
Epigenetics in the Diagnosis and Therapy of Malignant Melanoma

 , , , ,
, , , , 
Abstract
1. Introduction
2. Incidence and Etiology of Cutaneous Melanoma
3. Histone Modifications and Significance for Melanoma Progression
4. DNA Methylation Alterations in Melanoma
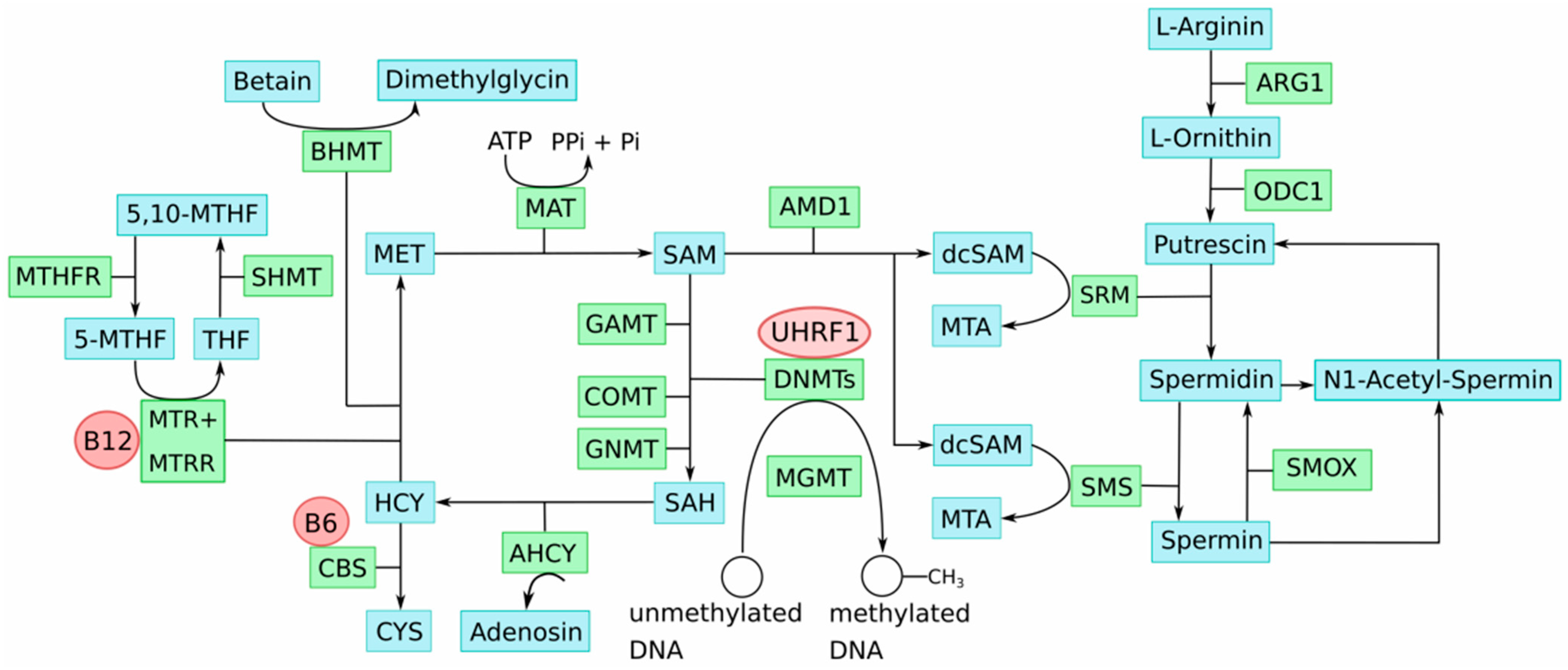
5. One-Carbon Metabolism in the Etiology of Epigenomic Aberrations in Melanoma
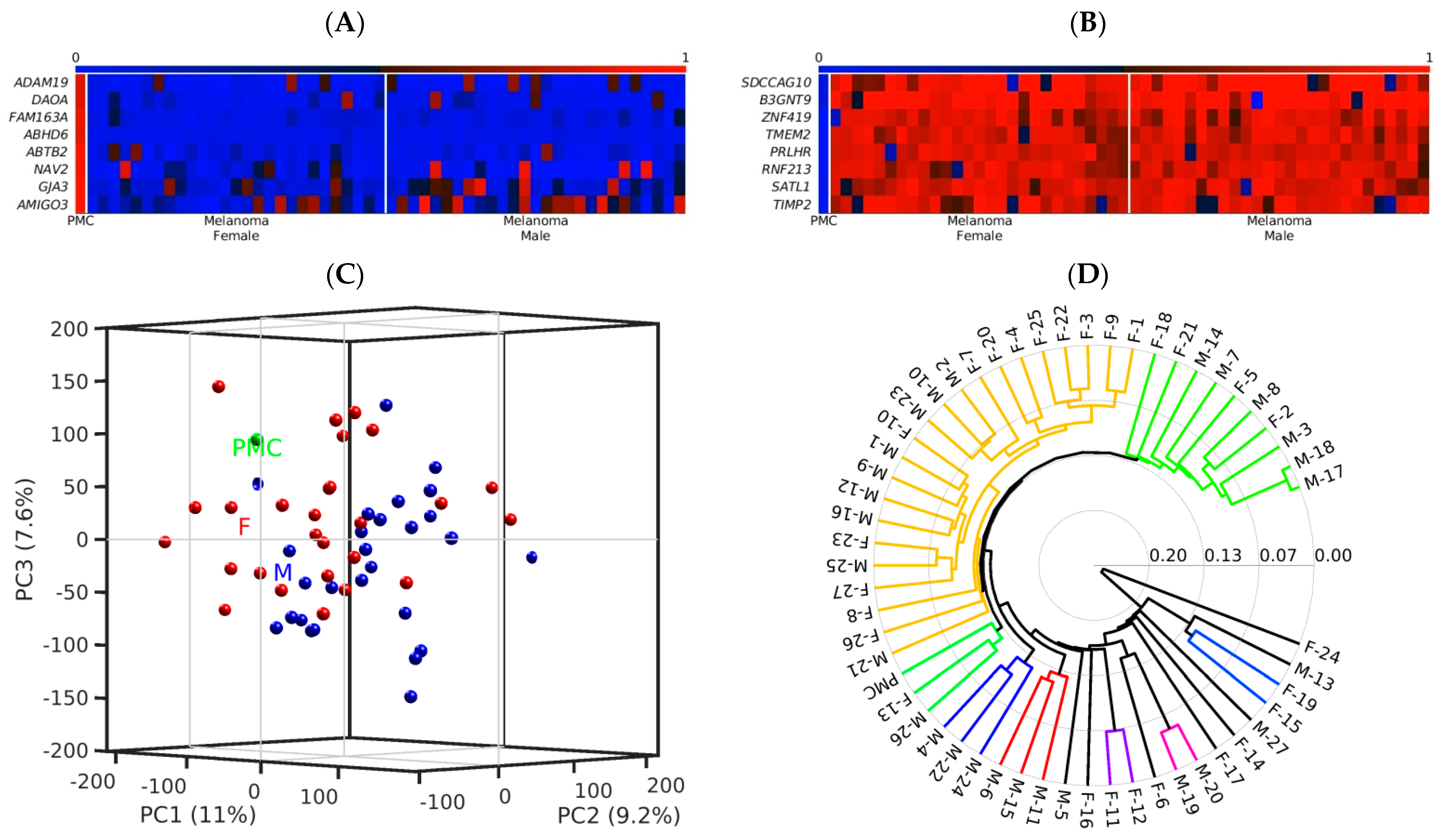
6. Epigenetic Diagnosis Based on Differential Chromatin Organization and DNA Methylation in Melanoma
7. Conclusions
8. Material and Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dietel, M. Boveri at 100: The life and times of Theodor Boveri. J. Pathol. 2014, 234, 135–137. [Google Scholar] [CrossRef] [PubMed]
- Papanicolaou, G.N. A new procedure for staining vaginal smears. Science 1942, 95, 438–439. [Google Scholar] [CrossRef] [PubMed]
- Bettigole, C. The thousand-dollar Pap smear. N. Engl. J. Med. 2013, 369, 1486–1487. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, G.R. Occurrence of 5-methylcytosine in nucleic acids. Nature 1950, 166, 237–238. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Sandru, A.; Voinea, S.; Panaitescu, E.; Blidaru, A. Survival rates of patients with metastatic malignant melanoma. J. Med. Life 2014, 7, 572–576. [Google Scholar]
- Sang, Y.; Deng, Y. Current insights into the epigenetic mechanisms of skin cancer. Dermatol. Ther. 2019, 32, e12964. [Google Scholar] [CrossRef]
- Lombardo, N.; Della Corte, M.; Pelaia, C.; Piazzetta, G.; Lobello, N.; Del Duca, E.; Bennardo, L.; Nisticò, S.P. Primary Mucosal Melanoma Presenting with a Unilateral Nasal Obstruction of the Left Inferior Turbinate. Medicina 2021, 8, 359. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Stratton, M.R.; et al. Signatures of mutational processes in human cancer. Nature 2013, 22, 415–421. [Google Scholar] [CrossRef]
- Dimitriou, F.; Krattinger, R.; Ramelyte, E.; Barysch, M.J.; Micaletto, S.; Dummer, R.; Goldinger, S.M. The World of Melanoma: Epidemiologic, Genetic, and Anatomic Differences of Melanoma Across the Globe. Curr. Oncol. Rep. 2018, 24, 87. [Google Scholar] [CrossRef] [PubMed]
- DeLeon, T.T.; Almquist, D.R.; Kipp, B.R.; Langlais, B.T.; Mangold, A.; Winters, J.L.; Kosiorek, H.E.; Joseph, R.W.; Dronca, S.R.; Bryce, A.H.; et al. Assessment of clinical outcomes with immune checkpoint inhibitor therapy in melanoma patients with CDKN2A and TP53 pathogenic mutations. PLoS ONE 2020, 20, e0230306. [Google Scholar] [CrossRef] [PubMed]
- Colombino, M.; Capone, M.; Lissia, A.; Cossu, A.; Rubino, C.; De Giorgi, V.; Massi, D.; Fonsatti, E.; Staiban, S.; Palmieri, G.; et al. BRAF/NRAS mutation frequencies among primary tumors and metastases in patients with melanoma. J. Clin. Oncol. 2012, 10, 2522–2529. [Google Scholar] [CrossRef] [PubMed]
- Ichihashi, M.; Ueda, M.; Budiyanto, A.; Bito, T.; Oka, M.; Fukunaga, M.; Tsuru, K.; Horikawa, T. UV-induced skin damage. Toxicology 2003, 189, 21–39. [Google Scholar] [CrossRef]
- Uzdensky, A.; Demyanenko, S.; Bibov, M.; Sharifulina, S.; Kit, O.; Przhedetski, Y.; Pozdnyakova, V. Expression of proteins involved in epigenetic regulation in human cutaneous melanoma and peritumoral skin. Tumor Biol. 2014, 35, 8225–8233. [Google Scholar] [CrossRef]
- Smallwood, A.; Estève, P.O.; Pradhan, S.; Carey, M. Functional cooperation between HP1 and DNMT1 mediates gene silencing. Genes Dev. 2007, 21, 1169–1178. [Google Scholar] [CrossRef]
- Orouji, E.; Utikal, J. Tackling malignant melanoma epigenetically: Histone lysine methylation. Clin. Epigenetics 2018, 10, 145. [Google Scholar] [CrossRef]
- Dang, N.N.; Jiao, J.; Meng, X.; An, Y.; Han, C.; Huang, S. Abnormal overexpression of G9a in melanoma cells promotes cancer progression via upregulation of the Notch1 signaling pathway. Aging 2020, 12, 2393–2407. [Google Scholar] [CrossRef]
- Miura, S.; Maesawa, C.; Shibazaki, M.; Yasuhira, S.; Kasai, S.; Tsunoda, K.; Maeda, F.; Takahashi, K.; Akasaka, T.; Masuda, T. Immunohistochemistry for histone h3 lysine 9 methyltransferase and demethylase proteins in human melanomas. Am. J. Dermatopathol. 2014, 36, 211–216. [Google Scholar] [CrossRef]
- Tan, Y.; Tajik, A.; Chen, J.; Jia, Q.; Chowdhury, F.; Wang, L.; Chen, J.; Zhang, S.; Hong, Y.; Yi, H.; et al. Matrix softness regulates plasticity of tumour-repopulating cells via H3K9 demethylation and Sox2 expression. Nat. Commun. 2014, 5, 4619. [Google Scholar] [CrossRef]
- Kelly, G.M.; Al-Ejeh, F.; McCuaig, R.; Casciello, F.; Ahmad Kamal, N.; Ferguson, B.; Pritchard, A.L.; Ali, S.; Silva, I.P.; Wilmott, J.S.; et al. G9a Inhibition Enhances Checkpoint Inhibitor Blockade Response in Melanoma. Clin. Cancer Res. 2021, 27, 2624–2635. [Google Scholar] [CrossRef] [PubMed]
- Ceol, C.J.; Houvras, Y.; Jane-Valbuena, J.; Bilodeau, S.; Orlando, D.A.; Battisti, V.; Fritsch, L.; Lin, W.M.; Hollmann, T.J.; Ferré, F.; et al. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature 2011, 471, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Kim, J.Y.; Lim, S.C.; Lee, K.Y.; Kim, O.; Choi, H.S. SUV39H1/DNMT3A-dependent methylation of the RB1 promoter stimulates PIN1 expression and melanoma development. FASEB J. 2018, 32, 5647–5660. [Google Scholar] [CrossRef] [PubMed]
- Vélez-Cruz, R.; Johnson, D.G. The Retinoblastoma (RB) Tumor Suppressor: Pushing Back against Genome Instability on Multiple Fronts. Int. J. Mol. Sci. 2017, 18, 1776. [Google Scholar] [CrossRef]
- Pützer, B.M.; Steder, M.; Alla, V. Predicting and preventing melanoma invasiveness: Advances in clarifying E2F1 function. Expert Rev. Anticancer Ther. 2010, 10, 1707–1720. [Google Scholar] [CrossRef]
- Soengas, M.S.; Lowe, S.W. Apoptosis and melanoma chemoresistance. Oncogene 2003, 22, 3138–3151. [Google Scholar] [CrossRef]
- Sarkar, D.; Leung, E.Y.; Baguley, B.C.; Finlay, G.J.; Askarian-Amiri, M.E. Epigeneticregulation in human melanoma: Past and future. Epigenetics 2015, 10, 103–121. [Google Scholar] [CrossRef]
- de Sá, B.C.S.; Fugimori, M.L.; Ribeiro, K.d.C.B.; Duprat Neto, J.P.; Neves, R.I.; Landman, G. Proteins involved in pRb and p53 pathways are differentially expressed in thin and thick superficial spreading melanomas. Melanoma Res. 2009, 19, 135–141. [Google Scholar] [CrossRef]
- Straume, O.; Smeds, J.; Kumar, R.; Hemminki, K.; Akslen, L.A. Significant impact of promoter hypermethylation and the 540 C>T polymorphism of CDKN2A in cutaneous melanoma of the vertical growth phase. Am. J. Pathol. 2002, 161, 229–237. [Google Scholar] [CrossRef]
- Tiffen, J.; Gallagher, S.J.; Filipp, F.; Gunatilake, D.; Emran, A.A.; Cullinane, C.; Dutton-Register, K.; Aoude, L.; Hayward, N.; Chatterjee, A.; et al. EZH2 Cooperates with DNA Methylation to Downregulate Key Tumor Suppressors and IFN Gene Signatures in Melanoma. J. Investig. Dermatol. 2020, 140, 2442–2454.e5. [Google Scholar] [CrossRef]
- Moran, B.; Silva, R.; Perry, A.S.; Gallagher, W.M. Epigenetics of malignant melanoma. Semin. Cancer Biol. 2018, 51, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, I.M.; Halvorsen, O.J.; Collett, K.; Stefansson, I.M.; Straume, O.; Haukaas, S.A.; Salvesen, H.B.; Otte, A.P.; Akslen, L.A. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J. Clin. Oncol. 2006, 24, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, D.; Byrum, S.D.; Avaritt, N.L.; Davis, L.; Shields, B.; Mahmoud, F.; Reynolds, M.; Orr, L.M.; Mackintosh, S.G.; Shalin, S.C.; et al. Quantitative Histone Mass Spectrometry Identifies Elevated Histone H3 Lysine27 (Lys27) Trimethylation in Melanoma. Mol. Cell Proteom. 2016, 15, 765–775. [Google Scholar] [CrossRef]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Pontes, K.C.; Heijkants, R.C.; Brouwer, N.J.; Groenewoud, A.; Jordanova, E.S.; Marinkovic, M.; van Duinen, S.; Teunisse, A.F.; Verdijk, R.M.; et al. Overexpression of EZH2 in conjunctival melanoma offers a new therapeutic target. J. Pathol. 2018, 245, 433–444. [Google Scholar] [CrossRef]
- Micevic, G.; Theodosakis, N.; Bosenberg, M. Aberrant DNA methylation in melanoma: Biomarker and therapeutic opportunities. Clin. Epigenet. 2017, 9, 34. [Google Scholar] [CrossRef]
- Koga, Y.; Pelizzola, M.; Cheng, E.; Krauthammer, M.; Sznol, M.; Ariyan, S.; Narayan, D.; Molinaro, A.M.; Halaban, R.; Weissman, S.M. Genome-wide screen of promoter methylation identifies novel markers in melanoma. Genome Res. 2009, 19, 1462–1470. [Google Scholar] [CrossRef]
- Tellez, C.S.; Shen, L.; Estécio, M.R.H.; Jelinek, J.; Gershenwald, J.E.; Issa, J.P.J. CpG island methylation profiling in human melanoma cell lines. Melanoma Res. 2009, 19, 146–155. [Google Scholar] [CrossRef]
- Conway, K.; Edmiston, S.N.; Khondker, Z.S.; Groben, P.A.; Zhou, X.; Chu, H.; Kuan, P.F.; Hao, H.; Carson, C.; Berwick, M.; et al. DNA-methylation profiling distinguishes malignant melanomas from benign nevi. Pigment Cell Melanoma Res. 2011, 24, 352–360. [Google Scholar] [CrossRef]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Kwong, L.N.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Goodier, J.L. Restricting retrotransposons: A review. Mob. DNA 2016, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Ecsedi, S.I.; Hernandez-Vargas, H.; Lima, S.C.; Herceg, Z.; Adany, R.; Balazs, M. Transposable hypomethylation is associated with metastatic capacity of primary melanomas. Int. J. Clin. Exp. Path. 2013, 6, 2943–2948. [Google Scholar]
- Cardelli, M.; Doorn, R.V.; Larcher, L.; Donato, M.D.; Piacenza, F.; Pierpaoli, E.; Giacconi, R.; Malavolta, M.; Rachakonda, S.; Gruis, N.A.; et al. Association of HERV-K and LINE-1 hypomethylation with reduced disease-free survival in melanoma patients. Epigenomics 2020, 12, 1689–1706. [Google Scholar] [CrossRef] [PubMed]
- Erichsen, L.; Ghanjati, F.; Beermann, A.; Poyet, C.; Hermanns, T.; Schulz, W.A.; Seifert, H.H.; Wild, P.J.; Buser, L.; Kröning, A.; et al. Aberrantmethylated key genes of methyl group metabolism within the molecularetiology of urothelial carcinogenesis. Sci. Rep. 2018, 8, 3477. [Google Scholar] [CrossRef]
- Erichsen, L.; Seifert, H.H.; Schulz, W.A.; Hoffmann, M.J.; Niegisch, G.; Araúzo-Bravo, M.J.; Bendhack, M.L.; Poyet, C.; Hermanns, T.; Beermann, A.; et al. Basic Hallmarks of Urothelial Cancer Unleashed in Primary Uroepithelium by Interference with the Epigenetic Master Regulator ODC1. Sci. Rep. 2020, 10, 3808. [Google Scholar] [CrossRef]
- Castanotto, D.; Tommasi, S.; Li, M.; Li, H.; Yanow, S.; Pfeifer, G.P.; Rossi, J.J. Short hairpin RNA-directed cytosine (CpG) methylation of the RASSF1A gene promoter in HeLa cells. Mol. Ther. 2005, 12, 179–183. [Google Scholar] [CrossRef]
- Grant, W.B. An ecologic study of cancer mortality rates in Spain with respect to indices of solar UVB irradiance and smoking. Int. J. Cancer 2007, 120, 1123–1128. [Google Scholar] [CrossRef]
- Hoffman, D.R.; Cornatzer, W.E.; Duerre, J.A. Relationship between tissue levels of S-adenosylmethionine, S-adenylhomocysteine, and transmethylation reactions. Can. J. Biochem. 1979, 57, 56–65. [Google Scholar] [CrossRef]
- Ulrey, C.L.; Liu, L.; Andrews, L.G.; Tollefsbol, T.O. The impact of metabolism on DNA methylation. Hum. Mol. Genet. 2005, 14, R139–R147. [Google Scholar] [CrossRef]
- Coppedè, F. One-carbon metabolism and Alzheimer’s disease: Focus on epigenetics. Curr. Genomics 2010, 11, 246–260. [Google Scholar] [CrossRef]
- Pegg, A.E. Spermidine/spermine-N(1)-acetyltransferase: A key metabolic regulator. Am. J. Physiol. Endocrinol. Metab. 2008, 29, E995–E1010. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, D.; Shima, K.; Matsuo, K.; Nishioka, T.; Chen, C.Y.; Hu, G.F.; Sasaki, A.; Tsuji, T. Ornithine decarboxylase antizyme induces hypomethylation of genome DNA and histone H3 lysine 9 dimethylation (H3K9me2) in human oral cancer cell line. PLoS ONE 2010, 5, e12554. [Google Scholar] [CrossRef] [PubMed]
- The Results Referred to Here Are in Whole or Part Based Upon Data Generated by the TCGA Research Network. Available online: https://www.cancer.gov/tcga (accessed on 17 November 2021).
- Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed]
- Graffmann, N.; Santourlidis, S.; Christ, J.; Wernet, P.; Uhrberg, M. Direct and quantitative analysis of chromatin accessibility by MIRECAL–a Micrococcusnuclease/real-time PCR chromatin accessibility assay with locus specificity. Anal. Biochem. 2006, 354, 308–310. [Google Scholar] [CrossRef] [PubMed]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation bytrans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Kuscu, C.; Arslan, S.; Singh, R.; Thorpe, J.; Adli, M. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat. Biotechnol. 2014, 32, 677–683. [Google Scholar] [CrossRef]
- O’Geen, H.; Henry, I.M.; Bhakta, M.S.; Meckler, J.F.; Segal, D.J. A genome-wide analysis of Cas9 binding specificity using ChIP-seq and targeted sequence capture. Nucleic Acids Res. 2015, 43, 3389–3404. [Google Scholar] [CrossRef]
- Wu, X.; Scott, D.A.; Kriz, A.J.; Chiu, A.C.; Hsu, P.D.; Dadon, D.B.; Cheng, A.W.; Trevino, A.E.; Konermann, S.; Chen, S.; et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat. Biotechnol. 2014, 32, 670–676. [Google Scholar] [CrossRef]
- Schulz, W.A. L1 retrotransposons in human cancers. J. Biomed. Biotechnol. 2006, 2006, 83672. [Google Scholar] [CrossRef]
- Schulz, W.A.; Steinhoff, C.; Florl, A.R. Methylation of endogenous human retroelements in health and disease. Curr. Top. Microbiol. Immunol. 2006, 310, 211–250. [Google Scholar]
- Wilson, A.S.; Power, B.E.; Molloy, P.L. DNA hypomethylation and human diseases. Biochim. Biophys. Acta 2007, 1775, 138–162. [Google Scholar] [CrossRef]
- Santourlidis, S.; Florl, A.; Ackermann, R.; Wirtz, H.C.; Schulz, W.A. Highfrequency of alterations in DNA methylation in adenocarcinoma of the prostate. Prostate 1999, 39, 166–174. [Google Scholar] [CrossRef]
- Florl, A.R.; Löwer, R.; Schmitz-Dräger, B.J.; Schulz, W.A. DNA methylation and expression of LINE-1 and HERV-K provirus sequences in urothelial and renal cell carcinomas. Br. J. Cancer 1999, 80, 1312–1321. [Google Scholar] [CrossRef]
- Jachowicz, J.W.; Bing, X.; Pontabry, J.; Bošković, A.; Rando, O.J.; Torres-Padilla, M.E. LINE-1 activation after fertilization regulates global chromatin accessibility in the early mouse embryo. Nat. Genet. 2017, 49, 1502–1510. [Google Scholar] [CrossRef]
- Ghanjati, F.; Beermann, A.; Hermanns, T.; Poyet, C.; Araúzo-Bravo, M.J.; Seifert, H.H.; Schmidtpeter, M.; Goering, W.; Sorg, R.; Wernet, P.; et al. Unreserved application of epigenetic methods to define differences of DNAmethylation between urinary cellular and cell-free DNA. Cancer Biomark. 2014, 14, 295–302. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Matsusaka, K.; Fukuyo, M.; Rahmutulla, B.; Matsue, H.; Kaneda, A. Higher methylation subtype of malignant melanoma and itscorrelation with thicker progression and worse prognosis. Cancer Med. 2020, 9, 7194–7204. [Google Scholar] [CrossRef]
- Giunta, E.F.; Arrichiello, G.; Curvietto, M.; Pappalardo, A.; Bosso, D.; Rosanova, M.; Diana, A.; Giordano, P.; Petrillo, A.; Ottaviano, M.; et al. Epigenetic Regulation in Melanoma: Facts and Hopes. Cells 2021, 10, 2048. [Google Scholar] [CrossRef]
- Dobre, E.G.; Constantin, C.; Costache, M.; Neagu, M. Interrogating Epigenome toward Personalized Approach in Cutaneous Melanoma. J. Pers. Med. 2021, 11, 901. [Google Scholar] [CrossRef]
- Santourlidis, S.; Ghanjati, F.; Beermann, A.; Hermanns, T.; Poyet, C. IDLN-MSP: Idiolocal normalization of real-time methylation-specific PCR for genetic imbalanced DNA specimens. Biotechniques 2016, 60, 84–87. [Google Scholar] [CrossRef]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef]
- Simon, R.; Bürger, H.; Brinkschmidt, C.; Böcker, W.; Hertle, L.; Terpe, H.J. Chromosomal aberrations associated with inasion in papillary superficial bladder cancer. J. Pathol. 1998, 185, 345–351. [Google Scholar] [CrossRef]
- Al-Quraishy, S.; Dkhil, M.; Abdel-Baki, A.A.; Ghanjati, F.; Erichsen, L.; Santourlidis, S.; Frank, W.; Araúzo-Bravo, M. Protective vaccination and blood-stage malaria modify DNA-methylation of gene promoters in the liver of Balb/c mice. Parasitol. Res. 2017, 116, 1463–1477. [Google Scholar] [CrossRef] [PubMed]
- Zaehres, H.; Kögler, G.; Arauzo-Bravo, M.J.; Bleidissel, M.; Santourlidis, S.; Weinhold, S.; Greber, B.; Kim, J.B.; Buchheiser, A.; Wernet, P.; et al. Induction of pluripotency in human cord blood unrestricted somatic stemcells. Exp. Hematol. 2010, 38, 809–818. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Gene | Fold Change | Gene | Fold Change |
|---|---|---|---|
| FOLR2 | 0.38 | DNMT3a | 0.919 |
| GNMT | 0.471 | AHCYL1 | 0.988 |
| BHMT2 | 0.48 | MTHFR | 0.99 |
| MAT2B | 0.489 | DNMT1 | 1.01 |
| ODC1 | 0.533 | BHMT | 1.08 |
| SHMT1 | 0.559 | MAT2A | 1.11 |
| MGMT | 0.612 | AHCYL2 | 1.13 |
| TCN2 | 0.654 | SMS | 1.34 |
| AMD1 | 0.706 | AHCY | 1.52 |
| UHRF1 | 0.877 | SMOX | 1.72 |
| DNMT3b | 0.893 | ARG1 | 2.11 |
| CBS | 0.904 | FOLR3 | 2.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santourlidis, S.; Schulz, W.A.; Araúzo-Bravo, M.J.; Gerovska, D.; Ott, P.; Bendhack, M.L.; Hassan, M.; Erichsen, L. Epigenetics in the Diagnosis and Therapy of Malignant Melanoma. Int. J. Mol. Sci. 2022, 23, 1531. https://doi.org/10.3390/ijms23031531
Santourlidis S, Schulz WA, Araúzo-Bravo MJ, Gerovska D, Ott P, Bendhack ML, Hassan M, Erichsen L. Epigenetics in the Diagnosis and Therapy of Malignant Melanoma. International Journal of Molecular Sciences. 2022; 23(3):1531. https://doi.org/10.3390/ijms23031531
Chicago/Turabian StyleSantourlidis, Simeon, Wolfgang A. Schulz, Marcos J. Araúzo-Bravo, Daniela Gerovska, Pauline Ott, Marcelo L. Bendhack, Mohamed Hassan, and Lars Erichsen. 2022. "Epigenetics in the Diagnosis and Therapy of Malignant Melanoma" International Journal of Molecular Sciences 23, no. 3: 1531. https://doi.org/10.3390/ijms23031531
APA StyleSantourlidis, S., Schulz, W. A., Araúzo-Bravo, M. J., Gerovska, D., Ott, P., Bendhack, M. L., Hassan, M., & Erichsen, L. (2022). Epigenetics in the Diagnosis and Therapy of Malignant Melanoma. International Journal of Molecular Sciences, 23(3), 1531. https://doi.org/10.3390/ijms23031531