
Allosterism in the PDZ Family

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Background
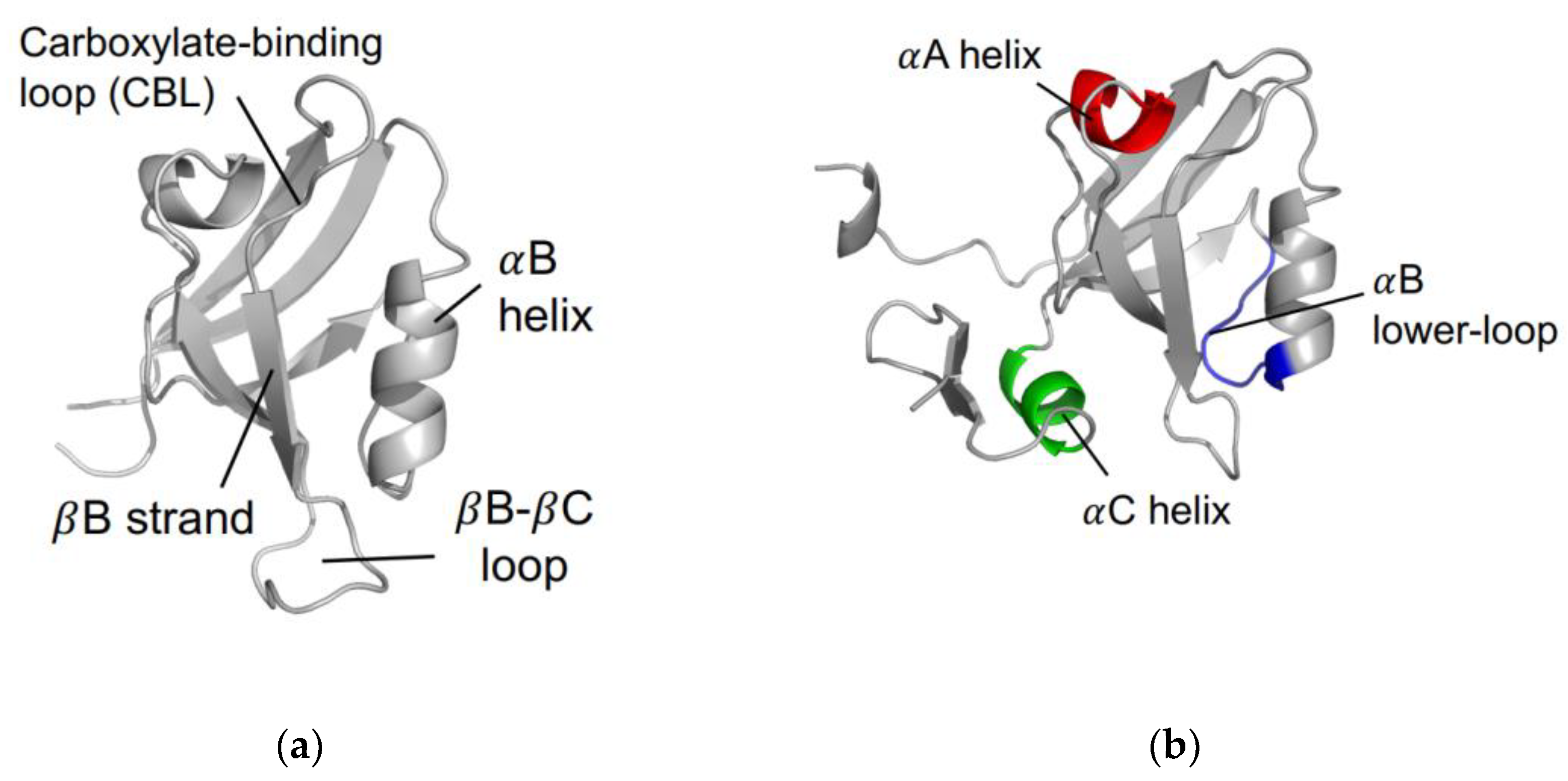
1.1. The αA Helix
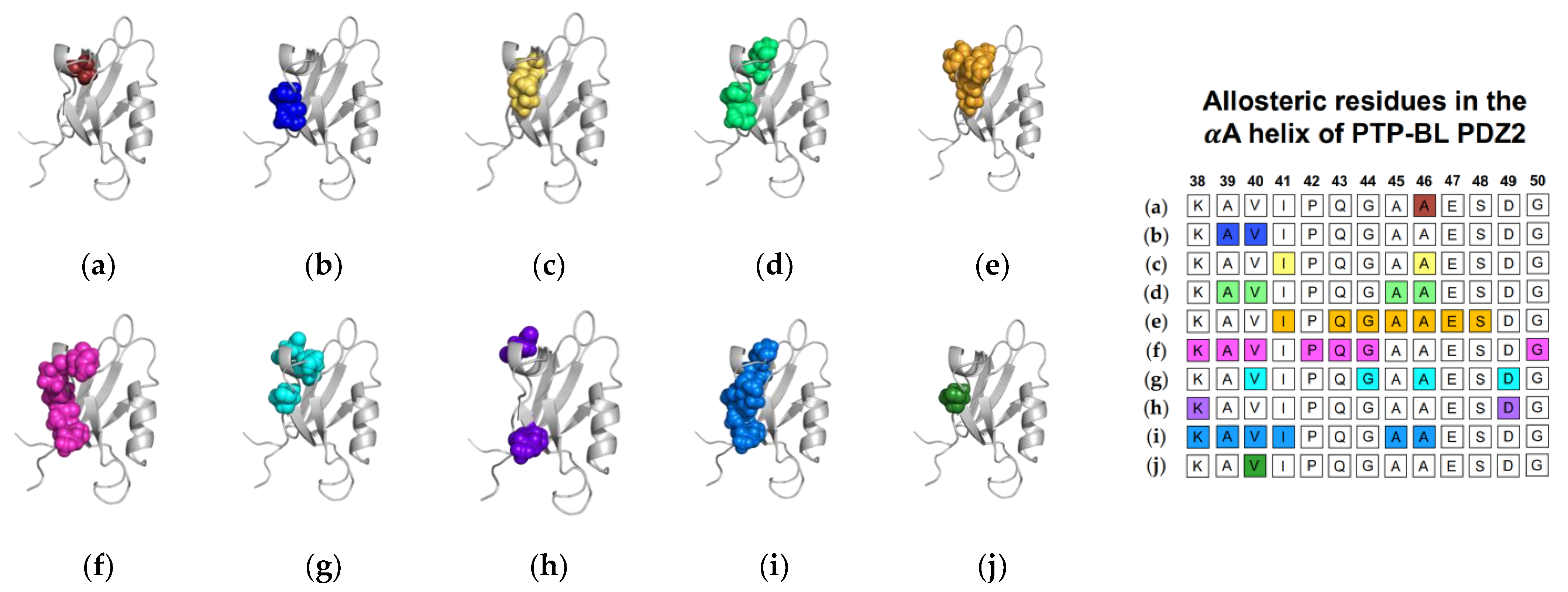
1.1.1. Agreement between Experimental and Computational Techniques: PTP-BL PDZ2
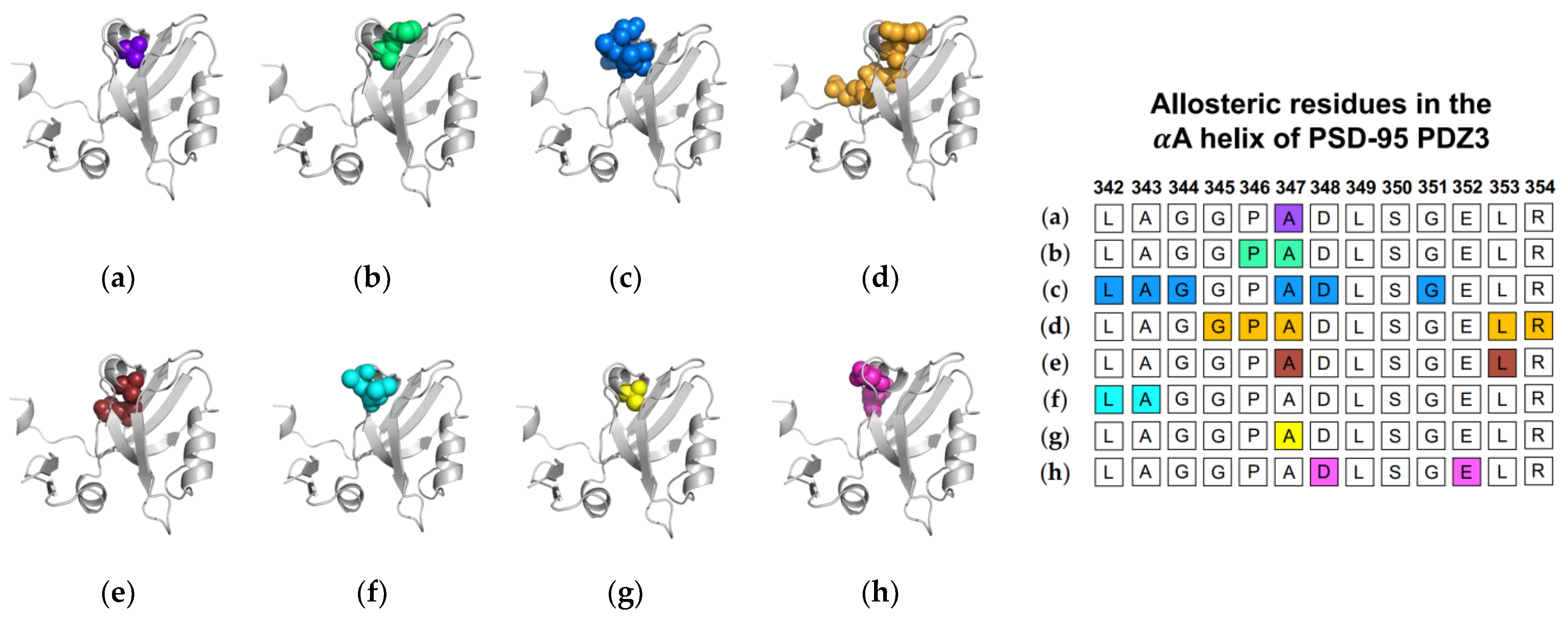
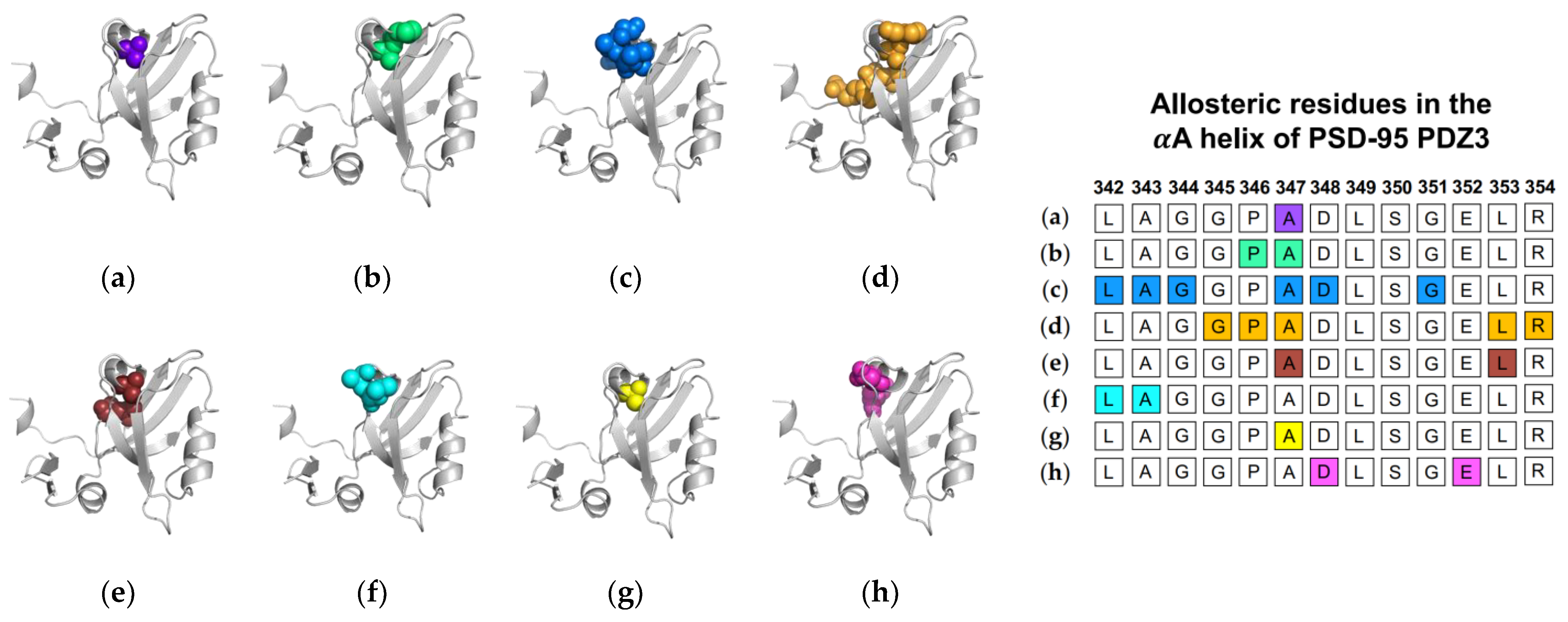
1.1.2. Computational Conclusions Lacking Experimental Support: PSD-95 PDZ3
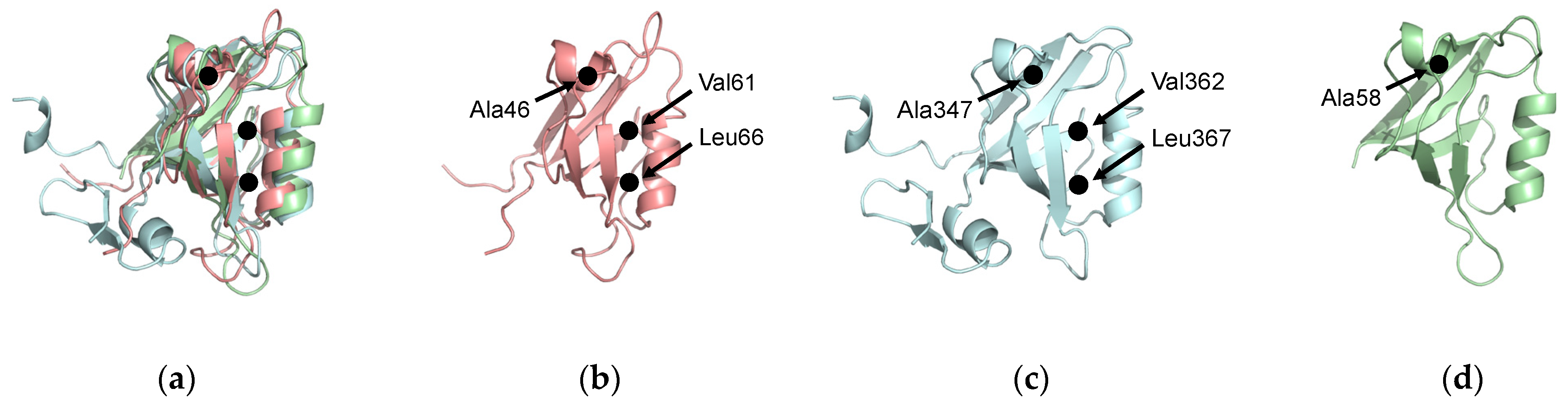
1.1.3. Two-Way Communication in Par-6 PDZ
1.2. The αB Lower-Loop
1.2.1. Agreement between Experimental and Computational Techniques: PTP-BL PDZ2
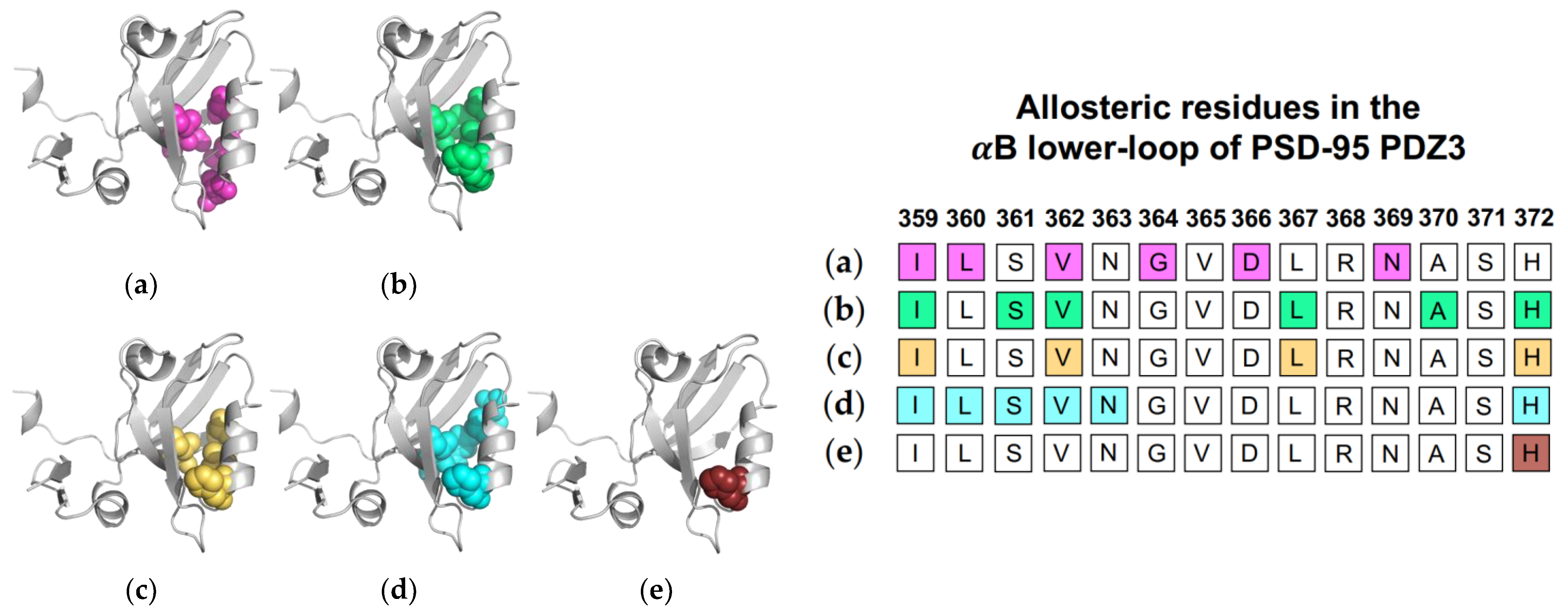
1.2.2. Computational Conclusions Lacking Experimental Support: PSD-95 PDZ3
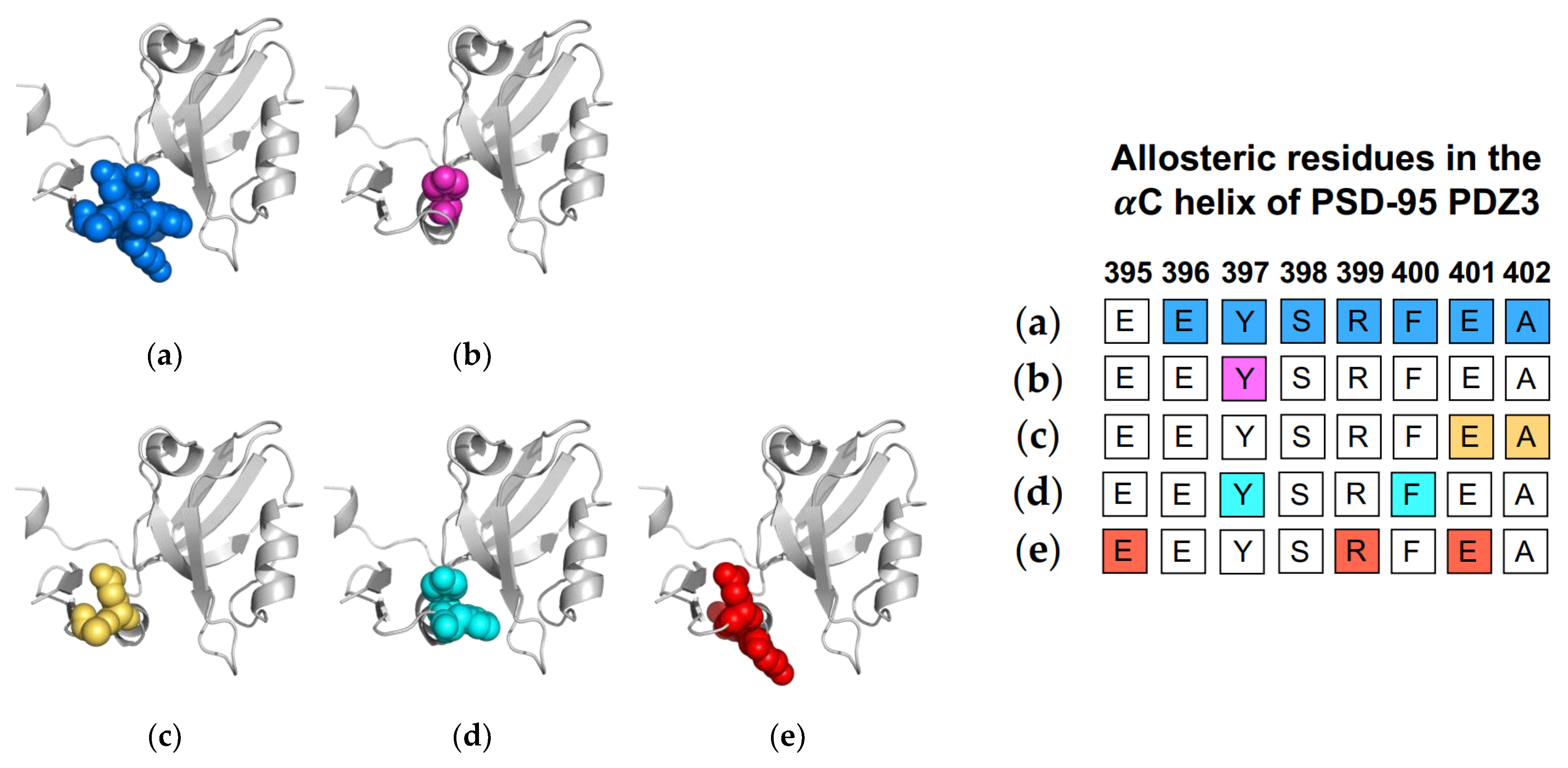
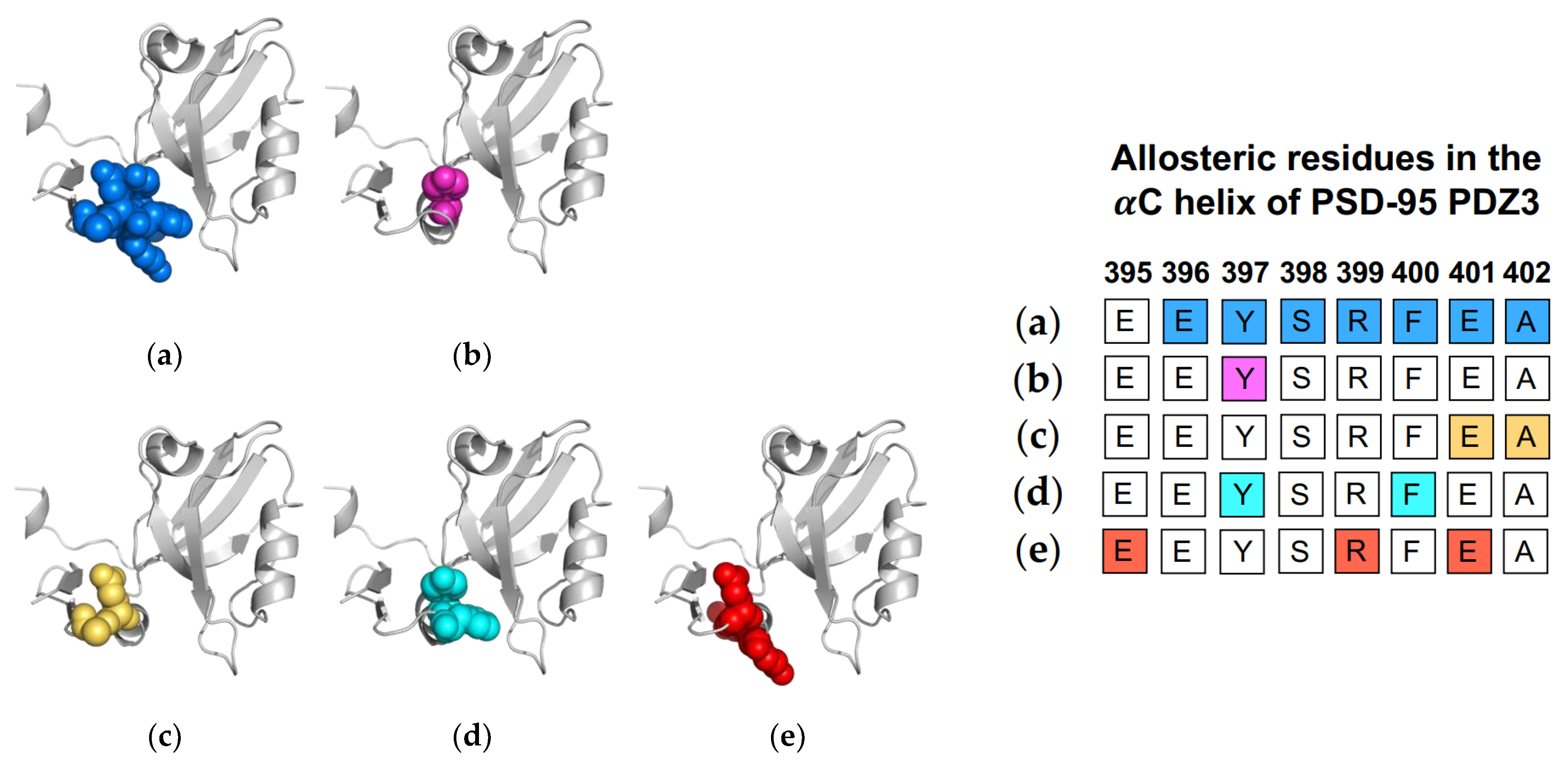
1.3. The αC Helix in PSD-95 PDZ3
2. Perspective
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Correction Statement
References
- Cooper, A.; Dryden, D.T.F. Allostery without conformational change. Eur. Biophys. J. 1984, 11, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Hilser, V.J.; Wrabl, J.O.; Motlagh, H.N. Structural and Energetic Basis of Allostery. Annu. Rev. Biophys. 2012, 41, 585–609. [Google Scholar] [CrossRef] [PubMed]
- Campitelli, P.; Modi, T.; Kumar, S.; Banu Ozkan, S. The Role of Conformational Dynamics and Allostery in Modulating Protein Evolution. Annu. Rev. Biophys. 2020, 49, 267–288. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.; Karplus, M. Allostery and cooperativity revisited. Protein Sci. 2008, 17, 1295–1307. [Google Scholar] [CrossRef]
- Gunasekaran, K.; Ma, B.; Nussinov, R. Is allostery an intrinsic property of all dynamic proteins? Proteins Struct. Funct. Bioinform. 2004, 57, 433–443. [Google Scholar] [CrossRef]
- Nussinov, R.; Tsai, C.-J.; Ma, B. The Underappreciated Role of Allostery in the Cellular Network. Annu. Rev. Biophys. 2013, 42, 169–189. [Google Scholar] [CrossRef]
- Christensen, N.R.; Čalyševa, J.; Fernandes, E.F.A.; Lüchow, S.; Clemmensen, L.S.; Haugaard-Kedström, L.M.; Strømgaard, K. PDZ Domains as Drug Targets. Adv. Ther. 2019, 2, 1800143. [Google Scholar] [CrossRef]
- Kennedy, M.B. Origin of PDZ (DHR, GLGF) domains. Trends Biochem. Sci. 1995, 20, 350. [Google Scholar] [CrossRef]
- Ponting, C.P. Evidence for PDZ domains in bacteria, yeast, and plants. Protein Sci. 2008, 6, 464–468. [Google Scholar] [CrossRef]
- Cabral, J.H.M.; Petosa, C.; Sutcliffe, M.J.; Raza, S.; Byron, O.; Poy, F.; Marfatia, S.M.; Chishti, A.H.; Liddington, R.C. Crystal structure of a PDZ domain. Nature 1996, 382, 649–652. [Google Scholar] [CrossRef]
- Van Ham, M.; Hendriks, W. PDZ domains-glue and guide. Mol. Biol. Rep. 2003, 30, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Sheng, M. PDZ domain proteins of synapses. Nat. Rev. Neurosci. 2004, 5, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Zhang, M. Structures and target recognition modes of PDZ domains: Recurring themes and emerging pictures. Biochem. J. 2013, 455, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Harris, B.Z.; Lim, W.A. Mechanisma and role of PDZ domains in signaling complex assembly. J. Cell Sci. 2001, 114, 3219–3231. [Google Scholar] [CrossRef]
- Brakeman, P.R.; Lanahan, A.A.; O’Brien, R.; Roche, K.; Barnes, C.A.; Huganir, R.L.; Worley, P.F. Homer: A protein that selectively binds metabotropic glutamate receptors. Nature 1997, 386, 284–288. [Google Scholar] [CrossRef]
- Romero, G.; Von Zastrow, M.; Friedman, P.A. Role of PDZ Proteins in Regulating Trafficking, Signaling, and Function of GPCRs. Means, Motif, and Opportunity. Adv. Pharmacol. 2011, 62, 279–314. [Google Scholar] [CrossRef]
- Doyle, D.A.; Lee, A.; Lewis, J.; Kim, E.; Sheng, M.; MacKinnon, R. Crystal structures of a complexed and peptide-free membrane protein- binding domain: Molecular basis of peptide recognition by PDZ. Cell 1996, 85, 1067–1076. [Google Scholar] [CrossRef]
- Pedersen, S.W.; Pedersen, S.B.; Anker, L.; Hultqvist, G.; Kristensen, A.S.; Jemth, P.; Strømgaard, K. Probing backbone hydrogen bonding in bePDZ/ligand interactions by protein amide-to-ester mutations. Nat. Commun. 2014, 5, 3215. [Google Scholar] [CrossRef]
- Walma, T.; Spronk, C.A.E.M.; Tessari, M.; Aelen, J.; Schepens, J.; Hendriks, W.; Vuister, G.W. Structure, dynamics and binding characteristics of the second PDZ domain of PTP-BL. J. Mol. Biol. 2002, 316, 1101–1110. [Google Scholar] [CrossRef]
- Lockless, S.W.; Ranganathan, R. Evolutionarily conserved pathways of energetic connectivity in protein families. Science 1999, 286, 295–299. [Google Scholar] [CrossRef]
- Kumawat, A.; Chakrabarty, S. Hidden electrostatic basis of dynamic allostery in a PDZ domain. Proc. Natl. Acad. Sci. USA 2017, 114, E5825–E5834. [Google Scholar] [CrossRef] [PubMed]
- De Los Rios, P.; Cecconi, F.; Pretre, A.; Dietler, G.; Michielin, O.; Piazza, F.; Juanico, B. Functional dynamics of PDZ binding domains: A normal-mode analysis. Biophys. J. 2005, 89, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Von Ossowski, I.; Oksanen, E.; von Ossowski, L.; Cai, C.; Sundberg, M.; Goldman, A.; Keinänen, K. Crystal structure of the second PDZ domain of SAP97 in complex with a GluR-AC-terminal peptide. FEBS J. 2006, 273, 5219–5229. [Google Scholar] [CrossRef]
- Grembecka, J.; Cierpicki, T.; Devedjiev, Y.; Derewenda, U.; Kang, B.S.; Bushweller, J.H.; Derewenda, Z.S. The Binding of the PDZ Tandem of Syntenin to Target Proteins. Biochemistry 2006, 45, 3674–3683. [Google Scholar] [CrossRef]
- Chen, Q.; Niu, X.; Xu, Y.; Wu, J.; Shi, Y. Solution structure and backbone dynamics of the AF-6 PDZ domain/Bcr peptide complex. Protein Sci. 2007, 16, 1053–1062. [Google Scholar] [CrossRef]
- Tochio, H.; Hung, F.; Li, M.; Bredt, D.S.; Zhang, M. Solution structure and backbone dynamics of the second PDZ domain of postsynaptic density-95. J. Mol. Biol. 2000, 295, 225–237. [Google Scholar] [CrossRef]
- Fuentes, E.J.; Der, C.J.; Lee, A.L. Ligand-dependent Dynamics and Intramolecular Signaling in a PDZ Domain. J. Mol. Biol. 2004, 335, 1105–1115. [Google Scholar] [CrossRef] [PubMed]
- Gianni, S.; Walma, T.; Arcovito, A.; Calosci, N.; Bellelli, A.; Engström, Å.; Travaglini-Allocatelli, C.; Brunori, M.; Jemth, P.; Vuister, G.W. Demonstration of Long-Range Interactions in a PDZ Domain by NMR, Kinetics, and Protein Engineering. Structure 2006, 14, 1801–1809. [Google Scholar] [CrossRef] [PubMed]
- Dhulesia, A.; Gsponer, J.; Vendruscolo, M. Mapping of two networks of residues that exhibit structural and dynamical changes upon binding in a PDZ domain protein. J. Am. Chem. Soc. 2008, 130, 8931–8939. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Chen, Q.; Zhang, J.; Shen, W.; Shi, Y.; Wu, J. Interesting structural and dynamical behaviors exhibited by the AF-6 PDZ domain upon Bcr peptide binding. Biochemistry 2007, 46, 15042–15053. [Google Scholar] [CrossRef]
- Morra, G.; Genoni, A.; Colombo, G. Mechanisms of differential allosteric modulation in homologous proteins: Insights from the analysis of internal dynamics and energetics of PDZ domains. J. Chem. Theory Comput. 2014, 10, 5677–5689. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Knecht, V.; Stock, G. Long-Range Conformational Response of a PDZ Domain to Ligand Binding and Release: A Molecular Dynamics Study. J. Chem. Theory Comput. 2016, 12, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Miño-Galaz, G.A. Allosteric communication pathways and thermal rectification in pdz-2 protein: A computational study. J. Phys. Chem. B 2015, 119, 6179–6189. [Google Scholar] [CrossRef]
- Peterson, F.C.; Penkert, R.R.; Volkman, B.F.; Prehoda, K.E. Cdc42 regulates the Par-6 PDZ domain through an allosteric CRIB-PDZ transition. Mol. Cell 2004, 13, 665–676. [Google Scholar] [CrossRef]
- Fuentes, E.J.; Gilmore, S.A.; Mauldin, R.V.; Lee, A.L. Evaluation of Energetic and Dynamic Coupling Networks in a PDZ Domain Protein. J. Mol. Biol. 2006, 364, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Petit, C.M.; Zhang, J.; Sapienza, P.J.; Fuentes, E.J.; Lee, A.L. Hidden dynamic allostery in a PDZ domain. Proc. Natl. Acad. Sci. USA 2009, 106, 18249–18254. [Google Scholar] [CrossRef]
- Zhang, J.; Petit, C.M.; King, D.S.; Lee, A.L. Phosphorylation of a PDZ domain extension modulates binding affinity and interdomain interactions in postsynaptic density-95 (PSD-95) protein, a membrane-associated guanylate kinase (MAGUK). J. Biol. Chem. 2011, 286, 41776–41785. [Google Scholar] [CrossRef]
- Swain, J.; Gierasch, L.M. The changing landscape of protein allostery. Curr. Opin. Struct. Biol. 2006, 16, 102–108. [Google Scholar] [CrossRef]
- Jemth, P.; Gianni, S. PDZ Domains: Folding and Binding. Biochemistry 2007, 46, 8701–8708. [Google Scholar] [CrossRef]
- Tsai, C.J.; Del Sol, A.; Nussinov, R. Protein allostery, signal transmission and dynamics: A classification scheme of allosteric mechanisms. Mol. Biosyst. 2009, 5, 207–216. [Google Scholar] [CrossRef]
- Smock, R.G.; Gierasch, L.M. Sending signals dynamically. Science 2009, 324, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Ivarsson, Y. Plasticity of PDZ domains in ligand recognition and signaling. FEBS Lett. 2012, 586, 2638–2647. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.L. Contrasting roles of dynamics in protein allostery: NMR and structural studies of CheY and the third PDZ domain from PSD-95. Biophys. Rev. 2015, 7, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Gautier, C.; Laursen, L.; Gianni, S. Seeking allosteric networks in PDZ domains. Protein Eng. Des. Sel. 2018, 31, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Van Den Berk, L.C.J.; Landi, E.; Walma, T.; Vuister, G.W.; Dente, L.; Hendriks, W.J.A.J. An allosteric intramolecular PDZ-PDZ interaction modulates PTP-BL PDZ2 binding specificity. Biochemistry 2007, 46, 13629–13637. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Karplus, M. Signaling pathways of PDZ2 domain: A molecular dynamics interaction correlation analysis. Proteins Struct. Funct. Bioinform. 2009, 74, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Kalescky, R.; Liu, J.; Tao, P. Identifying key residues for protein allostery through rigid residue scan. J. Phys. Chem. A 2015, 119, 1689–1700. [Google Scholar] [CrossRef]
- Cilia, E.; Vuister, G.W.; Lenaerts, T. Accurate Prediction of the Dynamical Changes within the Second PDZ Domain of PTP1e. PLoS Comput. Biol. 2012, 8, 1002794. [Google Scholar] [CrossRef]
- Ota, N.; Agard, D.A. Intramolecular signaling pathways revealed by modeling anisotropic thermal diffusion. J. Mol. Biol. 2005, 351, 345–354. [Google Scholar] [CrossRef]
- Gerek, Z.N.; Ozkan, S.B. Change in allosteric network affects binding affinities of PDZ domains: Analysis through perturbation response scanning. PLoS Comput. Biol. 2011, 7, 1002154. [Google Scholar] [CrossRef]
- Kozlov, G.; Gehring, K.; Ekiel, I. Solution Structure of the PDZ2 Domain from Human Phosphatase hPTP1E and Its Interactions with C-Terminal Peptides from the Fas Receptor. Biochemistry 2000, 39, 2572–2580. [Google Scholar] [CrossRef]
- Kozlov, G.; Banville, D.; Gehring, K.; Ekiel, I. Solution structure of the PDZ2 domain from cytosolic human phosphatase hPTP1E complexed with a peptide reveals contribution of the beta2-beta3 loop to PDZ domain-ligand interactions. J. Mol. Biol. 2002, 320, 813–820. [Google Scholar] [CrossRef]
- Raimondi, F.; Felline, A.; Seeber, M.; Mariani, S.; Fanelli, F. A mixed protein structure network and elastic network model approach to predict the structural communication in biomolecular systems: The PDZ2 domain from tyrosine phosphatase 1E as a case study. J. Chem. Theory Comput. 2013, 9, 2504–2518. [Google Scholar] [CrossRef]
- Kalescky, R.; Zhou, H.; Liu, J.; Tao, P. Rigid Residue Scan Simulations Systematically Reveal Residue Entropic Roles in Protein Allostery. PLoS Comput. Biol. 2016, 12, e1004893. [Google Scholar] [CrossRef]
- Bozovic, O.; Zanobini, C.; Gulzar, A.; Jankovic, B.; Buhrke, D.; Post, M.; Wolf, S.; Stock, G.; Hamm, P. Real-time observation of ligand-induced allosteric transitions in a PDZ domain. Proc. Natl. Acad. Sci. USA 2020, 117, 26031–26039. [Google Scholar] [CrossRef]
- McLaughlin, R.N., Jr.; Poelwijk, F.J.; Raman, A.; Gosal, W.S.; Ranganathan, R. The spatial architecture of protein function and adaptation. Nature 2012, 491, 138–142. [Google Scholar] [CrossRef]
- Gianni, S.; Haq, S.R.; Montemiglio, L.C.; Jürgens, M.C.; Engström, Å.; Chi, C.N.; Brunori, M.; Jemth, P. Sequence-specific long range networks in PSD-95/Discs large/ZO-1 (PDZ) domains tune their binding selectivity. J. Biol. Chem. 2011, 286, 27167–27175. [Google Scholar] [CrossRef]
- Du, Q.-S.; Wang, C.-H.; Liao, S.-M.; Huang, R.-B. Correlation Analysis for Protein Evolutionary Family Based on Amino Acid Position Mutations and Application in PDZ Domain. PLoS ONE 2010, 5, e13207. [Google Scholar] [CrossRef]
- Ho, B.K.; Agard, D.A. Conserved tertiary couplings stabilize elements in the PDZ fold, leading to characteristic patterns of domain conformational flexibility. Protein Sci. 2010, 19, 398–411. [Google Scholar] [CrossRef]
- Garrard, S.M.; Capaldo, C.T.; Gao, L.; Rosen, M.K.; Macara, I.G.; Tomchick, D.R. Structure of Cdc42 in a complex with the GTPase-binding domain of the cell polarity protein, Par6. EMBO J. 2003, 22, 1125–1133. [Google Scholar] [CrossRef]
- Penkert, R.; DiVittorio, H.M.; Prehoda, K.E. Internal recognition through PDZ domain plasticity in the Par-6-Pals1 complex. Nat. Struct. Mol. Biol. 2004, 11, 1122–1127. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Goda, N.; Tamashiro, T.; Narita, H.; Satomura, K.; Tenno, T.; Nakagawa, A.; Oda, M.; Suzuki, M.; Sakisaka, T.; et al. Crystal structure of afadin PDZ domain–nectin-3 complex shows the structural plasticity of the ligand-binding site. Protein Sci. 2015, 24, 376–385. [Google Scholar] [CrossRef]
- Bozovic, O.; Jankovic, B.; Hamm, P. Sensing the allosteric force. Nat. Commun. 2020, 11, 5841. [Google Scholar] [CrossRef]
- Stevens, A.O.; He, Y. The electrostatic allostery could be the trigger for the changes in dynamics for the PDZ domain of PICK1. bioRxiv 2020. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stevens, A.O.; He, Y. Allosterism in the PDZ Family. Int. J. Mol. Sci. 2022, 23, 1454. https://doi.org/10.3390/ijms23031454
Stevens AO, He Y. Allosterism in the PDZ Family. International Journal of Molecular Sciences. 2022; 23(3):1454. https://doi.org/10.3390/ijms23031454
Chicago/Turabian StyleStevens, Amy O., and Yi He. 2022. "Allosterism in the PDZ Family" International Journal of Molecular Sciences 23, no. 3: 1454. https://doi.org/10.3390/ijms23031454
APA StyleStevens, A. O., & He, Y. (2022). Allosterism in the PDZ Family. International Journal of Molecular Sciences, 23(3), 1454. https://doi.org/10.3390/ijms23031454