
SEC23B Loss-of-Function Suppresses Hepcidin Expression by Impairing Glycosylation Pathway in Human Hepatic Cells


 ,
, 
Abstract
1. Introduction
2. Results
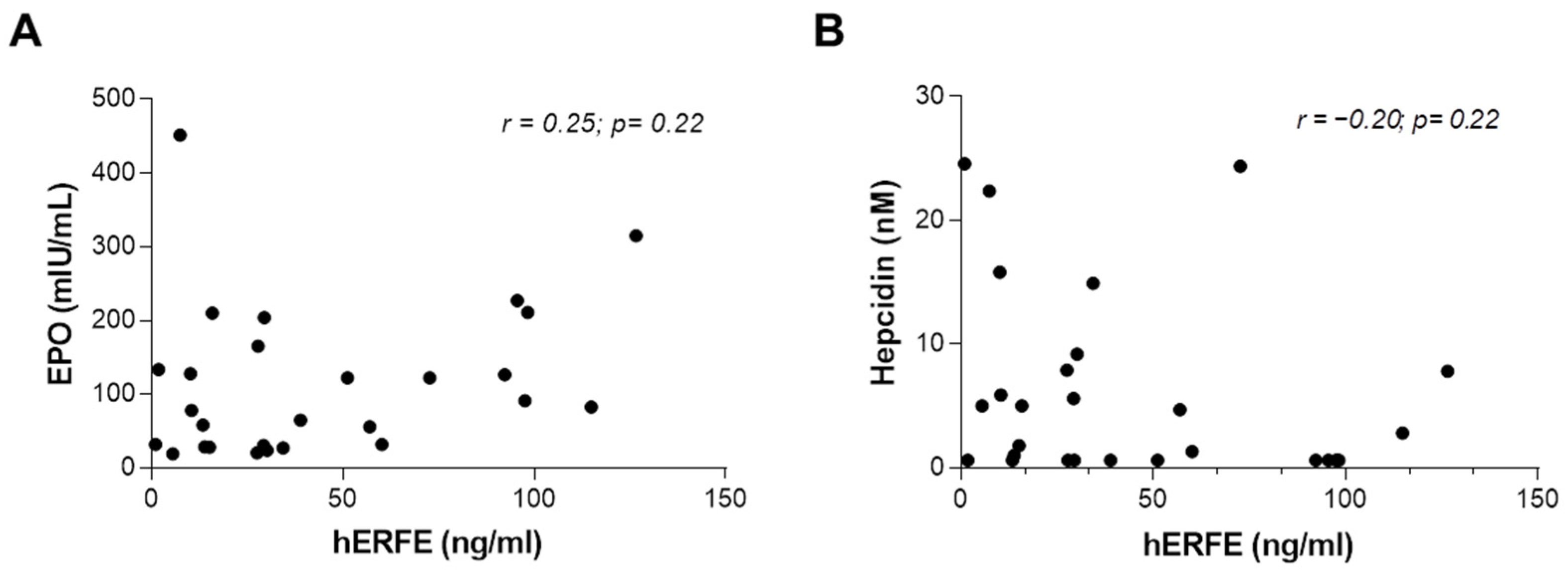
2.1. Analysis of Iron Balance in Patients with CDA II
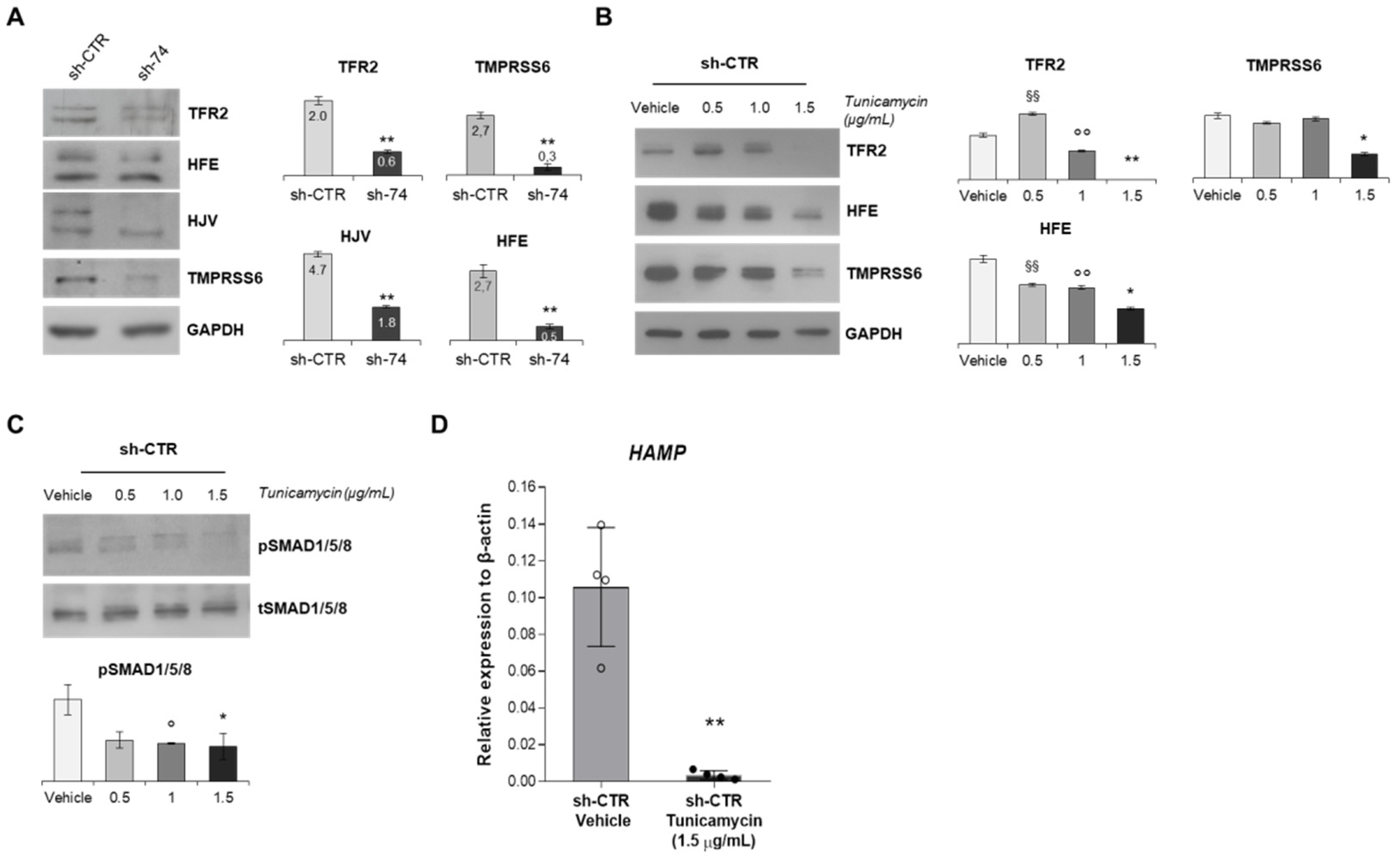
2.2. Assessment of Iron-Related Genes in HuH7 Cells Stably Silenced for SEC23B
2.3. SEC23B Loss-of-Function Alters BMP/SMADs Pathway and Fails to Activate the Signaling in Response to BMP6
2.4. Inhibition of N-Glycosylation Accounts for Reduced Activation of BMP/SMADs Pathway
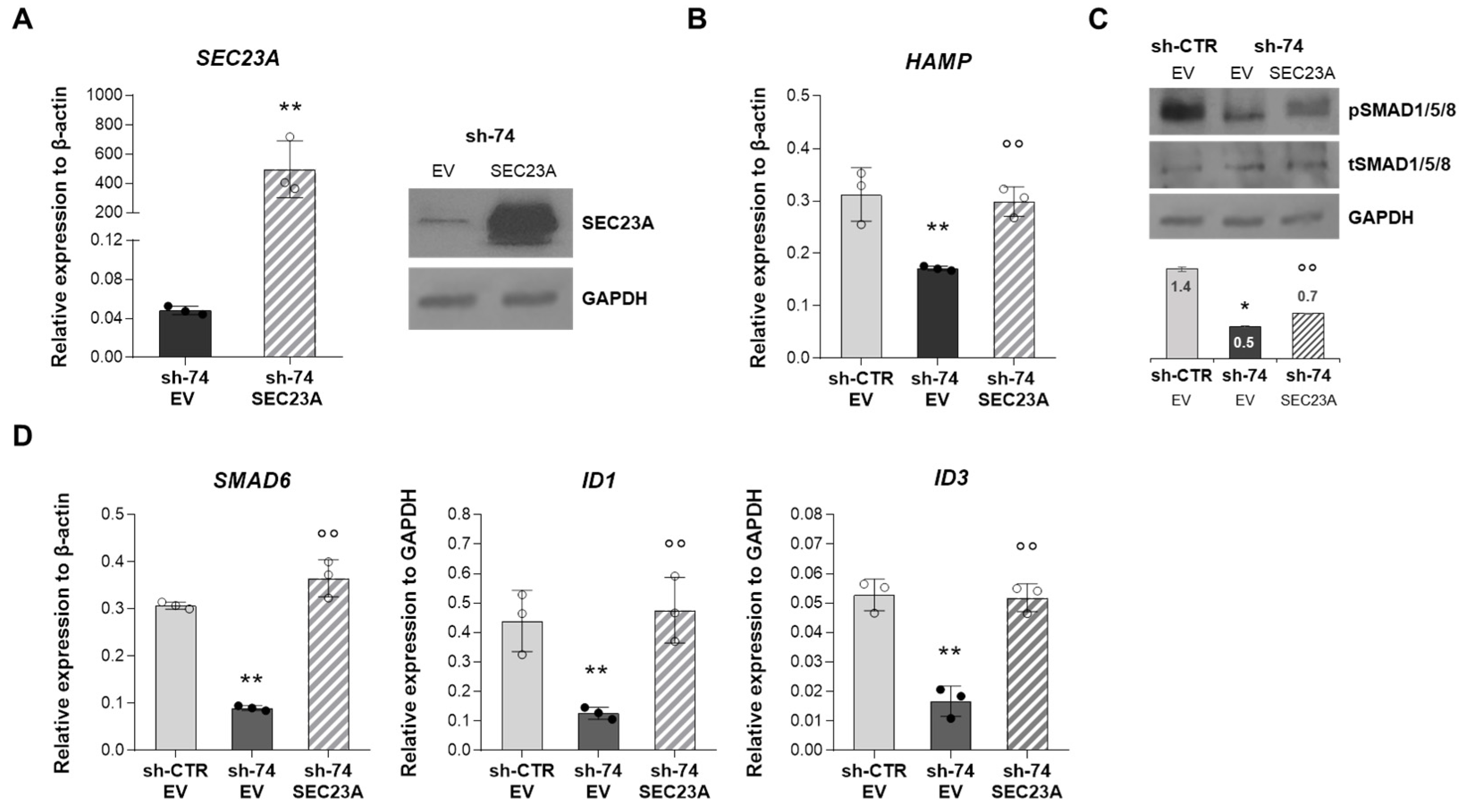
2.5. SEC23A Over-Expression Rescues the Impaired BMP/SMADs Signaling
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Hepcidin, ERFE, and Erythropoietin Levels in Plasma Samples
4.3. Production of Lentiviral Particles and Infection of the HuH7 and HepG2 Cell Line
4.4. Culture of Cell Lines and Drugs Treatments
4.5. Gene Expression Analysis
4.6. Protein Expression Analysis
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gambale, A.; Iolascon, A.; Andolfo, I.; Russo, R. Diagnosis and management of congenital dyserythropoietic anemias. Expert Rev. Hematol. 2016, 9, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Heimpel, H. Congenital dyserythropoietic anemias: Epidemiology, clinical significance, and progress in understanding their pathogenesis. Ann. Hematol. 2004, 83, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Iolascon, A.; Esposito, M.R.; Russo, R. Clinical aspects and pathogenesis of congenital dyserythropoietic anemias: From morphology to molecular approach. Haematologica 2012, 97, 1786–1794. [Google Scholar] [CrossRef] [PubMed]
- Heimpel, H.; Kellermann, K.; Neuschwander, N.; Hogel, J.; Schwarz, K. The morphological diagnosis of congenital dyserythropoietic anemia: Results of a quantitative analysis of peripheral blood and bone marrow cells. Haematologica 2010, 95, 1034–1036. [Google Scholar] [CrossRef] [PubMed]
- De Franceschi, L.; Turrini, F.; del Giudice, E.M.; Perrotta, S.; Olivieri, O.; Corrocher, R.; Mannu, F.; Iolascon, A. Decreased band 3 anion transport activity and band 3 clusterization in congenital dyserythropoietic anemia type II. Exp. Hematol. 1998, 26, 869–873. [Google Scholar] [PubMed]
- Mawby, W.J.; Tanner, M.J.; Anstee, D.J.; Clamp, J.R. Incomplete glycosylation of erythrocyte membrane proteins in congenital dyserythropoietic anaemia type II (CDA II). Br. J. Haematol. 1983, 55, 357–368. [Google Scholar] [CrossRef]
- Iolascon, A.; Delaunay, J. Close to unraveling the secrets of congenital dyserythropoietic anemia types I and II. Haematologica 2009, 94, 599–602. [Google Scholar] [CrossRef]
- Zdebska, E.; Iolascon, A.; Spychalska, J.; Perrotta, S.; Lanzara, C.; Smolenska-Sym, G.; Koscielak, J. Abnormalities of erythrocyte glycoconjugates are identical in two families with congenital dyserythropoietic anemia type II with different chromosomal localizations of the disease gene. Haematologica 2007, 92, 427–428. [Google Scholar] [CrossRef][Green Version]
- Bianchi, P.; Fermo, E.; Vercellati, C.; Boschetti, C.; Barcellini, W.; Iurlo, A.; Marcello, A.P.; Righetti, P.G.; Zanella, A. Congenital dyserythropoietic anemia type II (CDA II) is caused by mutations in the SEC23B gene. Hum. Mutat. 2009, 30, 1292–1298. [Google Scholar] [CrossRef]
- Russo, R.; Esposito, M.R.; Iolascon, A. Inherited hematological disorders due to defects in coat protein (COP)II complex. Am. J. Hematol. 2013, 88, 135–140. [Google Scholar] [CrossRef]
- Schwarz, K.; Iolascon, A.; Verissimo, F.; Trede, N.S.; Horsley, W.; Chen, W.; Paw, B.H.; Hopfner, K.P.; Holzmann, K.; Russo, R.; et al. Mutations affecting the secretory COPII coat component SEC23B cause congenital dyserythropoietic anemia type II. Nat. Genet. 2009, 41, 936–940. [Google Scholar] [CrossRef] [PubMed]
- Fromme, J.C.; Orci, L.; Schekman, R. Coordination of COPII vesicle trafficking by Sec23. Trends Cell Biol. 2008, 18, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Unal, S.; Russo, R.; Gumruk, F.; Kuskonmaz, B.; Cetin, M.; Sayli, T.; Tavil, B.; Langella, C.; Iolascon, A.; Uckan Cetinkaya, D. Successful hematopoietic stem cell transplantation in a patient with congenital dyserythropoietic anemia type II. Pediatr. Transplant. 2014, 18, E130–E133. [Google Scholar] [CrossRef] [PubMed]
- Uygun, V.; Russo, R.; Karasu, G.; Daloglu, H.; Iolascon, A.; Yesilipek, A. Hematopoietic Stem Cell Transplantation in Congenital Dyserythropetic Anemia Type II: A Case Report and Review of the Literature. J. Pediatr. Hematol. Oncol. 2019, 42, e507–e510. [Google Scholar] [CrossRef] [PubMed]
- Russo, R.; Gambale, A.; Langella, C.; Andolfo, I.; Unal, S.; Iolascon, A. Retrospective cohort study of 205 cases with congenital dyserythropoietic anemia type II: Definition of clinical and molecular spectrum and identification of new diagnostic scores. Am. J. Hematol. 2014, 89, E169–E175. [Google Scholar] [CrossRef] [PubMed]
- Kautz, L.; Jung, G.; Nemeth, E.; Ganz, T. Erythroferrone contributes to recovery from anemia of inflammation. Blood 2014, 124, 2569–2574. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Erythropoietic regulators of iron metabolism. Free Radic. Biol. Med. 2019, 133, 69–74. [Google Scholar] [CrossRef]
- Andolfo, I.; Rosato, B.E.; Marra, R.; De Rosa, G.; Manna, F.; Gambale, A.; Iolascon, A.; Russo, R. The BMP-SMAD pathway mediates the impaired hepatic iron metabolism associated with the ERFE-A260S variant. Am. J. Hematol. 2019, 94, 1227–1235. [Google Scholar] [CrossRef]
- Russo, R.; Andolfo, I.; Manna, F.; De Rosa, G.; De Falco, L.; Gambale, A.; Bruno, M.; Matte, A.; Ricchi, P.; Girelli, D.; et al. Increased levels of ERFE-encoding FAM132B in patients with congenital dyserythropoietic anemia type II. Blood 2016, 128, 1899–1902. [Google Scholar] [CrossRef]
- D’Alessio, F.; Hentze, M.W.; Muckenthaler, M.U. The hemochromatosis proteins HFE, TfR2, and HJV form a membrane-associated protein complex for hepcidin regulation. J. Hepatol. 2012, 57, 1052–1060. [Google Scholar] [CrossRef]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, L. The Iron Tale: If It Does Not Kill You, It Makes You Stronger (and Hepcidin Helps). Hemasphere 2018, 2, e27. [Google Scholar] [CrossRef]
- Silvestri, L.; Nai, A.; Dulja, A.; Pagani, A. Hepcidin and the BMP-SMAD pathway: An unexpected liaison. Vitam. Horm. 2019, 110, 71–99. [Google Scholar] [CrossRef] [PubMed]
- Iolascon, A.; Andolfo, I.; Russo, R. Congenital dyserythropoietic anemias. Blood 2020, 136, 1274–1283. [Google Scholar] [CrossRef] [PubMed]
- Andolfo, I.; Martone, S.; Rosato, B.E.; Marra, R.; Gambale, A.; Forni, G.L.; Pinto, V.; Goransson, M.; Papadopoulou, V.; Gavillet, M.; et al. Complex Modes of Inheritance in Hereditary Red Blood Cell Disorders: A Case Series Study of 155 Patients. Genes 2021, 12, 958. [Google Scholar] [CrossRef]
- Andolfo, I.; Russo, R.; Rosato, B.E.; Manna, F.; Gambale, A.; Brugnara, C.; Iolascon, A. Genotype-phenotype correlation and risk stratification in a cohort of 123 hereditary stomatocytosis patients. Am. J. Hematol. 2018, 93, 1509–1517. [Google Scholar] [CrossRef]
- Iolascon, A.; Russo, R.; Esposito, M.R.; Asci, R.; Piscopo, C.; Perrotta, S.; Feneant-Thibault, M.; Garcon, L.; Delaunay, J. Molecular analysis of 42 patients with congenital dyserythropoietic anemia type II: New mutations in the SEC23B gene and a search for a genotype-phenotype relationship. Haematologica 2010, 95, 708–715. [Google Scholar] [CrossRef]
- Russo, R.; Langella, C.; Esposito, M.R.; Gambale, A.; Vitiello, F.; Vallefuoco, F.; Ek, T.; Yang, E.; Iolascon, A. Hypomorphic mutations of SEC23B gene account for mild phenotypes of congenital dyserythropoietic anemia type II. Blood Cells Mol. Dis. 2013, 51, 17–21. [Google Scholar] [CrossRef]
- Wang, S.; Chen, C.; Yu, L.; Mueller, J.; Rausch, V.; Mueller, S. Bone morphogenetic protein 6-mediated crosstalk between endothelial cells and hepatocytes recapitulates the iron-sensing pathway in vitro. J. Biol. Chem. 2021, 297, 101378. [Google Scholar] [CrossRef]
- Ganz, T.; Nemeth, E. Iron metabolism: Interactions with normal and disordered erythropoiesis. Cold Spring Harb. Perspect. Med. 2012, 2, a011668. [Google Scholar] [CrossRef]
- Adamsky, K.; Weizer, O.; Amariglio, N.; Breda, L.; Harmelin, A.; Rivella, S.; Rachmilewitz, E.; Rechavi, G. Decreased hepcidin mRNA expression in thalassemic mice. Br. J. Haematol. 2004, 124, 123–124. [Google Scholar] [CrossRef] [PubMed]
- Papanikolaou, G.; Tzilianos, M.; Christakis, J.I.; Bogdanos, D.; Tsimirika, K.; MacFarlane, J.; Goldberg, Y.P.; Sakellaropoulos, N.; Ganz, T.; Nemeth, E. Hepcidin in iron overload disorders. Blood 2005, 105, 4103–4105. [Google Scholar] [CrossRef] [PubMed]
- Arezes, J.; Foy, N.; McHugh, K.; Sawant, A.; Quinkert, D.; Terraube, V.; Brinth, A.; Tam, M.; LaVallie, E.R.; Taylor, S.; et al. Erythroferrone inhibits the induction of hepcidin by BMP6. Blood 2018, 132, 1473–1477. [Google Scholar] [CrossRef] [PubMed]
- Coffey, R.; Ganz, T. Erythroferrone: An Erythroid Regulator of Hepcidin and Iron Metabolism. Hemasphere 2018, 2, e35. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Core, A.B.; Canali, S.; Zumbrennen-Bullough, K.B.; Ozer, S.; Umans, L.; Zwijsen, A.; Babitt, J.L. Smad1/5 is required for erythropoietin-mediated suppression of hepcidin in mice. Blood 2017, 130, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Andolfo, I.; Rosato, B.E.; Manna, F.; De Rosa, G.; Marra, R.; Gambale, A.; Girelli, D.; Russo, R.; Iolascon, A. Gain-of-function mutations in PIEZO1 directly impair hepatic iron metabolism via the inhibition of the BMP/SMADs pathway. Am. J. Hematol. 2020, 95, 188–197. [Google Scholar] [CrossRef]
- Casanovas, G.; Mleczko-Sanecka, K.; Altamura, S.; Hentze, M.W.; Muckenthaler, M.U. Bone morphogenetic protein (BMP)-responsive elements located in the proximal and distal hepcidin promoter are critical for its response to HJV/BMP/SMAD. J. Mol. Med. 2009, 87, 471–480. [Google Scholar] [CrossRef]
- Verga Falzacappa, M.V.; Casanovas, G.; Hentze, M.W.; Muckenthaler, M.U. A bone morphogenetic protein (BMP)-responsive element in the hepcidin promoter controls HFE2-mediated hepatic hepcidin expression and its response to IL-6 in cultured cells. J. Mol. Med. 2008, 86, 531–540. [Google Scholar] [CrossRef]
- Gao, J.; Chen, J.; Kramer, M.; Tsukamoto, H.; Zhang, A.S.; Enns, C.A. Interaction of the hereditary hemochromatosis protein HFE with transferrin receptor 2 is required for transferrin-induced hepcidin expression. Cell Metab. 2009, 9, 217–227. [Google Scholar] [CrossRef]
- Schmidt, P.J.; Andrews, N.C.; Fleming, M.D. Hepcidin induction by transgenic overexpression of Hfe does not require the Hfe cytoplasmic tail, but does require hemojuvelin. Blood 2010, 116, 5679–5687. [Google Scholar] [CrossRef]
- Wallace, D.F.; Summerville, L.; Crampton, E.M.; Frazer, D.M.; Anderson, G.J.; Subramaniam, V.N. Combined deletion of Hfe and transferrin receptor 2 in mice leads to marked dysregulation of hepcidin and iron overload. Hepatology 2009, 50, 1992–2000. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, L.; Guillem, F.; Pagani, A.; Nai, A.; Oudin, C.; Silva, M.; Toutain, F.; Kannengiesser, C.; Beaumont, C.; Camaschella, C.; et al. Molecular mechanisms of the defective hepcidin inhibition in TMPRSS6 mutations associated with iron-refractory iron deficiency anemia. Blood 2009, 113, 5605–5608. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, L.; Pagani, A.; Nai, A.; De Domenico, I.; Kaplan, J.; Camaschella, C. The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metab. 2008, 8, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, L.; Murphy, C.; O’Driscoll, L.S.; Carmo-Fonseca, M.; McCaffrey, M.W.; Fleming, J.V. N-glycosylation is important for the correct intracellular localization of HFE and its ability to decrease cell surface transferrin binding. FEBS J. 2010, 277, 3219–3234. [Google Scholar] [CrossRef]
- Jiang, J.; Yang, J.; Feng, P.; Zuo, B.; Dong, N.; Wu, Q.; He, Y. N-glycosylation is required for matriptase-2 autoactivation and ectodomain shedding. J. Biol. Chem. 2014, 289, 19500–19507. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Enns, C.A. N-linked glycosylation is required for transferrin-induced stabilization of transferrin receptor 2, but not for transferrin binding or trafficking to the cell surface. Biochemistry 2013, 52, 3310–3319. [Google Scholar] [CrossRef] [PubMed]
- Salzberger, W.; Garcia-Beltran, W.F.; Dugan, H.; Gubbala, S.; Simoneau, C.; Gressens, S.B.; Jost, S.; Altfeld, M. Influence of Glycosylation Inhibition on the Binding of KIR3DL1 to HLA-B*57:01. PLoS ONE 2015, 10, e0145324. [Google Scholar] [CrossRef] [PubMed]
- King, R.; Lin, Z.; Balbin-Cuesta, G.; Myers, G.; Friedman, A.; Zhu, G.; McGee, B.; Saunders, T.L.; Kurita, R.; Nakamura, Y.; et al. SEC23A rescues SEC23B-deficient congenital dyserythropoietic anemia type II. Sci. Adv. 2021, 7, eabj5293. [Google Scholar] [CrossRef]
- Pellegrin, S.; Haydn-Smith, K.L.; Hampton-O’Neil, L.A.; Hawley, B.R.; Heesom, K.J.; Fermo, E.; Bianchi, P.; Toye, A.M. Transduction with BBF2H7/CREB3L2 upregulates SEC23A protein in erythroblasts and partially corrects the hypo-glycosylation phenotype associated with CDA II. Br. J. Haematol. 2019, 184, 876–881. [Google Scholar] [CrossRef]
- De Rosa, G.; Andolfo, I.; Marra, R.; Manna, F.; Rosato, B.E.; Iolascon, A.; Russo, R. RAP-011 Rescues the Disease Phenotype in a Cellular Model of Congenital Dyserythropoietic Anemia Type II by Inhibiting the SMAD2-3 Pathway. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Russo, R.; Marra, R.; Andolfo, I.; De Rosa, G.; Rosato, B.E.; Manna, F.; Gambale, A.; Raia, M.; Unal, S.; Barella, S.; et al. Characterization of Two Cases of Congenital Dyserythropoietic Anemia Type I Shed Light on the Uncharacterized C15orf41 Protein. Front Physiol 2019, 10, 621. [Google Scholar] [CrossRef] [PubMed]
- Lunde, N.N.; Haugen, M.H.; Bodin Larsen, K.B.; Damgaard, I.; Pettersen, S.J.; Kasem, R.; Rut, W.; Drag, M.; Poreba, M.; Johansen, H.T.; et al. Glycosylation is important for legumain localization and processing to active forms but not for cystatin E/M inhibitory functions. Biochimie 2017, 139, 27–37. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analysis | Units | Chronic Anemia | p-Value † | Reference | |
|---|---|---|---|---|---|
| Mild | Moderate/Severe | Range | |||
| Hb ≥ 10.0 g/dL | Hb < 10.0 g/dL | ||||
| N | 10 | 18 | - | - | |
| Gender | male/female | 3 (0.3)/7 (0.7) | 10 (0.6)/8 (0.4) | 0.19 | - |
| Age at sampling | years | 28.6 ± 4.3 | 17.0 ± 5.7 | 0.19 | - |
| Hemoglobin | g/dL | 10.9 ± 0.2 | 8.6 ± 0.3 | 0.00001 | 11.5–15.5 |
| ARC | ×103/µL | 51.5 ± 6.0 | 77.7 ± 14.7 | 0.21 | 20–90 |
| TSAT | % | 90.5 ± 6.5 | 76.7 ± 4.6 | 0.09 | 15–39 |
| hERFE | ng/mL | 40.5 ± 11.9 | 43.2 ± 9.3 | 0.86 | 0.1–3.8 |
| EPO | mIU/mL | 51.6 ± 11.1 | 151.1 ± 27.4 | 0.01 | 3.1–14.9 |
| sTfR | mg/L | 3.8 ± 0.5 | 4.2 ± 0.5 | 0.59 | 0.78–1.89 |
| Hepcidin | nM | 5.6 ± 2.3 | 6.2 ± 1.9 | 0.85 | male: 40.10 |
| female: 23.27 | |||||
| Hepcidin/ferritin | - | 0.02 ± 0.01 | 0.03 ± 0.01 | 0.46 | - |
| Ferritin | ng/mL | 559.7 ± 234.4 | 369.2 ± 115.2 | 0.42 | 22.0–275.0 |
| Ferritin/age § | - | 20.5 ± 6.9 | 57.9 ± 19.9 | 0.21 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosato, B.E.; Marra, R.; D’Onofrio, V.; Del Giudice, F.; Della Monica, S.; Iolascon, A.; Andolfo, I.; Russo, R. SEC23B Loss-of-Function Suppresses Hepcidin Expression by Impairing Glycosylation Pathway in Human Hepatic Cells. Int. J. Mol. Sci. 2022, 23, 1304. https://doi.org/10.3390/ijms23031304
Rosato BE, Marra R, D’Onofrio V, Del Giudice F, Della Monica S, Iolascon A, Andolfo I, Russo R. SEC23B Loss-of-Function Suppresses Hepcidin Expression by Impairing Glycosylation Pathway in Human Hepatic Cells. International Journal of Molecular Sciences. 2022; 23(3):1304. https://doi.org/10.3390/ijms23031304
Chicago/Turabian StyleRosato, Barbara Eleni, Roberta Marra, Vanessa D’Onofrio, Federica Del Giudice, Simone Della Monica, Achille Iolascon, Immacolata Andolfo, and Roberta Russo. 2022. "SEC23B Loss-of-Function Suppresses Hepcidin Expression by Impairing Glycosylation Pathway in Human Hepatic Cells" International Journal of Molecular Sciences 23, no. 3: 1304. https://doi.org/10.3390/ijms23031304
APA StyleRosato, B. E., Marra, R., D’Onofrio, V., Del Giudice, F., Della Monica, S., Iolascon, A., Andolfo, I., & Russo, R. (2022). SEC23B Loss-of-Function Suppresses Hepcidin Expression by Impairing Glycosylation Pathway in Human Hepatic Cells. International Journal of Molecular Sciences, 23(3), 1304. https://doi.org/10.3390/ijms23031304