELs | No. of CNVs | No. of Fusions | Total Positive Variants Including SNVs/MNVs/CNVs/Fusions | No. of SNVs/MNVs/INDELs with COSMIC IDs | No. of Non-Synonymous Variants | Actionable Mutations |
|---|---|---|---|---|---|---|---|---|
| FJU01/Metastatic lymph node | 154 | 19 | 11 | - | 184 | 1 | 59 | FGFR2 p.C382R |
| FJU02/Malignant effusion | 179 | 4 | 13 | - | 196 | 3 | 83 | KRAS p.G12R/ |
| ATM deletion | ||||||||
| FJU03/Metastatic lymph node | 137 | 10 | 1 | - | 148 | 2 | 44 | KRAS p.G12V/ |
| TP53 | ||||||||
| p.R273C | ||||||||
| FJU05/Omentum metastasis | 136 | 13 | 10 | - | 159 | 1 | 43 | KRAS |
| p.G12D | ||||||||
| FJU06/Malignant effusion | 135 | 10 | 2 | - | 147 | 2 | 44 | KRAS |
| p.G12V/ | ||||||||
| TP53 | ||||||||
| p.248Q |
| Gene Symbol | No. of Variants | No. of PAC Samples |
|---|---|---|
| TP53 | 29 | 11 |
| KRAS | 23 | 23 |
| PDGFRA | 10 | 10 |
| KIT | 6 | 6 |
| PTEN | 6 | 4 |
| SMARCB1 | 6 | 6 |
| GNAS | 4 | 2 |
| MET | 4 | 4 |
| CDKN2A | 2 | 2 |
| CTNNB1 | 2 | 2 |
| EGFR | 2 | 2 |
| IDH1 | 2 | 2 |
| STK11 | 2 | 2 |
| SMO | 1 | 1 |
| HRAS | 1 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, C.-C.; Liu, C.-Y.; Huang, C.-J.; Hsu, Y.-C.; Lien, H.-H.; Wong, J.-U.; Tai, F.-C.; Ku, W.-H.; Hung, C.-F.; Lin, J.-T.; et al. Deciphering Genetic Alterations of Taiwanese Patients with Pancreatic Adenocarcinoma through Targeted Sequencing. Int. J. Mol. Sci. 2022, 23, 1579. https://doi.org/10.3390/ijms23031579
Huang C-C, Liu C-Y, Huang C-J, Hsu Y-C, Lien H-H, Wong J-U, Tai F-C, Ku W-H, Hung C-F, Lin J-T, et al. Deciphering Genetic Alterations of Taiwanese Patients with Pancreatic Adenocarcinoma through Targeted Sequencing. International Journal of Molecular Sciences. 2022; 23(3):1579. https://doi.org/10.3390/ijms23031579
Chicago/Turabian StyleHuang, Chi-Cheng, Chih-Yi Liu, Chi-Jung Huang, Yao-Chun Hsu, Heng-Hui Lien, Jia-Uei Wong, Feng-Chuan Tai, Wen-Hui Ku, Chi-Feng Hung, Jaw-Town Lin, and et al. 2022. "Deciphering Genetic Alterations of Taiwanese Patients with Pancreatic Adenocarcinoma through Targeted Sequencing" International Journal of Molecular Sciences 23, no. 3: 1579. https://doi.org/10.3390/ijms23031579
APA StyleHuang, C.-C., Liu, C.-Y., Huang, C.-J., Hsu, Y.-C., Lien, H.-H., Wong, J.-U., Tai, F.-C., Ku, W.-H., Hung, C.-F., Lin, J.-T., Huang, C.-S., & Chiang, H.-S. (2022). Deciphering Genetic Alterations of Taiwanese Patients with Pancreatic Adenocarcinoma through Targeted Sequencing. International Journal of Molecular Sciences, 23(3), 1579. https://doi.org/10.3390/ijms23031579