
Histamine H4 Receptor Agonism Induces Antitumor Effects in Human T-Cell Lymphoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
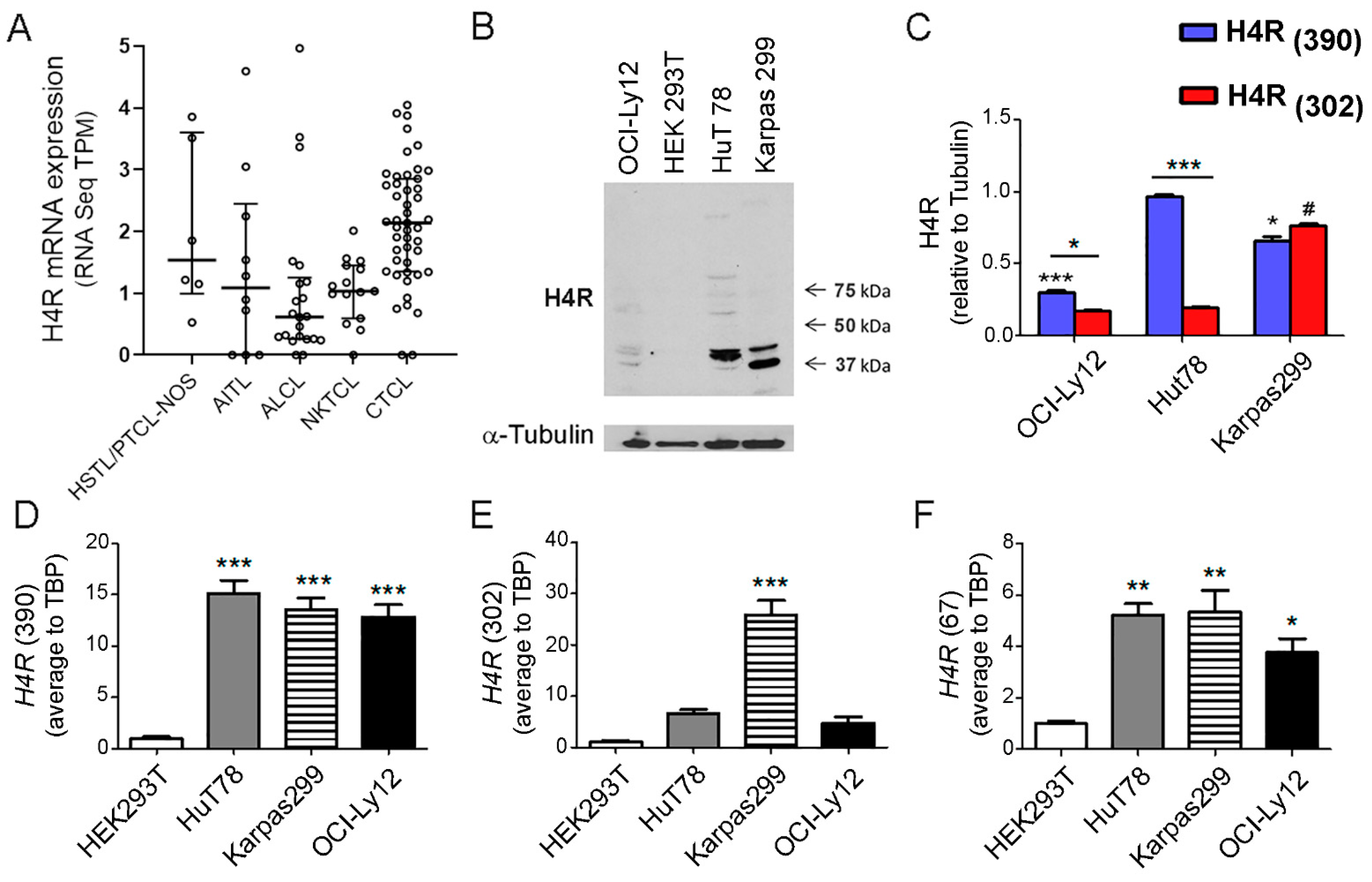
2.1. H4R Expression in TCL
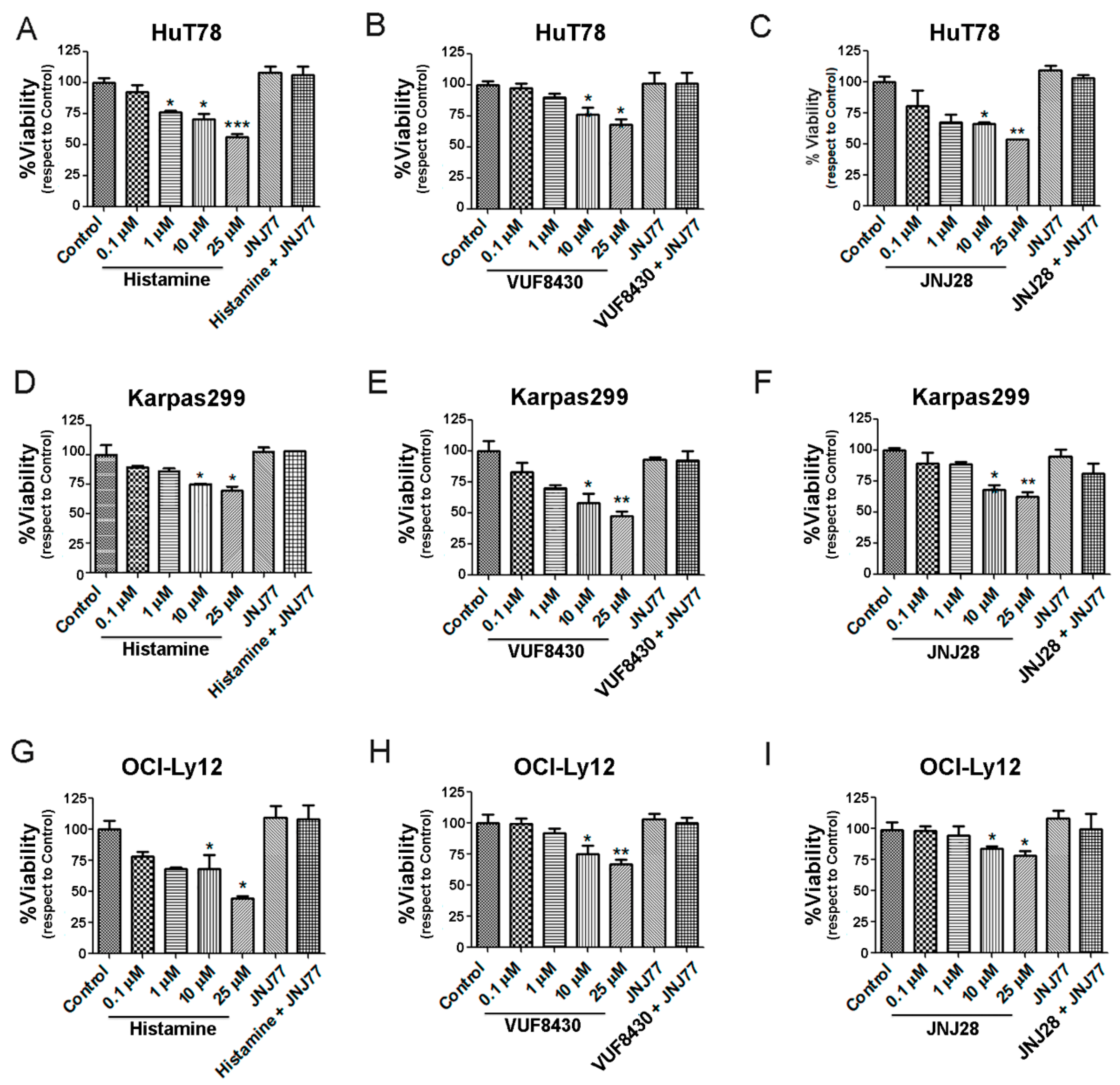
2.2. Effect of Histamine and H4R Ligands on the Viability of TCL Cell Lines
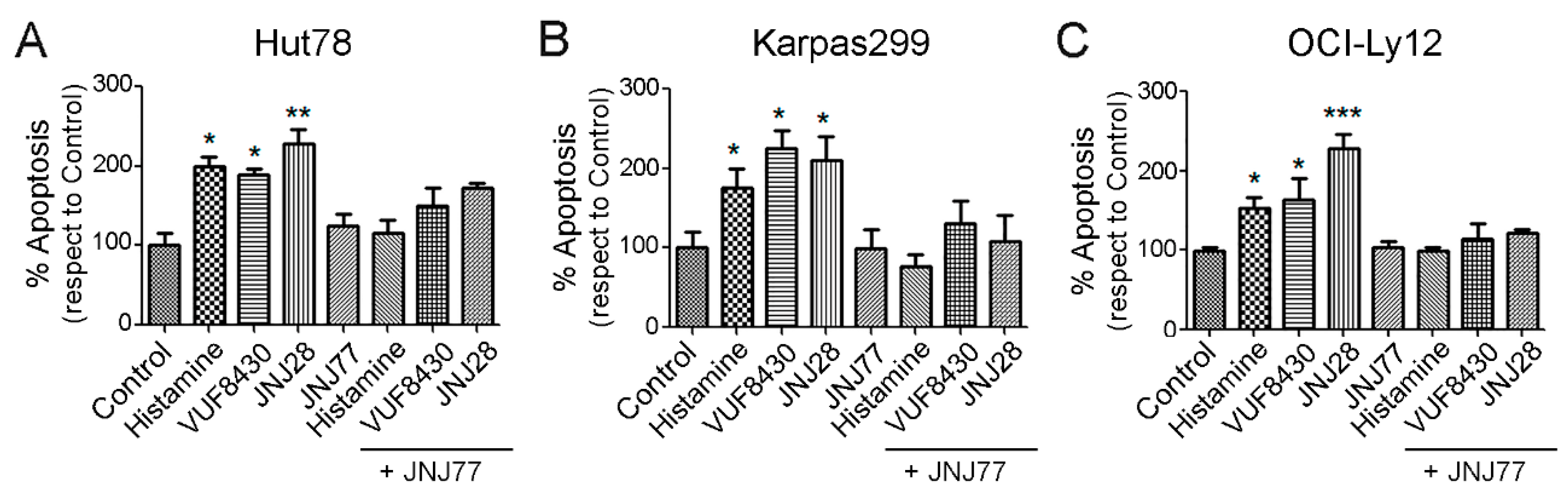
2.3. Effect of Histamine and H4R Ligands on the Apoptosis of TCL Cell Lines
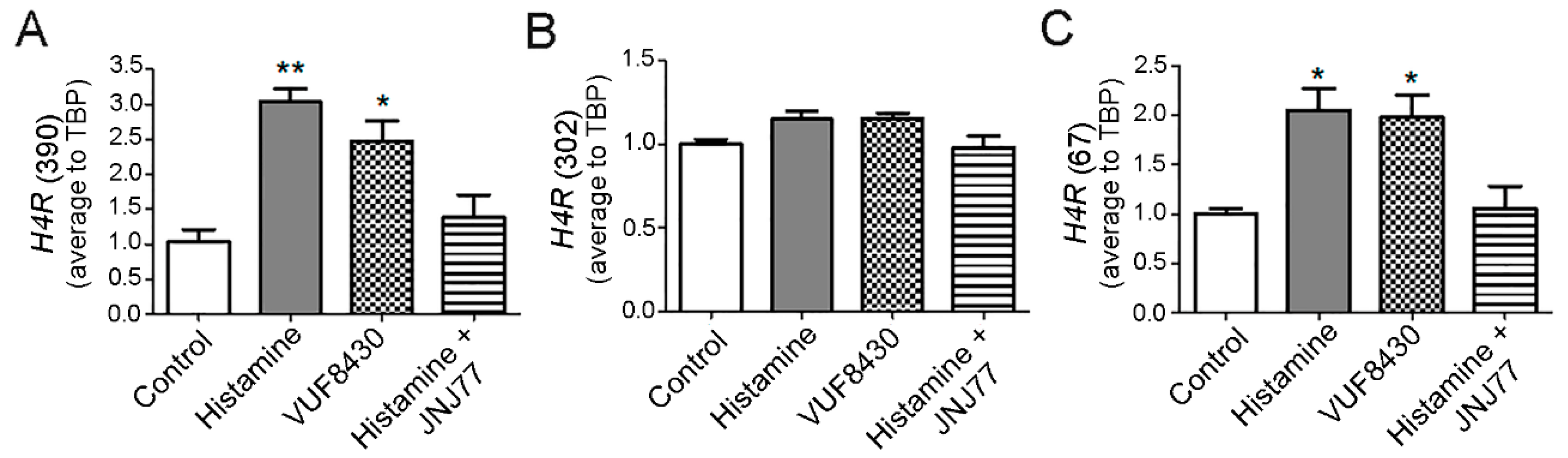
2.4. Modulation of H4R mRNA Expression in HuT78 Cells by Histamine and Specific H4R Agonists
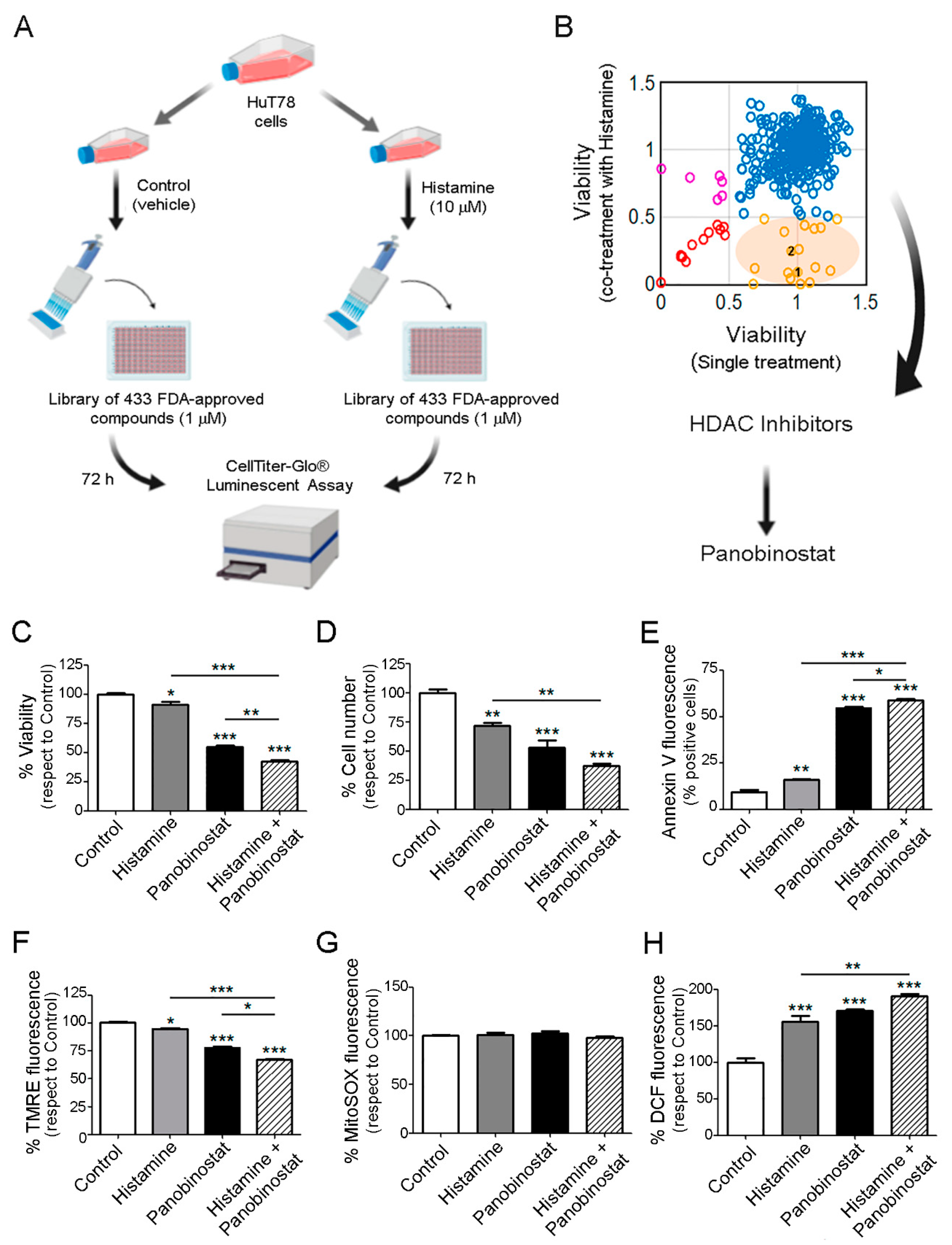
2.5. Therapeutic Benefit for the Combination Therapy with Histamine and Histone Deacetylase Inhibitors
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Transcriptional Datasets Analyses
4.3. Cultured Cells
4.4. Reverse Transcription and Quantitative Real-Time PCR (qRT-PCR) for H4R Isoforms
4.5. Protein Extraction
4.6. Immunoblotting
4.7. Cell Viability Assay
4.8. Apoptosis Determinations
4.9. Drug Screening Library
4.10. Measurement of Mitochondrial and Cellular ROS Levels
4.11. Mitochondrial Transmembrane Potential Determination
4.12. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Willemze, R.; Cerroni, L.; Kempf, W.; Berti, E.; Facchetti, F.; Swerdlow, S.H.; Jaffe, E.S. The 2018 Update of the WHO-EORTC Classification for Primary Cutaneous Lymphomas. Blood 2019, 133, 1703–1714. [Google Scholar] [CrossRef]
- Kamijo, H.; Miyagaki, T. Mycosis Fungoides and Sézary Syndrome: Updates and Review of Current Therapy. Curr. Treat. Options Oncol. 2021, 22, 10. [Google Scholar] [CrossRef]
- Jiang, M.; Bennani, N.N.; Feldman, A.L. Lymphoma Classification Update: T-Cell Lymphomas, Hodgkin Lymphomas, and Histiocytic/Dendritic Cell Neoplasms. Expert Rev. Hematol. 2017, 10, 239–249. [Google Scholar] [CrossRef] [Green Version]
- Pera, B.; Krumsiek, J.; Assouline, S.E.; Marullo, R.; Patel, J.; Phillip, J.M.; Román, L.; Mann, K.K.; Cerchietti, L. Metabolomic Profiling Reveals Cellular Reprogramming of B-Cell Lymphoma by a Lysine Deacetylase Inhibitor through the Choline Pathway. EBioMedicine 2018, 28, 80–89. [Google Scholar] [CrossRef] [Green Version]
- Mehrpouri, M.; Pourbagheri-Sigaroodi, A.; Bashash, D. The Contributory Roles of Histone Deacetylases (HDACs) in Hematopoiesis Regulation and Possibilities for Pharmacologic Interventions in Hematologic Malignancies. Int. Immunopharmacol. 2021, 100, 108–114. [Google Scholar] [CrossRef]
- Hristov, A.C.; Tejasvi, T.; Wilcox, R.A. Cutaneous T-Cell Lymphomas: 2021 Update on Diagnosis, Risk-Stratification, and Management. Am. J. Hematol. 2021, 96, 1313–1328. [Google Scholar] [CrossRef] [PubMed]
- Yeruva, S.L.H.; Zhao, F.; Miller, K.D.; Tevaarwerk, A.J.; Wagner, L.I.; Gray, R.J.; Sparano, J.A.; Connolly, R.M. E2112: Randomized Phase III Trial of Endocrine Therapy plus Entinostat/Placebo in Patients with Hormone Receptor-Positive Advanced Breast Cancer. Npj Breast Cancer 2018, 4, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terranova-Barberio, M.; Thomas, S.; Ali, N.; Pawlowska, N.; Park, J.; Krings, G.; Rosenblum, M.D.; Budillon, A.; Munster, P.N. HDAC Inhibition Potentiates Immunotherapy in Triple Negative Breast Cancer. Oncotarget 2017, 8, 114156–114172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byron, J.W. Mechanism for Histamine H2-Receptor Induced Cell-Cycle Changes in the Bone Marrow Stem Cell. Agents Actions 1977, 7, 209–213. [Google Scholar] [CrossRef]
- Stark, H. Histamine H4 Receptor: A Novel Drug Target in Immunoregulary and Inflammation Diseases; Stark, H., Ed.; Versita Ltd.: London, UK, 2013; ISBN 978-83-7656-056-4. [Google Scholar]
- Massari, N.A.; Nicoud, M.B.; Medina, V.A. Histamine Receptors and Cancer Pharmacology: An Update. Br. J. Pharmacol. 2020, 177, 516–538. [Google Scholar] [CrossRef] [Green Version]
- Nicoud, M.B.; Formoso, K.; Medina, V.A. Pathophysiological Role of Histamine H4 Receptor in Cancer: Therapeutic Implications. Front. Pharmacol. 2019, 10, 556. [Google Scholar] [CrossRef] [Green Version]
- Deiteren, A.; de Man, J.G.; Pelckmans, P.A.; de Winter, B.Y. Histamine H4 Receptors in the Gastrointestinal Tract. Br. J. Pharmacol. 2015, 172, 1165–1178. [Google Scholar] [CrossRef] [Green Version]
- Panula, P.; Chazot, P.L.; Cowart, M.; Gutzmer, R.; Leurs, R.; Liu, W.L.S.; Stark, H.; Thurmond, R.L.; Haas, H.L. International Union of Basic and Clinical Pharmacology. XCVIII. Histamine Receptors. Pharmacol. Rev. 2015, 67, 601–655. [Google Scholar] [CrossRef] [Green Version]
- Leurs, R.; Chazot, P.L.; Shenton, F.C.; Lim, H.D.; de Esch, I.J.P. Molecular and Biochemical Pharmacology of the Histamine H4 Receptor. Br. J. Pharmacol. 2009, 157, 14–23. [Google Scholar] [CrossRef] [PubMed]
- van Rijn, R.M.; Chazot, P.L.; Shenton, F.C.; Sansuk, K.; Bakker, R.A.; Leurs, R. Oligomerization of Recombinant and Endogenously Expressed Human Histamine H4 Receptors. Mol. Pharmacol. 2006, 70, 604–615. [Google Scholar] [CrossRef] [PubMed]
- van Rijn, R.M.; van Marle, A.; Chazot, P.L.; Langemeijer, E.; Qin, Y.; Shenton, F.C.; Lim, H.D.; Zuiderveld, O.P.; Sansuk, K.; Dy, M.; et al. Cloning and Characterization of Dominant Negative Splice Variants of the Human Histamine H4 Receptor. Biochem. J. 2008, 414, 121–131. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.K.; Hu, J.; Li, T.; Meng, J.R.; Ma, X.; Yin, S.J.; Zhao, C.H.; He, G.H.; Xu, G.L. Activation of Histamine H4 Receptors Decreases Epithelial-to-Mesenchymal Transition Progress by Inhibiting Transforming Growth Factor-Β1 Signalling Pathway in Non-Small Cell Lung Cancer. Eur. J. Cancer 2014, 50, 1195–1206. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.K.; Zhang, J.B.; Chen, J.H.; Meng, J.R.; Ma, X.; Zhang, J.; Zhou, Y.L.; Xu, G.L.; He, G.H. The HRH4 Rs11662595 Mutation Is Associated with Histamine H4 Receptor Dysfunction and with Increased Epithelial-to-Mesenchymal Transition Progress in Non-Small Cell Lung Cancer. Biochim. Biophys. Acta—Mol. Basis Dis. 2017, 1863, 2954–2963. [Google Scholar] [CrossRef]
- Massari, N.A.; Nicoud, M.B.; Sambuco, L.; Cricco, G.P.; Lamas, D.J.M.; Herrero Ducloux, M.V.; Blanco, H.; Rivera, E.S.; Medina, V.A. Histamine Therapeutic Efficacy in Metastatic Melanoma: Role of Histamine H4 Receptor Agonists and Opportunity for Combination with Radiation. Oncotarget 2017, 8, 26471–26491. [Google Scholar] [CrossRef] [Green Version]
- Martinel-Lamas, D.; Rivera, E.S.; Medina, V.A. Histamine H4 Receptor: Insights into a Potential Therapeutic Target in Breast Cancer. Front. Biosci.–Sch. 2015, 7, 1–9. [Google Scholar] [CrossRef]
- Martinel-Lamas, D.; Nicoud, M.; Sterle, H.; Carabajal, E.; Tesan, F.; Perazzo, J.; Cremaschi, G.; Rivera, E.; Medina, V. Selective Cytoprotective Effect of Histamine on Doxorubicin-Induced Hepatic and Cardiac Toxicity in Animal Models. Cell Death Discov. 2015, 1, 15059. [Google Scholar] [CrossRef] [Green Version]
- Martinel-Lamas, D.J.; Cortina, J.E.; Ventura, C.; Sterle, H.A.; Valli, E.; Balestrasse, K.B.; Blanco, H.; Cremaschi, G.A.; Rivera, E.S.; Medina, V.A. Enhancement of Ionizing Radiation Response by Histamine in Vitro and in Vivo in Human Breast Cancer. Cancer Biol. Ther. 2015, 16, 137–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, V.; Cricco, G.; Nuñez, M.; Martín, G.; Mohamad, N.; Correa-Fiz, F.; Sanchez-Jimenez, F.; Bergoc, R.; Rivera, E.S. Histamine-Mediated Signaling Processes in Human Malignant Mammary Cells. Cancer Biol. Ther. 2006, 5, 1462–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martner, A.; Thorén, F.B.; Aurelius, J.; Sderholm, J.; Brune, M.; Hellstrand, K. Immunotherapy with Histamine Dihydrochloride for the Prevention of Relapse in Acute Myeloid Leukemia. Expert Rev. Hematol. 2010, 3, 381–391. [Google Scholar] [CrossRef]
- Rydström, A.; Hallner, A.; Aurelius, J.; Sander, F.E.; Bernson, E.; Kiffin, R.; Thoren, F.B.; Hellstrand, K.; Martner, A. Dynamics of Myeloid Cell Populations during Relapse-Preventive Immunotherapy in Acute Myeloid Leukemia. J. Leukoc. Biol. 2017, 102, 467–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belcheva, A.; Mishkova, R. Histamine Content in Lymph Nodes from Patients with Malignant Lymphomas. Inflamm. Res. 1995, 44, s86–s87. [Google Scholar] [CrossRef]
- Yoo, H.Y.; Sung, M.K.; Lee, S.H.; Kim, S.; Lee, H.; Park, S.; Kim, S.C.; Lee, B.; Rho, K.; Lee, J.-E.; et al. A Recurrent Inactivating Mutation in RHOA GTPase in Angioimmunoblastic T Cell Lymphoma. Nat. Genet. 2014, 46, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Crescenzo, R.; Abate, F.; Lasorsa, E.; Tabbo’, F.; Gaudiano, M.; Chiesa, N.; Di Giacomo, F.; Spaccarotella, E.; Barbarossa, L.; Ercole, E.; et al. Convergent Mutations and Kinase Fusions Lead to Oncogenic STAT3 Activation in Anaplastic Large Cell Lymphoma. Cancer Cell 2015, 27, 516–532. [Google Scholar] [CrossRef] [Green Version]
- Finalet Ferreiro, J.; Rouhigharabaei, L.; Urbankova, H.; van der Krogt, J.-A.; Michaux, L.; Shetty, S.; Krenacs, L.; Tousseyn, T.; de Paepe, P.; Uyttebroeck, A.; et al. Integrative Genomic and Transcriptomic Analysis Identified Candidate Genes Implicated in the Pathogenesis of Hepatosplenic T-Cell Lymphoma. PLoS ONE 2014, 9, e102977. [Google Scholar] [CrossRef] [Green Version]
- Küçük, C.; Jiang, B.; Hu, X.; Zhang, W.; Chan, J.K.C.; Xiao, W.; Lack, N.; Alkan, C.; Williams, J.C.; Avery, K.N.; et al. Activating Mutations of STAT5B and STAT3 in Lymphomas Derived from Γδ-T or NK Cells. Nat. Commun. 2015, 6, 6025. [Google Scholar] [CrossRef] [Green Version]
- Querfeld, C.; Leung, S.; Myskowski, P.L.; Curran, S.A.; Goldman, D.A.; Heller, G.; Wu, X.; Kil, S.H.; Sharma, S.; Finn, K.J.; et al. Primary T Cells from Cutaneous T-cell Lymphoma Skin Explants Display an Exhausted Immune Checkpoint Profile. Cancer Immunolology Res. 2018, 6, 900–909. [Google Scholar] [CrossRef] [Green Version]
- Beermann, S.; Seifert, R.; Neumann, D. Commercially Available Antibodies against Human and Murine Histamine H4-Receptor Lack Specificity. Naunyn-Schmiedebergs Arch. Pharmacol. 2012, 385, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Thurmond, R.L. The Histamine H4 Receptor: From Orphan to the Clinic. Front. Pharmacol. 2015, 6, 65. [Google Scholar] [CrossRef]
- Hansen Selnø, A.T.; Sumbayev, V.V.; Raap, U.; Gibbs, B.F. Role of Histamine in Inflammatory Diseases. In Immunopharmacology and Inflammation; Springer Science & Business Media: New York, NY, USA, 2018; pp. 88–106. [Google Scholar]
- Cogé, F.; Guénin, S.P.; Rique, H.; Boutin, J.A.; Galizzi, J.P. Structure and Expression of the Human Histamine H4-Receptor Gene. Biochem. Biophys. Res. Commun. 2001, 284, 301–309. [Google Scholar] [CrossRef]
- Hodge, E.; Chang, W.Y.C.; Selby, K.A.; Hall, I.P.; Sayers, I. Effects of Atopy and Grass Pollen Season on Histamine H4 Receptor Expression in Human Leukocytes. Ann. Allergy Asthma Immunol. 2013, 111, 38–44. [Google Scholar] [CrossRef]
- Cianchi, F.; Cortesini, C.; Schiavone, N.; Perna, F.; Magnelli, L.; Fanti, E.; Bani, D.; Messerini, L.; Fabbroni, V.; Perigli, G.; et al. The Role of Cyclooxygenase-2 in Mediating the Effects of Histamine on Cell Proliferation and Vascular Endothelial Growth Factor Production in Colorectal Cancer. Clin. Cancer Res. 2005, 11, 6807–6815. [Google Scholar] [CrossRef] [Green Version]
- He, G.H.; Ding, J.Q.; Zhang, X.; Xu, W.M.; Lin, X.Q.; Huang, M.J.; Feng, J.; Wang, P.; Cai, W.K. Activation of Histamine H4 Receptor Suppresses the Proliferation and Invasion of Esophageal Squamous Cell Carcinoma via Both Metabolism and Non-Metabolism Signaling Pathways. J. Mol. Med. 2018, 96, 951–964. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Xiong, Y.; Li, J.; Yang, Y.; Liu, L.; Wang, W.; Wang, L.; Li, M.; Fang, Z. Deletion and Down-Regulation of HRH4 Gene in Gastric Carcinomas: A Potential Correlation with Tumor Progression. PLoS ONE 2012, 7, e31207. [Google Scholar] [CrossRef]
- Martinel-Lamas, D.J.; Croci, M.; Carabajal, E.; Crescenti, E.J.V.; Sambuco, L.; Massari, N.A.; Bergoc, R.M.; Rivera, E.S.; Medina, V.A. Therapeutic Potential of Histamine H4 Receptor Agonists in Triple-Negative Human Breast Cancer Experimental Model. Br. J. Pharmacol. 2013, 170, 188–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salem, A.; Salo, T. Nothing to Sneeze at: Histamine and Histamine Receptors in Oral Carcinogenesis. Oral Dis. 2021, 27, 1090–1096. [Google Scholar] [CrossRef]
- Martner, A.; Wiktorin, H.G.; Lenox, B.; Ewald Sander, F.; Aydin, E.; Aurelius, J.; Thorén, F.B.; Ståhlberg, A.; Hermodsson, S.; Hellstrand, K. Histamine Promotes the Development of Monocyte-Derived Dendritic Cells and Reduces Tumor Growth by Targeting the Myeloid NADPH Oxidase. J. Immunol. 2015, 194, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Grauers Wiktorin, H.; Nilsson, M.S.; Kiffin, R.; Sander, F.E.; Lenox, B.; Rydström, A.; Hellstrand, K.; Martner, A. Histamine Targets Myeloid-Derived Suppressor Cells and Improves the Anti-Tumor Efficacy of PD-1/PD-L1 Checkpoint Blockade. Cancer Immunol. Immunother. 2019, 68, 163–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paudel, S.; Mehtani, D.; Puri, N. Mast Cells May Differentially Regulate Growth of Lymphoid Neoplasms by Opposite Modulation of Histamine Receptors. Front. Oncol. 2019, 9, 1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellstrand, K.; Brune, M.; Naredi, P.; Mellqvist, U.H.; Hansson, M.; Gehlsen, K.R.; Hermodsson, S. Histamine: A Novel Approach to Cancer Immunotherapy. Cancer Investig. 2000, 18, 347–355. [Google Scholar] [CrossRef]
- Brune, M.; Castaigne, S.; Catalano, J.; Gehlsen, K.; Ho, A.D.; Hofmann, W.K.; Hogge, D.E.; Nilsson, B.; Or, R.; Romero, A.I.; et al. Improved Leukemia-Free Survival after Postconsolidation Immunotherapy with Histamine Dihydrochloride and Interleukin-2 in Acute Myeloid Leukemia: Results of a Randomized Phase 3 Trial. Blood 2006, 108, 88–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, S.M.; Broglio, K.R.; Berry, D.A. Addressing the Incremental Benefit of Histamine Dihydrochloride When Added to Interleukin-2 in Treating Acute Myeloid Leukemia: A Bayesian Meta-Analysis. Cancer Investig. 2011, 29, 293–299. [Google Scholar] [CrossRef]
- Buyse, M.; Squifflet, P.; Lange, B.J.; Alonzo, T.A.; Larson, R.A.; Kolitz, J.E.; George, S.L.; Bloomfield, C.D.; Castaigne, S.; Chevret, S.; et al. Individual Patient Data Meta-Analysis of Randomized Trials Evaluating IL-2 Monotherapy as Remission Maintenance Therapy in Acute Myeloid Leukemia. Blood 2011, 117, 7007–7013. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.P.H.; Perry, C.M. Histamine Dihydrochloride: In the Management of Acute Myeloid Leukaemia. Drugs 2011, 71, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Nicoud, M.B.; Táquez Delgado, M.A.; de la Paz Sarasola, M.; Vidal, A.; Speisky, D.; Cremaschi, G.A.; Sterle, H.A.; Medina, V.A. Impact of Histamine H4 Receptor Deficiency on the Modulation of T Cells in a Murine Breast Cancer Model. Cancer Immunol. Immunother. 2021, 70, 233–244. [Google Scholar] [CrossRef]
- Li, J.; Huang, X.; Wang, Q.; Jing, S.; Jiang, H.; Wei, Z.; Zang, Y.; Liu, Y.; Zhao, L.; Fang, Y.; et al. Pharmacokinetic Properties and Safety Profile of Histamine Dihydrochloride Injection in Chinese Healthy Volunteers: A Phase I, Single-Center, Open-Label, Randomized Study. Clin. Ther. 2015, 37, 2352–2364. [Google Scholar] [CrossRef]
- Sarasola, M.D.L.P.; Táquez Delgado, M.A.; Nicoud, M.B.; Medina, V.A. Histamine in Cancer Immunology and Immunotherapy. Current Status and New Perspectives. Pharmacol. Res. Perspect. 2021, 9, 778. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug Repurposing: Progress, Challenges and Recommendations. Nat. Rev. Drug Discov. 2018, 18, 41–58. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, S.; Chen, J.; Yu, Z. Histone Deacetylases (HDACs) Guided Novel Therapies for T-Cell Lymphomas. Int. J. Med. Sci. 2019, 16, 424–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, M.; Kijima, M.; Akita, M.; Beppu, T. Potent and Specific Inhibition of Mammalian Histone Deacetylase Both in Vivo and in Vitro by Trichostatin A. J. Biol. Chem. 1990, 265, 1788–1789. [Google Scholar] [CrossRef]
- Yoon, S.; Eom, G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med. J. 2016, 52, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA Approval Summary: Vorinostat for Treatment of Advanced Primary Cutaneous T-Cell Lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, S.J.; Demierre, M.F.; Kim, E.J.; Rook, A.H.; Lerner, A.; Duvic, M.; Scarisbrick, J.; Reddy, S.; Robak, T.; Becker, J.C.; et al. Final Results from a Multicenter, International, Pivotal Study of Romidepsin in Refractory Cutaneous T-Cell Lymphoma. J. Clin. Oncol. 2010, 28, 4485–4491. [Google Scholar] [CrossRef]
- Coiffier, B.; Pro, B.; Prince, H.M.; Foss, F.; Sokol, L.; Greenwood, M.; Caballero, D.; Borchmann, P.; Morschhauser, F.; Wilhelm, M.; et al. Results from a Pivotal, Open-Label, Phase II Study of Romidepsin in Relapsed or Refractory Peripheral T-Cell Lymphoma after Prior Systemic Therapy. J. Clin. Oncol. 2012, 30, 631–636. [Google Scholar] [CrossRef]
- Tate, C.R.; Rhodes, L.V.; Segar, H.C.; Driver, J.L.; Pounder, F.N.; Burow, M.E.; Collins-Burow, B.M. Targeting Triple-Negative Breast Cancer Cells with the Histone Deacetylase Inhibitor Panobinostat. Breast Cancer Res. 2012, 14, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clauzure, M.; Valdivieso, A.G.; Massip Copiz, M.M.; Schulman, G.; Teiber, M.L.; Santa-Coloma, T.A. Disruption of Interleukin-1β Autocrine Signaling Rescues Complex I Activity and Improves ROS Levels in Immortalized Epithelial Cells with Impaired Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Function. PLoS ONE 2014, 9, e99257. [Google Scholar] [CrossRef] [PubMed]
- Clauzure, M.; Valdivieso, Á.G.; Dugour, A.V.; Mori, C.; Massip-Copiz, M.M.; Aguilar, M.; Sotomayor, V.; Asensio, C.J.A.; Figueroa, J.M.; Santa-Coloma, T.A. NLR Family Pyrin Domain Containing 3 (NLRP3) and Caspase 1 (CASP1) Modulation by Intracellular Cl– Concentration. Immunology 2021, 163, 493–511. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clauzure, M.; Táquez Delgado, M.A.; Phillip, J.M.; Revuelta, M.V.; Cerchietti, L.; Medina, V.A. Histamine H4 Receptor Agonism Induces Antitumor Effects in Human T-Cell Lymphoma. Int. J. Mol. Sci. 2022, 23, 1378. https://doi.org/10.3390/ijms23031378
Clauzure M, Táquez Delgado MA, Phillip JM, Revuelta MV, Cerchietti L, Medina VA. Histamine H4 Receptor Agonism Induces Antitumor Effects in Human T-Cell Lymphoma. International Journal of Molecular Sciences. 2022; 23(3):1378. https://doi.org/10.3390/ijms23031378
Chicago/Turabian StyleClauzure, Mariángeles, Mónica A. Táquez Delgado, Jude M. Phillip, Maria V. Revuelta, Leandro Cerchietti, and Vanina A. Medina. 2022. "Histamine H4 Receptor Agonism Induces Antitumor Effects in Human T-Cell Lymphoma" International Journal of Molecular Sciences 23, no. 3: 1378. https://doi.org/10.3390/ijms23031378
APA StyleClauzure, M., Táquez Delgado, M. A., Phillip, J. M., Revuelta, M. V., Cerchietti, L., & Medina, V. A. (2022). Histamine H4 Receptor Agonism Induces Antitumor Effects in Human T-Cell Lymphoma. International Journal of Molecular Sciences, 23(3), 1378. https://doi.org/10.3390/ijms23031378