
Revisiting miRNA Association with Melanoma Recurrence and Metastasis from a Machine Learning Point of View

 ,
,  , ,
, , 
Abstract
1. Introduction
2. Datasets and miRNA Signatures from CM Patients
3. Transcriptomics and Gene Signatures from CM Patients
4. AI-Based miRNA Signatures for Prediction of Melanoma Recurrence and Metastasis
4.1. Specific miRNAs Expression Patterns Regulate Melanoma Related Genes
4.2. Contribution of AI in the Prediction of Melanoma Recurrence and Metastasis
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Keung, E.Z.; Gershenwald, J.E. The eighth edition American Joint Committee on Cancer (AJCC) melanoma staging system: Implications for melanoma treatment and care. Expert Rev. Anticancer Ther. 2018, 18, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Rebecca, V.W.; Somasundaram, R.; Herlyn, M. Pre-clinical modeling of cutaneous melanoma. Nat. Commun. 2020, 11, 2858. [Google Scholar] [CrossRef] [PubMed]
- Dimitriou, F.; Krattinger, R.; Ramelyte, E.; Barysch, M.J.; Micaletto, S.; Dummer, R.; Goldinger, S.M. The World of Melanoma: Epidemiologic, Genetic, and Anatomic Differences of Melanoma Across the Globe. Curr. Oncol. Rep. 2018, 20, 87. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.M.; Vétizou, M.; Daillère, R.; Roberti, M.P.; Yamazaki, T.; Routy, B.; Lepage, P.; Boneca, I.G.; Chamaillard, M.; Kroemer, G.; et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and—Extrinsic Factors. Immunity 2016, 44, 1255–1269. [Google Scholar] [CrossRef]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed]
- Lauss, M.; Nsengimana, J.; Staaf, J.; Newton-Bishop, J.; Jönsson, G. Consensus of Melanoma Gene Expression Subtypes Converges on Biological Entities. J. Investig. Dermatol. 2016, 136, 2502–2505. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, S.; Kaur, H.; Dhall, A.; Raghava, G.P.S. Prediction and Analysis of Skin Cancer Progression using Genomics Profiles of Patients. Sci. Rep. 2019, 9, 15790. [Google Scholar] [CrossRef]
- Marie, K.L.; Sassano, A.; Yang, H.H.; Michalowski, A.M.; Michael, H.T.; Guo, T.; Tsai, Y.C.; Weissman, A.M.; Lee, M.P.; Jenkins, L.M.; et al. Melanoblast transcriptome analysis reveals pathways promoting melanoma metastasis. Nat. Commun. 2020, 11, 333. [Google Scholar] [CrossRef]
- Varrone, F.; Caputo, E. The miRNAs Role in Melanoma and in Its Resistance to Therapy. Int. J. Mol. Sci. 2020, 21, 878. [Google Scholar] [CrossRef]
- Jiang, Y.; Shi, X.; Zhao, Q.; Krauthammer, M.; Rothberg, B.E.G.; Ma, S. Integrated analysis of multidimensional omics data on cutaneous melanoma prognosis. Genomics 2016, 107, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Cheng, P.; Jiang, J.; Ren, Y.; Wu, D.; Xue, D. Epigenomic and genomic analysis of transcriptome modulation in skin cutaneous melanoma. Aging 2020, 12, 12703–12725. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Huang, B.; Zhao, X.-Y.; Shen, G.-L. Data mining of immune-related prognostic genes in metastatic melanoma microenvironment. Biosci. Rep. 2020, 40, BSR20201704. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; He, X.; Zhang, X.; Zhang, X.; Wei, Y.; Wu, B.; Li, W.; Li, J.; Xiao, Y. Predicting the clinical outcome of melanoma using an immune-related gene pairs signature. PLoS ONE 2020, 15, e0240331. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Schaafsma, E.; Gorlov, I.P.; Hernando, E.; Thomas, N.E.; Shen, R.; Turk, M.J.; Berwick, M.; Amos, C.I.; Cheng, C. A Leukocyte Infiltration Score Defined by a Gene Signature Predicts Melanoma Patient Prognosis. Mol. Cancer Res. 2019, 17, 109–119. [Google Scholar] [CrossRef]
- Zeng, Y.; Zeng, Y.; Yin, H.; Chen, F.; Wang, Q.; Yu, X.; Zhou, Y. Exploration of the immune cell infiltration-related gene signature in the prognosis of melanoma. Aging 2021, 13, 3459–3482. [Google Scholar] [CrossRef]
- Sheng, Y.; Tong, L.; Geyu, L. An immune risk score with potential implications in prognosis and immunotherapy of metastatic melanoma. Int. Immunopharmacol. 2020, 88, 106921. [Google Scholar] [CrossRef]
- Emran, A.A.; Nsengimana, J.; Punnia-Moorthy, G.; Schmitz, U.; Gallagher, S.J.; Newton-Bishop, J.; Tiffen, J.C.; Hersey, P. Study of the Female Sex Survival Advantage in Melanoma—A Focus on X-Linked Epigenetic Regulators and Immune Responses in Two Cohorts. Cancers 2020, 12, 2082. [Google Scholar] [CrossRef]
- Alkallas, R.; Lajoie, M.; Moldoveanu, D.; Hoang, K.V.; Lefrançois, P.; Lingrand, M.; Ahanfeshar-Adams, M.; Watters, K.; Spatz, A.; Zippin, J.H.; et al. Multi-omic analysis reveals significantly mutated genes and DDX3X as a sex-specific tumor suppressor in cutaneous melanoma. Nat. Cancer 2020, 1, 635–652. [Google Scholar] [CrossRef]
- Corthésy, J.; Theofilatos, K.; Mavroudi, S.; Macron, C.; Cominetti, O.; Remlawi, M.; Ferraro, F.; Núñez Galindo, A.; Kussmann, M.; Likothanassis, S.; et al. An Adaptive Pipeline to Maximize Isobaric Tagging Data in Large-Scale MS-Based Proteomics. J. Proteome Res. 2018, 17, 2165–2173. [Google Scholar] [CrossRef]
- Neagu, M.; Constantin, C.; Cretoiu, S.M.; Zurac, S. miRNAs in the Diagnosis and Prognosis of Skin Cancer. Front. Cell Dev. Biol. 2020, 8, 71. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.L.; Kaushik, S.; Valdes-Rodriguez, R.; Anvekar, R. MicroRNAs in cutaneous melanoma: Role as diagnostic and prognostic biomarkers. J. Cell. Physiol. 2018, 233, 5133–5141. [Google Scholar] [CrossRef] [PubMed]
- Mione, M.; Liebig, J.; Munoz, L.; Bosserhoff, A. MiRNAs in Malignant Melanoma. In Melanoma Development; Springer International Publishing: Cham, Switzerland, 2017; pp. 119–175. [Google Scholar]
- Thyagarajan, A.; Tsai, K.Y.; Sahu, R.P. MicroRNA heterogeneity in melanoma progression. Semin. Cancer Biol. 2019, 59, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Fattore, L.; Costantini, S.; Malpicci, D.; Ruggiero, C.F.; Ascierto, P.A.; Croce, C.M.; Mancini, R.; Ciliberto, G. MicroRNAs in melanoma development and resistance to target therapy. Oncotarget 2017, 8, 22262–22278. [Google Scholar] [CrossRef] [PubMed]
- Ghafouri-Fard, S.; Gholipour, M.; Taheri, M. MicroRNA Signature in Melanoma: Biomarkers and Therapeutic Targets. Front. Oncol. 2021, 11, 8987. [Google Scholar] [CrossRef] [PubMed]
- Lorusso, C.; De Summa, S.; Pinto, R.; Danza, K.; Tommasi, S. miRNAs as Key Players in the Management of Cutaneous Melanoma. Cells 2020, 9, 415. [Google Scholar] [CrossRef] [PubMed]
- Mumford, S.; Towler, B.; Pashler, A.; Gilleard, O.; Martin, Y.; Newbury, S. Circulating MicroRNA Biomarkers in Melanoma: Tools and Challenges in Personalised Medicine. Biomolecules 2018, 8, 21. [Google Scholar] [CrossRef]
- Adler, N.R.; Haydon, A.; McLean, C.A.; Kelly, J.W.; Mar, V.J. Metastatic pathways in patients with cutaneous melanoma. Pigment Cell Melanoma Res. 2017, 30, 13–27. [Google Scholar] [CrossRef]
- Gajos-Michniewicz, A.; Czyz, M. Role of miRNAs in Melanoma Metastasis. Cancers 2019, 11, 326. [Google Scholar] [CrossRef]
- Bustos, M.A.; Gross, R.; Rahimzadeh, N.; Cole, H.; Tran, L.T.; Tran, K.D.; Takeshima, L.; Stern, S.L.; O’Day, S.; Hoon, D.S.B. A Pilot Study Comparing the Efficacy of Lactate Dehydrogenase Levels Versus Circulating Cell-Free microRNAs in Monitoring Responses to Checkpoint Inhibitor Immunotherapy in Metastatic Melanoma Patients. Cancers 2020, 12, 3361. [Google Scholar] [CrossRef]
- Bustos, M.A.; Tran, K.D.; Rahimzadeh, N.; Gross, R.; Lin, S.Y.; Shoji, Y.; Murakami, T.; Boley, C.L.; Tran, L.T.; Cole, H.; et al. Integrated Assessment of Circulating Cell-Free MicroRNA Signatures in Plasma of Patients with Melanoma Brain Metastasis. Cancers 2020, 12, 1692. [Google Scholar] [CrossRef] [PubMed]
- Schneegans, S.; Lück, L.; Besler, K.; Bluhm, L.; Stadler, J.; Staub, J.; Greinert, R.; Volkmer, B.; Kubista, M.; Gebhardt, C.; et al. Pre-analytical factors affecting the establishment of a single tube assay for multiparameter liquid biopsy detection in melanoma patients. Mol. Oncol. 2020, 14, 1001–1015. [Google Scholar] [CrossRef] [PubMed]
- Ning, M.S.; Kim, A.S.; Prasad, N.; Levy, S.E.; Zhang, H.; Andl, T. Characterization of the Merkel Cell Carcinoma miRNome. J. Skin Cancer 2014, 2014, 289548. [Google Scholar] [CrossRef] [PubMed]
- Meng, W.; McElroy, J.P.; Volinia, S.; Palatini, J.; Warner, S.; Ayers, L.W.; Palanichamy, K.; Chakravarti, A.; Lautenschlaeger, T. Comparison of MicroRNA Deep Sequencing of Matched Formalin-Fixed Paraffin-Embedded and Fresh Frozen Cancer Tissues. PLoS ONE 2013, 8, e64393. [Google Scholar] [CrossRef]
- Babapoor, S.; Fleming, E.; Wu, R.; Dadras, S.S. A Novel miR-451a isomiR, Associated with Amelanotypic Phenotype, Acts as a Tumor Suppressor in Melanoma by Retarding Cell Migration and Invasion. PLoS ONE 2014, 9, e107502. [Google Scholar] [CrossRef] [PubMed]
- Torres, R.; Lang, U.E.; Hejna, M.; Shelton, S.J.; Joseph, N.M.; Shain, A.H.; Yeh, I.; Wei, M.L.; Oldham, M.C.; Bastian, B.C.; et al. MicroRNA Ratios Distinguish Melanomas from Nevi. J. Investig. Dermatol. 2020, 140, 164–173. [Google Scholar] [CrossRef]
- Gencia, I.; Baderca, F.; Avram, S.; Gogulescu, A.; Marcu, A.; Seclaman, E.; Marian, C.; Solovan, C. A preliminary study of microRNA expression in different types of primary melanoma. Bosn. J. Basic Med. Sci. 2019, 20, 197. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, L.; Jiang, L.; Zhang, X. Novel MicroRNA Biomarkers, miR-142-5p, miR-550a, miR-1826, and miR-1201, Were Identified for Primary Melanoma. J. Comput. Biol. 2020, 27, 815–824. [Google Scholar] [CrossRef]
- Watt, K.; Tyryshkin, K.; Renwick, N.; Craig, A.W.B. Distinguishing Tumor and Stromal Sources of MicroRNAs Linked to Metastasis in Cutaneous Melanoma. Transl. Oncol. 2020, 13, 100802. [Google Scholar] [CrossRef]
- Hanniford, D.; Zhong, J.; Koetz, L.; Gaziel-Sovran, A.; Lackaye, D.J.; Shang, S.; Pavlick, A.; Shapiro, R.; Berman, R.; Darvishian, F.; et al. A miRNA-Based Signature Detected in Primary Melanoma Tissue Predicts Development of Brain Metastasis. Clin. Cancer Res. 2015, 21, 4903–4912. [Google Scholar] [CrossRef]
- Li, H.; Song, J.-B.; Chen, H.-X.; Wang, Q.-Q.; Meng, L.-X.; Li, Y. MiR-155 inhibits proliferation, invasion and migration of melanoma via targeting CBL. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 9525–9534. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Sendra, B.; Serna, E.; Navarro, L.; González-Muñoz, J.F.; Portero, J.; Ramos, A.; Murgui, A.; Monteagudo, C. Transcriptomic identification of miR-205 target genes potentially involved in metastasis and survival of cutaneous malignant melanoma. Sci. Rep. 2020, 10, 4771. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Willems, E.; Singh, A.; Ong, I.M.; Verma, A.K. Ultraviolet radiation-induced differential microRNA expression in the skin of hairless SKH1 mice, a widely used mouse model for dermatology research. Oncotarget 2016, 7, 84924–84937. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, Z.-W. Expression of miR-203 is decreased and associated with the prognosis of melanoma patients. Int. J. Clin. Exp. Pathol. 2015, 8, 13249–13254. [Google Scholar] [PubMed]
- Lohcharoenkal, W.; Das Mahapatra, K.; Pasquali, L.; Crudden, C.; Kular, L.; Akkaya Ulum, Y.Z.; Zhang, L.; Xu Landén, N.; Girnita, L.; Jagodic, M.; et al. Genome-Wide Screen for MicroRNAs Reveals a Role for miR-203 in Melanoma Metastasis. J. Investig. Dermatol. 2018, 138, 882–892. [Google Scholar] [CrossRef]
- Yang, Z.; Liao, B.; Xiang, X.; Ke, S. miR-21-5p promotes cell proliferation and G1/S transition in melanoma by targeting CDKN2C. FEBS Open Bio 2020, 10, 752–760. [Google Scholar] [CrossRef]
- Díaz-Martínez, M.; Benito-Jardón, L.; Alonso, L.; Koetz-Ploch, L.; Hernando, E.; Teixidó, J. miR-204-5p and miR-211-5p Contribute to BRAF Inhibitor Resistance in Melanoma. Cancer Res. 2018, 78, 1017–1030. [Google Scholar] [CrossRef] [PubMed]
- Nyholm, A.M.; Lerche, C.M.; Manfé, V.; Biskup, E.; Johansen, P.; Morling, N.; Thomsen, B.M.; Glud, M.; Gniadecki, R. miR-125b induces cellular senescence in malignant melanoma. BMC Dermatol. 2014, 14, 8. [Google Scholar] [CrossRef]
- Sun, X.; Zhang, C.; Cao, Y.; Liu, E. miR-150 Suppresses Tumor Growth in Melanoma Through Downregulation of MYB. Oncol. Res. 2019, 27, 317–323. [Google Scholar] [CrossRef]
- Sánchez-Sendra, B.; Martinez-Ciarpaglini, C.; González-Muñoz, J.F.; Murgui, A.; Terrádez, L.; Monteagudo, C. Downregulation of intratumoral expression of miR-205, miR-200c and miR-125b in primary human cutaneous melanomas predicts shorter survival. Sci. Rep. 2018, 8, 17076. [Google Scholar] [CrossRef]
- Guo, J.; Yang, M.; Zhang, W.; Lu, H.; Li, J. A panel of miRNAs as prognostic indicators for clinical outcome of skin cutaneous melanoma. Int. J. Clin. Exp. Med. 2016, 9, 28–39. [Google Scholar]
- Stark, M.S.; Klein, K.; Weide, B.; Haydu, L.E.; Pflugfelder, A.; Tang, Y.H.; Palmer, J.M.; Whiteman, D.C.; Scolyer, R.A.; Mann, G.J.; et al. The Prognostic and Predictive Value of Melanoma-related MicroRNAs Using Tissue and Serum: A MicroRNA Expression Analysis. EBioMedicine 2015, 2, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Segura, M.F.; Belitskaya-Lévy, I.; Rose, A.E.; Zakrzewski, J.; Gaziel, A.; Hanniford, D.; Darvishian, F.; Berman, R.S.; Shapiro, R.L.; Pavlick, A.C.; et al. Melanoma MicroRNA Signature Predicts Post-Recurrence Survival. Clin. Cancer Res. 2010, 16, 1577–1586. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, S.; Qu, L.; Wang, Y.; Chen, H.; He, C. Identification of a five-miRNA signature predicting survival in cutaneous melanoma cancer patients. PeerJ 2019, 7, e7831. [Google Scholar] [CrossRef] [PubMed]
- Saldanha, G.; Elshaw, S.; Sachs, P.; Alharbi, H.; Shah, P.; Jothi, A.; Pringle, J.H. microRNA-10b is a prognostic biomarker for melanoma. Mod. Pathol. 2016, 29, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Caramuta, S.; Egyházi, S.; Rodolfo, M.; Witten, D.; Hansson, J.; Larsson, C.; Lui, W.-O. MicroRNA Expression Profiles Associated with Mutational Status and Survival in Malignant Melanoma. J. Investig. Dermatol. 2010, 130, 2062–2070. [Google Scholar] [CrossRef]
- Tembe, V.; Schramm, S.-J.; Stark, M.S.; Patrick, E.; Jayaswal, V.; Tang, Y.H.; Barbour, A.; Hayward, N.K.; Thompson, J.F.; Scolyer, R.A.; et al. MicroRNA and mRNA expression profiling in metastatic melanoma reveal associations with BRAF mutation and patient prognosis. Pigment Cell Melanoma Res. 2015, 28, 254–266. [Google Scholar] [CrossRef]
- Babapoor, S.; Wu, R.; Kozubek, J.; Auidi, D.; Grant-Kels, J.M.; Dadras, S.S. Identification of microRNAs associated with invasive and aggressive phenotype in cutaneous melanoma by next-generation sequencing. Lab. Investig. 2017, 97, 636–648. [Google Scholar] [CrossRef][Green Version]
- Lin, G.; Yin, G.; Yan, Y.; Lin, B. Identification of prognostic biomarkers for malignant melanoma using microarray datasets. Oncol. Lett. 2019, 18, 5243–5254. [Google Scholar] [CrossRef]
- Wang, L.; Wei, C.; Xu, Y.; Deng, X.; Wang, Q.; Ying, J.; Zhang, S.; Yuan, X.; Xuan, T.; Pan, Y.; et al. Prognostic genes of melanoma identified by weighted gene co-expression network analysis and drug repositioning using a network-based method. Oncol. Lett. 2019, 18, 6066–6078. [Google Scholar] [CrossRef]
- Lee, S.; Suh, H.B.; Choi, S.J.; Kang, J.; Kang, J.W.; Kwon, E.J.; Kim, H.-J.; Kim, Y.H.; Shin, K. Identification of prognostic mRNAs in metastatic cutaneous melanoma. Melanoma Res. 2020, 30, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Yingjuan, W.; Li, Z.; Wei, C.; Xiaoyuan, W. Identification of prognostic genes and construction of a novel gene signature in the skin melanoma based on the tumor microenvironment. Medicine 2021, 100, e26017. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Qu, X.; Wang, M. A Four-Gene-Based Prognostic Model Predicts Overall Survival in Patients with Cutaneous Melanoma. Front. Oncol. 2021, 11, 9874. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Xu, Y.; Yan, Y.; Luo, P.; Chen, S.; Zheng, B.; Yan, W.; Chen, Y.; Wang, C. Common Nevus and Skin Cutaneous Melanoma: Prognostic Genes Identified by Gene Co-Expression Network Analysis. Genes 2019, 10, 747. [Google Scholar] [CrossRef] [PubMed]
- Brunner, G.; Reitz, M.; Heinecke, A.; Lippold, A.; Berking, C.; Suter, L.; Atzpodien, J. A nine-gene signature predicting clinical outcome in cutaneous melanoma. J. Cancer Res. Clin. Oncol. 2013, 139, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Gerami, P.; Cook, R.W.; Wilkinson, J.; Russell, M.C.; Dhillon, N.; Amaria, R.N.; Gonzalez, R.; Lyle, S.; Johnson, C.E.; Oelschlager, K.M.; et al. Development of a Prognostic Genetic Signature to Predict the Metastatic Risk Associated with Cutaneous Melanoma. Clin. Cancer Res. 2015, 21, 175–183. [Google Scholar] [CrossRef]
- Sheng, Z.; Han, W.; Huang, B.; Shen, G. Screening and identification of potential prognostic biomarkers in metastatic skin cutaneous melanoma by bioinformatics analysis. J. Cell. Mol. Med. 2020, 24, 11613–11618. [Google Scholar] [CrossRef]
- Metri, R.; Mohan, A.; Nsengimana, J.; Pozniak, J.; Molina-Paris, C.; Newton-Bishop, J.; Bishop, D.; Chandra, N. Identification of a gene signature for discriminating metastatic from primary melanoma using a molecular interaction network approach. Sci. Rep. 2017, 7, 17314. [Google Scholar] [CrossRef]
- Thakur, R.; Laye, J.P.; Lauss, M.; Diaz, J.M.S.; O’Shea, S.J.; Poźniak, J.; Filia, A.; Harland, M.; Gascoyne, J.; Randerson-Moor, J.A.; et al. Transcriptomic Analysis Reveals Prognostic Molecular Signatures of Stage I Melanoma. Clin. Cancer Res. 2019, 25, 7424–7435. [Google Scholar] [CrossRef]
- Sun, L.; Li, P.; Ren, H.; Liu, G.; Sun, L. A four-gene expression-based signature predicts the clinical outcome of melanoma. J. BUON 2019, 24, 2161–2167. [Google Scholar]
- Wan, Q.; Liu, C.; Liu, C.; Liu, W.; Wang, X.; Wang, Z. Discovery and Validation of a Metastasis-Related Prognostic and Diagnostic Biomarker for Melanoma Based on Single Cell and Gene Expression Datasets. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Li, Y.; Niu, X.; Wu, Y.; Guan, X.; Hong, Y.; Chen, H.; Song, B. Identification and Validation of Prognostically Relevant Gene Signature in Melanoma. BioMed Res. Int. 2020, 2020, 5323614. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-X.; Wan, C.; Dong, Z.-B.; Wang, B.-H.; Liu, H.-Y.; Li, Y. Integrative Analysis of Long Noncoding RNA (lncRNA), microRNA (miRNA) and mRNA Expression and Construction of a Competing Endogenous RNA (ceRNA) Network in Metastatic Melanoma. Med. Sci. Monit. 2019, 25, 2896–2907. [Google Scholar] [CrossRef] [PubMed]
- Fortis, S.P.; Anastasopoulou, E.A.; Voutsas, I.F.; Baxevanis, C.N.; Perez, S.A.; Mahaira, L.G. Potential Prognostic Molecular Signatures in a Preclinical Model of Melanoma. Anticancer Res. 2017, 37, 143–148. [Google Scholar] [CrossRef]
- De Oliveira Pessoa, D.; Rius, F.E.; Papaiz, D.D.; Ayub, A.L.P.; Morais, A.S.; de Souza, C.F.; da Paixão, V.F.; Setubal, J.C.; Newton-Bishop, J.; Nsengimana, J.; et al. Transcriptional signatures underlying dynamic phenotypic switching and novel disease biomarkers in a linear cellular model of melanoma progression. Neoplasia 2021, 23, 439–455. [Google Scholar] [CrossRef]
- Pérez-Guijarro, E.; Yang, H.H.; Araya, R.E.; El Meskini, R.; Michael, H.T.; Vodnala, S.K.; Marie, K.L.; Smith, C.; Chin, S.; Lam, K.C.; et al. Multimodel preclinical platform predicts clinical response of melanoma to immunotherapy. Nat. Med. 2020, 26, 781–791. [Google Scholar] [CrossRef]
- Jayawardana, K.; Schramm, S.-J.; Tembe, V.; Mueller, S.; Thompson, J.F.; Scolyer, R.A.; Mann, G.J.; Yang, J. Identification, Review, and Systematic Cross-Validation of microRNA Prognostic Signatures in Metastatic Melanoma. J. Investig. Dermatol. 2016, 136, 245–254. [Google Scholar] [CrossRef]
- Xiong, J.; Bing, Z.; Guo, S. Observed Survival Interval: A Supplement to TCGA Pan-Cancer Clinical Data Resource. Cancers 2019, 11, 280. [Google Scholar] [CrossRef]
- Korkmaz, G.; le Sage, C.; Tekirdag, K.A.; Agami, R.; Gozuacik, D. miR-376b controls starvation and mTOR inhibition-related autophagy by targeting ATG4C and BECN1. Autophagy 2012, 8, 165–176. [Google Scholar] [CrossRef]
- Linck, L.; Liebig, J.; Völler, D.; Eichner, N.; Lehmann, G.; Meister, G.; Bosserhoff, A. MicroRNA-sequencing data analyzing melanoma development and progression. Exp. Mol. Pathol. 2018, 105, 371–379. [Google Scholar] [CrossRef]
- Jin, C.; Rajabi, H.; Kufe, D. miR-1226 targets expression of the mucin 1 oncoprotein and induces cell death. Int. J. Oncol. 2010, 37, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Du, X.; Liu, L.; Cao, Q.; Pan, Z.; Li, Q. miR-1306 Mediates the Feedback Regulation of the TGF-β/SMAD Signaling Pathway in Granulosa Cells. Cells 2019, 8, 298. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Sui, J.; Yang, S.; Liu, Y.; Wang, Y.; Liang, G. Integrative analysis of competing endogenous RNA network focusing on long noncoding RNA associated with progression of cutaneous melanoma. Cancer Med. 2018, 7, 1019–1029. [Google Scholar] [CrossRef] [PubMed]
- Bayat, Z.; Ghaemi, Z.; Behmanesh, M.; Soltani, B.M. Hsa-miR-186-5p regulates TGFβ signaling pathway through expression suppression of SMAD6 and SMAD7 genes in colorectal cancer. Biol. Chem. 2021, 402, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Zhou, S.; Gan, C.; Zhang, X. MiR-186 inhibits cell proliferation and invasion in human cutaneous malignant melanoma. J. Cancer Res. Ther. 2018, 14, 60. [Google Scholar] [CrossRef] [PubMed]
- Harrison, E.B.; Porrello, A.; Bowman, B.M.; Belanger, A.R.; Yacovone, G.; Azam, S.H.; Windham, I.A.; Ghosh, S.K.; Wang, M.; Mckenzie, N.; et al. A Circle RNA Regulatory Axis Promotes Lung Squamous Metastasis via CDR1-Mediated Regulation of Golgi Trafficking. Cancer Res. 2020, 80, 4972–4985. [Google Scholar] [CrossRef]
- Zhang, B. Guizhi Fuling pills inhibit the proliferation, migration and invasion of human cutaneous malignant melanoma cells by regulating the molecular axis of LncRNA TPT1-AS1/miR-671-5p. Cell. Mol. Biol. 2020, 66, 148–154. [Google Scholar] [CrossRef]
- Manvati, M.K.S.; Khan, J.; Verma, N.; Dhar, P.K. Association of miR-760 with cancer: An overview. Gene 2020, 747, 144648. [Google Scholar] [CrossRef]
- Tang, J.; Gao, W.; Liu, G.; Sheng, W.; Zhou, J.; Dong, Q.; Dong, M. miR-944 Suppresses EGF-Induced EMT in Colorectal Cancer Cells by Directly Targeting GATA6. OncoTargets Ther. 2021, 14, 2311–2325. [Google Scholar] [CrossRef]
- Lv, L.; Wang, X.; Ma, T. microRNA-944 inhibits the malignancy of hepatocellular carcinoma by directly targeting IGF-1R and deactivating the PI3K/Akt signaling pathway. Cancer Manag. Res. 2019, 11, 2531–2543. [Google Scholar] [CrossRef]
- Park, S.; Kim, J.; Eom, K.; Oh, S.; Kim, S.; Kim, G.; Ahn, S.; Park, K.H.; Chung, D.; Lee, H. microRNA-944 overexpression is a biomarker for poor prognosis of advanced cervical cancer. BMC Cancer 2019, 19, 419. [Google Scholar] [CrossRef]
- Chen, G.; Hu, J.; Huang, Z.; Yang, L.; Chen, M. MicroRNA-1976 functions as a tumor suppressor and serves as a prognostic indicator in non-small cell lung cancer by directly targeting PLCE1. Biochem. Biophys. Res. Commun. 2016, 473, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, M.; Han, X.; Wang, H.; Wang, X.; Ma, G.; Xia, T.; Wang, S. MiR-1976 knockdown promotes epithelial–mesenchymal transition and cancer stem cell properties inducing triple-negative breast cancer metastasis. Cell Death Dis. 2020, 11, 500. [Google Scholar] [CrossRef] [PubMed]
- Islam, T.; Rahman, R.; Gov, E.; Turanli, B.; Gulfidan, G.; Haque, A.; Arga, K.Y.; Haque Mollah, N. Drug Targeting and Biomarkers in Head and Neck Cancers: Insights from Systems Biology Analyses. Omics J. Integr. Biol. 2018, 22, 422–436. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Zhang, Y.; Yu, Z. Expression and clinical significance of miR-3615 in hepatocellular carcinoma. J. Int. Med. Res. 2021, 49, 030006052098154. [Google Scholar] [CrossRef]
- Theofilatos, K.; Korfiati, A.; Mavroudi, S.; Cowperthwaite, M.C.; Shpak, M. Discovery of stroke-related blood biomarkers from gene expression network models. BMC Med. Genom. 2019, 12, 118. [Google Scholar] [CrossRef]
- Shiiyama, R.; Fukushima, S.; Jinnin, M.; Yamashita, J.; Miyashita, A.; Nakahara, S.; Kogi, A.; Aoi, J.; Masuguchi, S.; Inoue, Y.; et al. Sensitive detection of melanoma metastasis using circulating microRNA expression profiles. Melanoma Res. 2013, 23, 366–372. [Google Scholar] [CrossRef]
- Shellman, M.H.; Shellman, Y.G. Human against Machine? Machine Learning Identifies MicroRNA Ratios as Biomarkers for Melanoma. J. Investig. Dermatol. 2020, 140, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Höhn, J.; Krieghoff-Henning, E.; Jutzi, T.B.; von Kalle, C.; Utikal, J.S.; Meier, F.; Gellrich, F.F.; Hobelsberger, S.; Hauschild, A.; Schlager, J.G.; et al. Combining CNN-based histologic whole slide image analysis and patient data to improve skin cancer classification. Eur. J. Cancer 2021, 149, 94–101. [Google Scholar] [CrossRef]
- Fattore, L.; Ruggiero, C.F.; Liguoro, D.; Mancini, R.; Ciliberto, G. Single cell analysis to dissect molecular heterogeneity and disease evolution in metastatic melanoma. Cell Death Dis. 2019, 10, 827. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| miRNASeq Dataset | Melanoma Samples | Control Samples | Other Diseases Samples | Biomaterial |
|---|---|---|---|---|
| TCGA-SKCM [6] | 448 melanoma patients | - | - | Tissue |
| GSE157370 [31] | 47 stage III and IV melanoma patients (pre-treatment samples) and 111 CII post-treatment samples from the same patients | 73 healthy donors | - | Plasma |
| GSE150956 [32] | 36 + 24 pre-operative MBM patients’ plasma samples | 48 Normal (cancer-free) donor plasma and serum plasma | 49 other cancer types that had brain metastasis and glioblastomas | Plasma |
| 24 MBM tissues | - | - | Tissue | |
| 20 pre-and post-treatment plasma and 14 urine samples collected from metastatic melanoma patients receiving CII | 8 Normal (cancer-free) urine samples | - | plasma and urine | |
| GSE143231 [33] | 10 metastatic melanoma AJCC stage IV patients | five HDs | - | plasma and EVs |
| GSE53600 [34] | 1 melanoma lymph node metastases | 1 normal skin | 6 MCC lymph node metastases, SCC and BCC primary cutaneous lesions | Frozen tissue |
| GSE45740 [35] | 1 metastatic melanoma | 7 breast invasive ductal carcinoma, renal clear cell carcinoma, lung adenocarcinoma, prostate adenocarcinoma and sarcoma of thigh | paired FFPE and fresh frozen samples | |
| GSE36236 [36] | 19 primary cutaneous melanomas biopsies/excisions | matched normal skin and common nevi | - | FFPE tissue |
| phs001550.v2.p1 [37] | 8 melanomas | 7 intact adjacent benign nevi | - | FFPE microdissected regions |
| miRNA Signature | Significance | Datasets | Samples | Reference |
|---|---|---|---|---|
| miR-31-5p, miR-21-5p, miR-211-5p, miR-125a-5p, miR-125b-5p and miR-100-5p (miRNA ratios) | distinction of melanomas from nevi | FFPE phs001550.v2.p1 (miRNASeq) | 41 nevi and 41 melanomas | [37] |
| miR-155-5p, miR-9-5p, miR-142-5p, miR-19a-3p, miR-134-5p, miR-301a-3p, miR-205-5p, miR-203a-3p, miR-27b-3p, miR-218-5p, and miR-23b-3p | FFPE (microarray) | 5 cutaneous nevi and 27 primary melanomas | [38] | |
| miR-142-5p, miR-550a, miR-1826, and miR-1201 | GSE62370 (microarray) | 9 congenital nevi and 92 primary melanomas | [39] | |
| miR-205, miR-203, miR-200a-c, and miR-141 | distinction of metastatic from primary melanomas | TCGA (miRNASeq) | 97 primary and 350 metastatic melanomas | [40] |
| miR-150-5p, miR-15b-5p, miR-16-5p, and miR-374b-3p | prediction of brain metastases | IMCG GSE62372 (microarray) | 256 primary melanomas | [41] |
| miR-125b, miR-200c and miR-205 | prediction of overall survival | FF (RT-qPCR) | 65 primary and 67 metastatic melanomas | [51] |
| miR-202, miR-206, miR-3681, miR-122 and miR-1246 | TCGA (miRNASeq) | 448 melanomas | [52] | |
| miR-16, miR-211, miR-4487, miR-4706, miR-4731, miR-509-3p and miR-509-5p | FFPE (RT-qPCR) | 86 melanomas | [53] | |
| miR-497, miR-145, miR-342-5p, miR-150, miR-155 and miR-455-5p | prediction of post-recurrence survival | FFPE (microarray) | 59 melanomas | [54] |
| miR-25, miR-204, miR-211, miR-510 and miR-513c | prognostic biomarker in cutaneous melanoma | GSE35579 (microarray) | 11 benign nevi and 41 melanomas | [55] |
| miR-10b | FF (microarray) | 20 non-metastasizing and 20 metastasizing primary melanomas | [56] | |
| miR-338, let-7, miR-365, miR-191, miR-193b-3p and miR-193a-3p | FF, GSE19387 (microarray) | 32 samples from regional lymph node metastases | [57] | |
| miR-150-5p, miR-142-3p and miR-142-5p | FF (microarray) | 84 samples from lymph node metastases | [58] | |
| miR-21-5p, miR-424-5p and let-7b | associated with invasive and aggressive phenotype | FF, GSE36236 (miRNASeq) | 12 normal skin, 13 common nevi, 17 dysplastic nevi, 45 melanomas in situ and 80 primary cutaneous melanomas | [59] |
| Gene Signature | Significance | Datasets | Samples | Reference |
|---|---|---|---|---|
| BAX, CALM1, CALM3, FN1, PRKCA, RB1, VEGFA, IGF1 | prognostic biomarker in cutaneous melanoma | GSE3189, GSE4570 and GSE4587 | 28 nevi and 58 melanoma samples | [60] |
| CXCR4, IL7R, PIK3CG | GSE65904 | 214 melanoma samples | [61] | |
| IGF2BP1, PTMA, MYC, MITF | elevated levels in more aggressive phenotypes | mouse model | [75] | |
| KRT9, KBTBD10, DCD, ECRG2, PIP, SCGB1D2, SCGB2A2, COL6A6, HES6 | prediction of clinical outcome | FF | 135 melanomas | [66] |
| RHBDL3, GPR64, ANKRD30A, PRKCD | TCGA, GSE22138, GSE54467, GSE65904 and E-MTAB-4725 | 102 melanomas + 565 samples (for confirmation) | [71] | |
| DSG3, DSC3, PKP1, EVPL, IVL, FLG, SPRR1A, SPRR1B | distinction of metastatic from primary melanomas | GSE46517, GSE15605, GSE8401 | 109 primary and 136 metastatic skin melanomas | [68] |
| ALDH1A1, HSP90AB1, KIT, KRT16, SPRR3, TMEM45B | GSE15605, GSE7553, LMC and TCGA | 20 normal samples, 867 primary and 419 metastatic melanomas | [69] | |
| ABCC3, CAPS2, CCR6, CDCA8, CLU, DPF1, PTK2B, SATB1, SYNE1 | prognostic biomarker in metastatic melanoma | TCGA, GSE19234, and GSE22153 | 556 cutaneous melanomas | [62] |
| STK26, KCNT2, CASP12 | GSE98394 | 27 common required nevi and 51 primary melanomas | [65] | |
| BAP1b, MGP, SPP1, CXCL14, CLCA2, S100A8, BTG1, SAP130, ARG1, KRT6B, GJA1, ID2, EIF1B, S100A9, CRABP2, KRT14, ROBO1, RBM23, TACSTD2, DSC1, SPRR1B, TRIM29, AQP3, TYRP1, PPL, LTA4H, CST6 | FFPE | 268 melanoma samples | [67] | |
| A2M, DUSP6, HLA-B, SERPINE2, SLC26A2 | GSE115978 | 31 melanoma samples | [72] | |
| CDKN2A, CDKN2B, ZBTB16/PLZF, CDKN1A, TYR, ARNT2, MDM2, GPR143, RAB38, ANGPT2, MGAT5, POU4F1, SIX1 | GSE149884 | murine melanoma cell lines | [76] | |
| SERPINH1, HOXC10, MYH10, EPHB2, SRPX2, CGREF1, DDR2, P4HA2, IGSF10, OSM, ADORA3, RECK, KDELR3, TMEM8, SMARCA1, JAZF1, FKBP7, ZFP449, TRIQK, REN1, IGF2BP2, GRB10, DPYSL4, CMBL, PDE3B, DAB2, PPP1R9A, QPRT, PEG10, NID1, EFNB3, COLGALT2, DBN1, C1QTNF3, CDC7, MDK, GULP1, HOXD13, EYA4, DEPDC1A, CRABP2, ATP10B, TTYH1, SLITRK2, ELOVL2, STK32B | prediction of overall survival | GSE140193, GSE25164 | genetically engineered mouse model | [9] |
| IL15, CCL8, CLIC2, SAMD9L, TLR2, HLA.DQB1, IGHV1-18, RARRES3, GBP4, APOBEC3G | TCGA | 470 melanomas | [63] | |
| ADAMDEC1, GNLY, HSPA13, TRIM29 | GSE7553, GSE46517, and GSE15605 | 17 normal skin and 202 melanomas | [64] | |
| IQCE, RFX6, GPAA1, BAHCC1, CLEC2B, AGAP2 | TCGA, GSE19234 and GES65094 | 485 melanomas | [73] | |
| CCR9, CNR2, DIRAS2, ESRP2, FAM83C, KCNT2, USH1G | TCGA | 103 primary and 368 metastatic melanomas | [74] | |
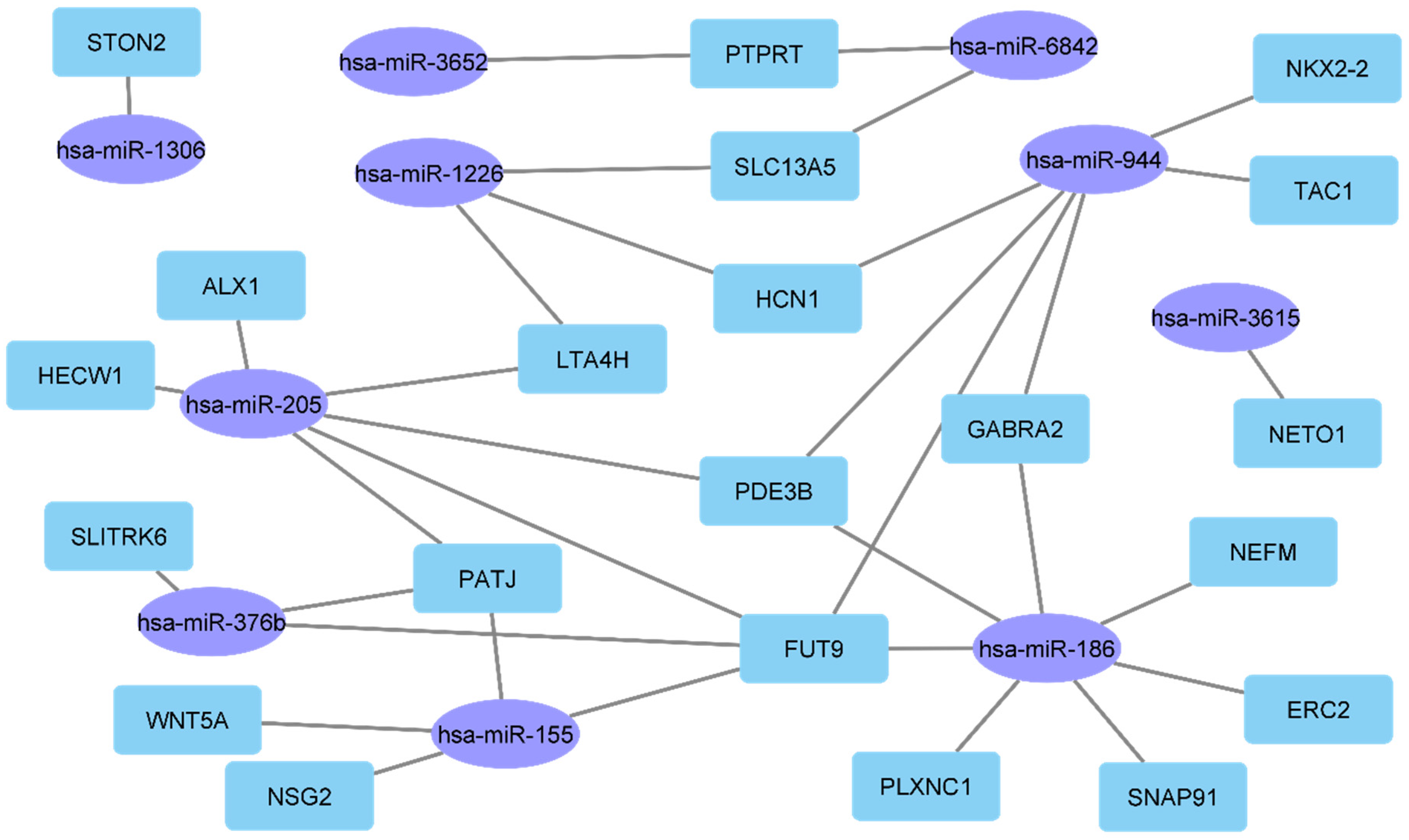
| AKR1C3, BMP1, CRTAC1, ECEL1, ERC2, FAM110C, FUT9, GABRA2, GAP43, GREM1, HECW1, KLHL1, KRT12, LHFPL4, NEFL, NEFM, NETO1, NKX2-2, NSG2, OCIAD2, OTOP1, PDE3B, PTPRN2, PTPRT, SIGLEC15, SLC13A5, SLC9A2, SLITRK6, SNAP91, STON2, TAC1, VAT1L, WNT5A, ALX1, BRD7, DTD1, GRSF1, HCN1, LTA4H, OXCT1, PATJ, PLXNC1, SSBP4, TELO2, TMEM177 | prediction of clinical response to ICB | GSE144946 | genetically engineered mouse model | [77] |
| JUN, AXL | prediction of poor prognosis and response to immunotherapy | LMC | 687 primary melanomas | [70] |
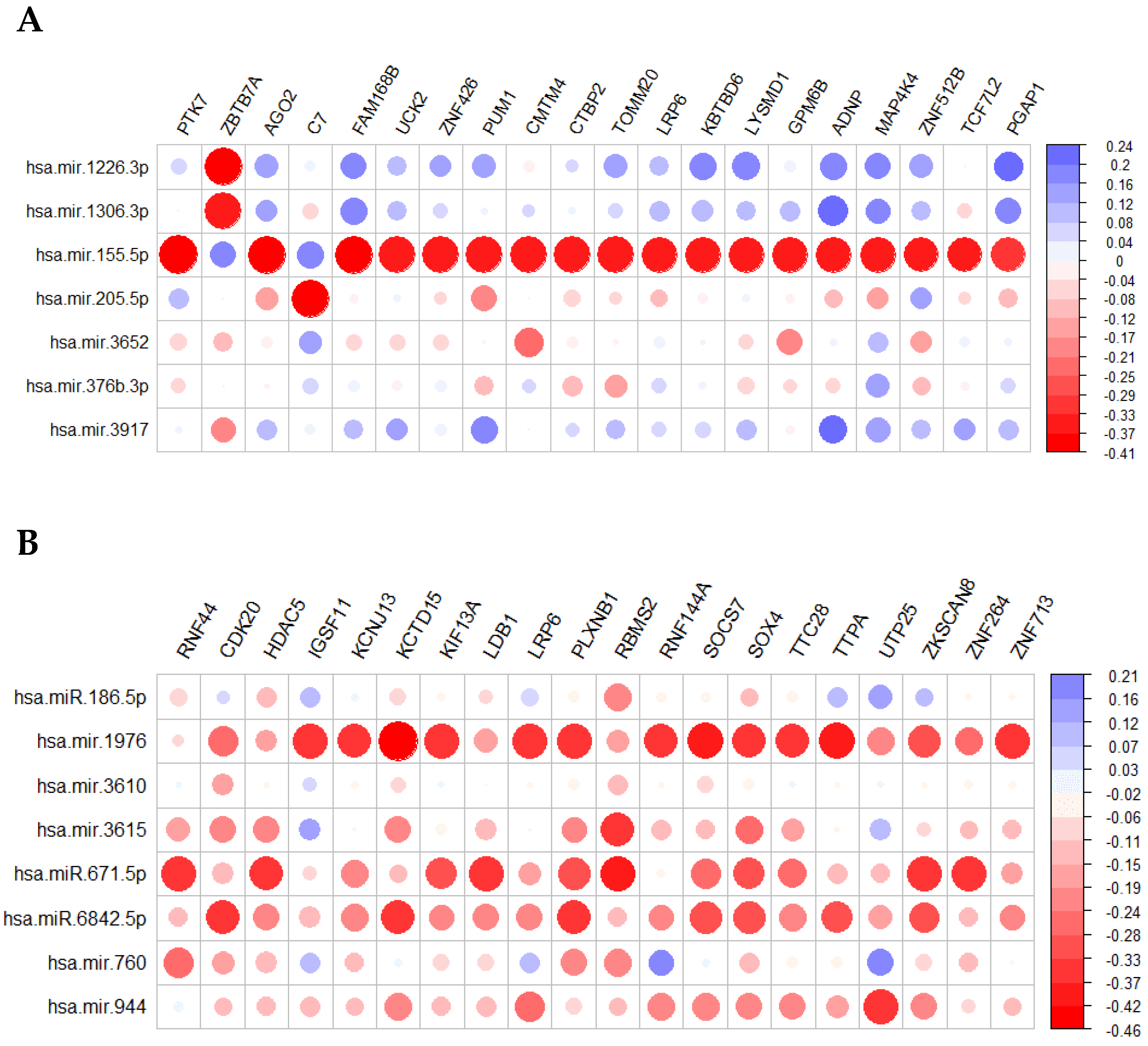
| miRNA | log2 Fold Change | Adjusted p Value | Role in the Literature |
|---|---|---|---|
| mir-155 | 0.441282 | 0.046899 | Associated with tumor prognosis. Its inhibition causes retarded glucose metabolism and thus, reduces in vivo tumor growth [78,79]. |
| mir-205 | −3.69183 | 1.03 × 10−14 | Is a tumor suppressor miRNA in breast cancer which inhibits cell proliferation and anchorage independent growth as well as cell invasion [79]. |
| mir-376b | 1.057248 | 0.002396 | Controls autophagy by directly regulating intracellular levels of two key autophagy proteins, ATG4C and BECN1 [80]. |
| mir-1226 | 0.393576 | 0.010158 | Regulates MUC1 and thus, dendritic cells resting which in turn play an important role in STS recurrence [81]. Targets expression of the mucin 1 oncoprotein and induces cell death [82]. |
| mir-1306 | 0.254205 | 0.027816 | Promotes apoptosis of granulosa cells (GCs) as well as attenuates the TGF-β/SMAD signaling pathway targeting and impairing TGFBR2 [83]. |
| mir-3652 | 0.549545 | 0.002342 | N/A |
| mir-3917 | 0.388593 | 0.020348 | Has been recognized as biomarker and used for the construction of a stomach adenocarcinoma (STAD) prognostic signature [84]. |
| miRNA | log2 Fold Change | Adjusted p Value | Role in the Literature |
|---|---|---|---|
| mir-186 | 0.290805 | 0.000389 | Regulates TGFβ by suppressing SMAD6-7 in colorectal cancer and inhibits cell proliferation in melanoma [85,86]. |
| mir-671 | −0.24244 | 0.027927 | miR-671-5p reduces NSCLC (squamous carcinoma) metastasis [87]. Its upregulation slows down proliferation and metastasis of A375 melanoma cells [88]. |
| mir-760 | 0.503684 | 0.012509 | It has been found downregulated in several cancers that can act both as tumor suppressor and as oncomir [89]. |
| mir-944 | −3.41097 | 8.62 × 10−37 | Suppresses EMT in colorectal cancer [90]. It has been reported as downregulated in hepatocellular carcinoma (HCC) and suppresses the malignancy of HCC by deactivating PI3K [91]. Its overexpression is correlated with poor prognosis in cervical cancer [92]. |
| mir-1976 | 0.444564 | 0.000327 | It has been identified as tumor suppressor in NSCLC [93]. Its downregulation has been correlated with worse overall survival in triple-negative breast cancer (TNBC) from TCGA [94]. |
| mir-3610 | 0.339103 | 0.049814 | It has been associated with sumoylation, a molecular signature in head and neck cancer [95]. |
| mir-3615 | 0.245396 | 0.036379 | Its upregulation is correlated with high TNM stage and high proliferation in HCC [96]. |
| mir-6842 | 0.450524 | 0.003272 | N/A |
| Metrics | Cross-Validation | Unseen Test Samples | ||||
|---|---|---|---|---|---|---|
| ACC | SP | SEN | ACC | SP | SEN | |
| recurrence signature | 91.51% | 92.65% | 91.29% | 73.85% | 79.09% | 88.78% |
| recurrence signature + clinical data | 96.51% | 97.13% | 96.07% | 85.38% | 88.35% | 92.86% |
| metastasis signature | 97.39% | 96.67% | 98.38% | 82.09% | 82.40% | 98.10% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korfiati, A.; Grafanaki, K.; Kyriakopoulos, G.C.; Skeparnias, I.; Georgiou, S.; Sakellaropoulos, G.; Stathopoulos, C. Revisiting miRNA Association with Melanoma Recurrence and Metastasis from a Machine Learning Point of View. Int. J. Mol. Sci. 2022, 23, 1299. https://doi.org/10.3390/ijms23031299
Korfiati A, Grafanaki K, Kyriakopoulos GC, Skeparnias I, Georgiou S, Sakellaropoulos G, Stathopoulos C. Revisiting miRNA Association with Melanoma Recurrence and Metastasis from a Machine Learning Point of View. International Journal of Molecular Sciences. 2022; 23(3):1299. https://doi.org/10.3390/ijms23031299
Chicago/Turabian StyleKorfiati, Aigli, Katerina Grafanaki, George C. Kyriakopoulos, Ilias Skeparnias, Sophia Georgiou, George Sakellaropoulos, and Constantinos Stathopoulos. 2022. "Revisiting miRNA Association with Melanoma Recurrence and Metastasis from a Machine Learning Point of View" International Journal of Molecular Sciences 23, no. 3: 1299. https://doi.org/10.3390/ijms23031299
APA StyleKorfiati, A., Grafanaki, K., Kyriakopoulos, G. C., Skeparnias, I., Georgiou, S., Sakellaropoulos, G., & Stathopoulos, C. (2022). Revisiting miRNA Association with Melanoma Recurrence and Metastasis from a Machine Learning Point of View. International Journal of Molecular Sciences, 23(3), 1299. https://doi.org/10.3390/ijms23031299