
N-Acetyl-Aspartyl-Glutamate in Brain Health and Disease


Abstract
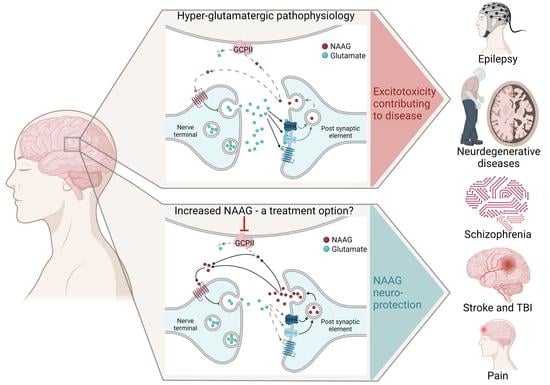
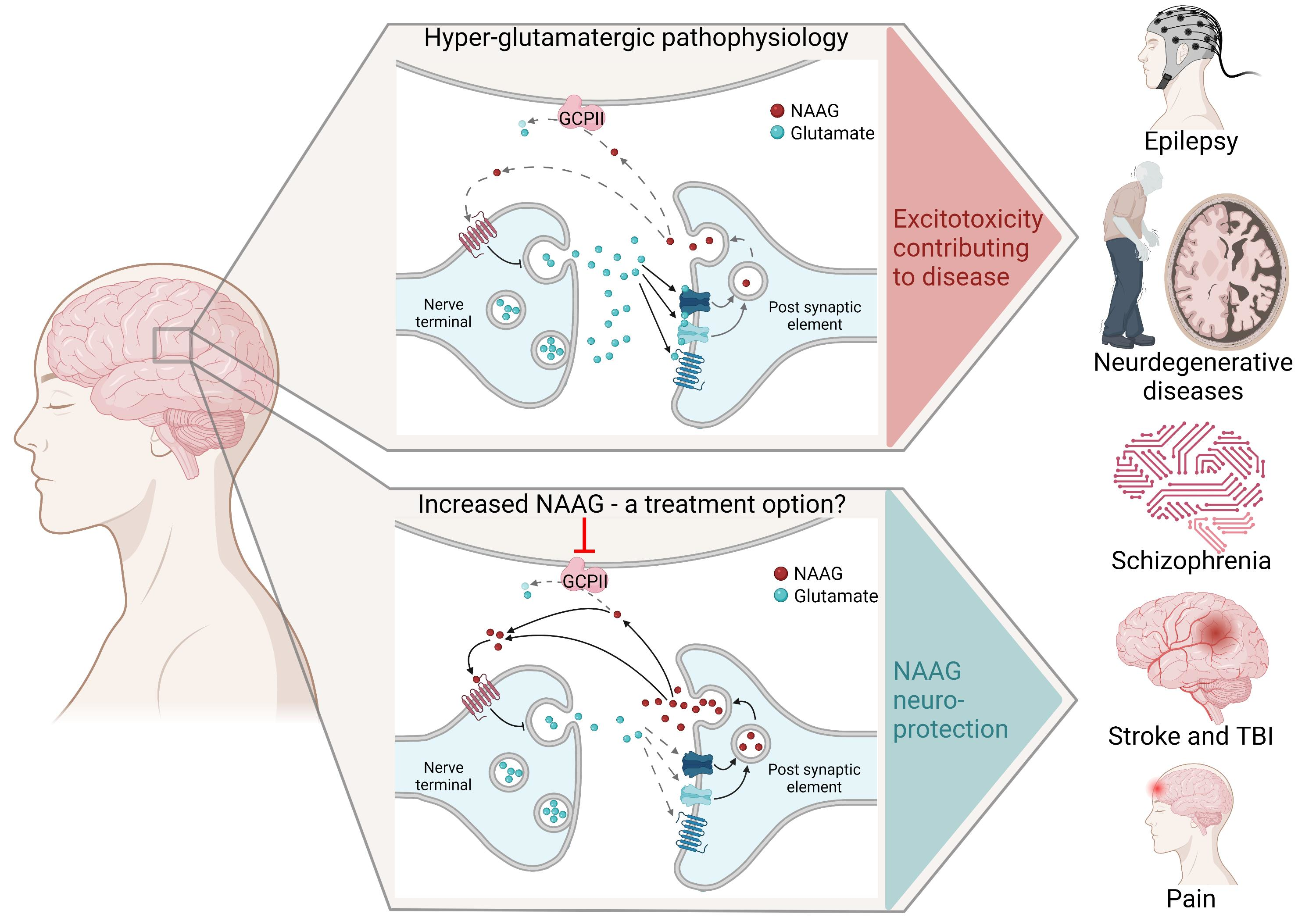
1. NAAG in Health
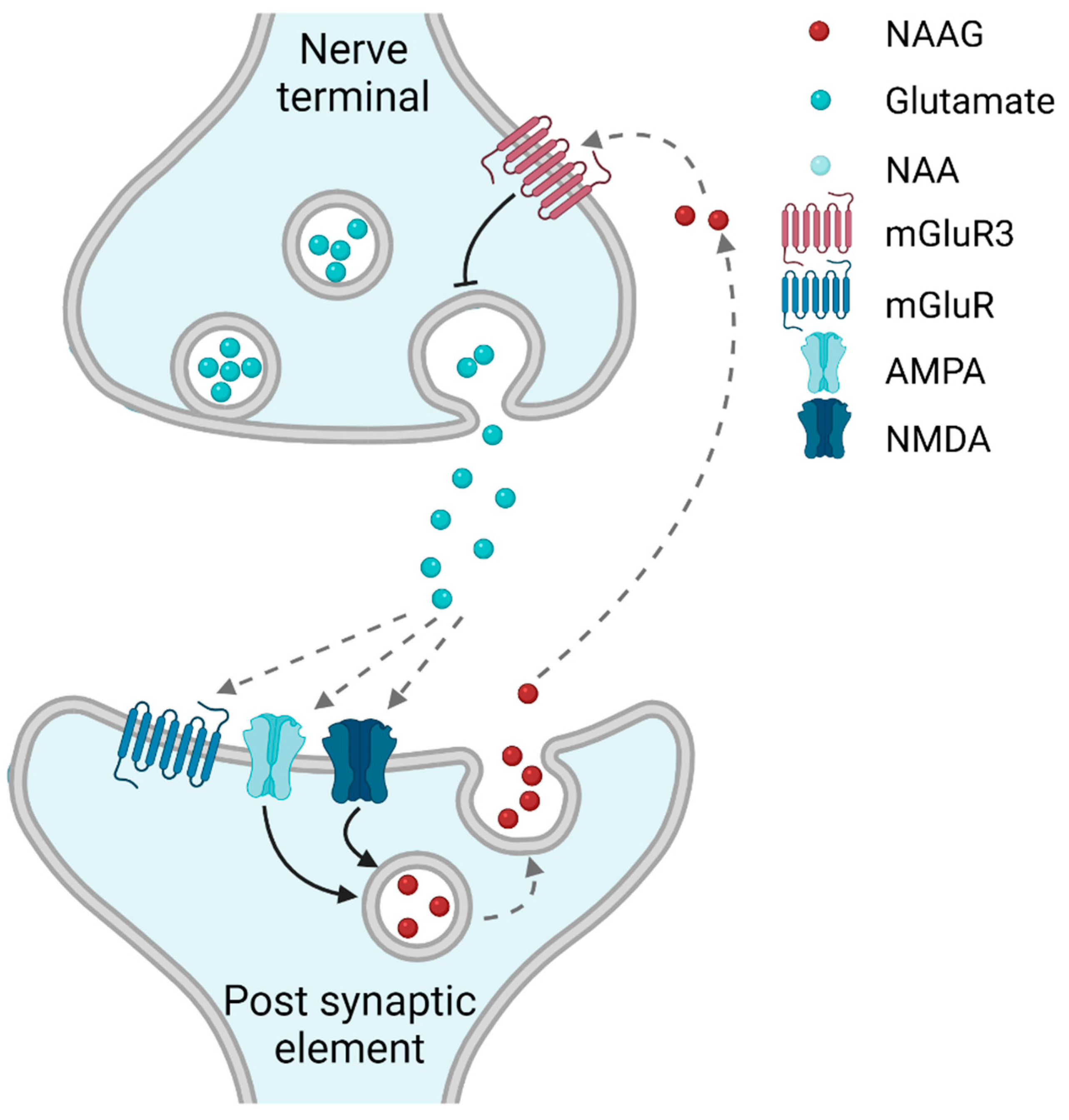
1.1. NAAG-Sensitive Receptors
1.2. Is NAAG a Classical (Anterograde) or an Retrograde Neurotransmitter?
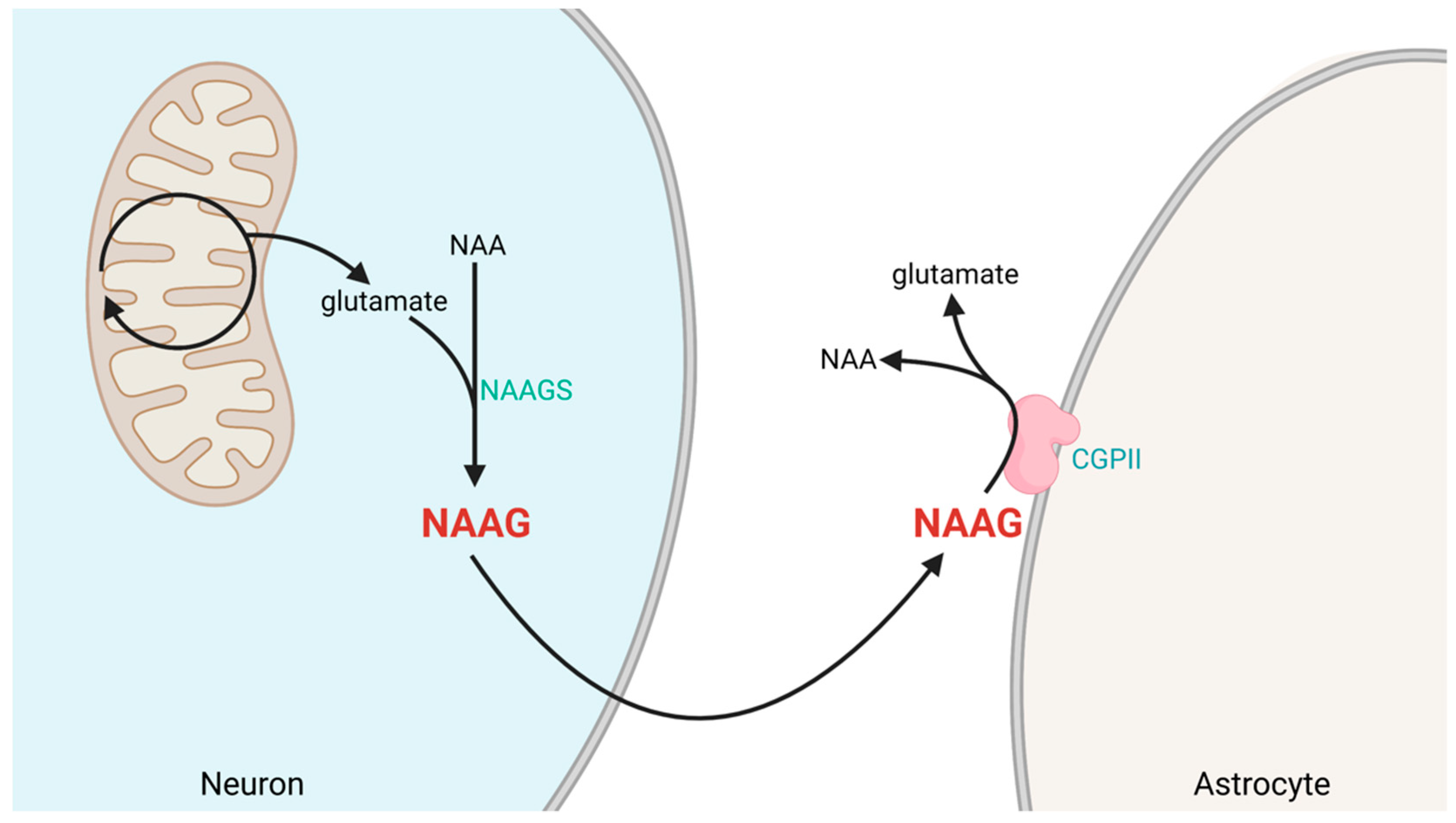
1.3. Synthesis and Degradation of NAAG
1.4. The Role of NAAG in Cognition
2. NAAG in Brain Disorders
2.1. NAAG in Neurodegenerative Diseases
2.2. NAAG in Epilepsy
2.3. NAAG in Stroke
2.4. NAAG in Traumatic Brain Injury
2.5. NAAG in Pain
2.6. NAAG in Schizophrenia
3. Translational Considerations
Funding
Conflicts of Interest
References
- Guarda, A.S.; Robinson, M.B.; Ory-Lavollee, L.; Forloni, G.L.; Blakely, R.D.; Coyle, J.T. Quantitation of N-acetyl-aspartyl-glutamate in microdissected rat brain nuclei and peripheral tissues: Findings with a novel liquid phase radioimmunoassay. Brain Res. 1988, 427, 223–231. [Google Scholar] [CrossRef]
- Vallianatou, T.; Lin, W.; Bèchet, N.B.; Correia, M.S.; Shanbhag, N.C.; Lundgaard, I.; Globisch, D. Differential regulation of oxidative stress, microbiota-derived, and energy metabolites in the mouse brain during sleep. J. Cereb. Blood Flow Metab. 2021, 41, 3324–3338. [Google Scholar] [CrossRef] [PubMed]
- Curatolo, A.; d’Arcangelo, P.; Lino, A.; Brancati, A. Distribution of N-acetyl-aspartic and N-acetyl-aspartyl-glutamic acids in nervous tissue. J. Neurochem. 1965, 12, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Fuhrman, S.; Palkovits, M.; Cassidy, M.; Neale, J.H. The regional distribution of N-acetylaspartylglutamate (NAAG) and peptidase activity against NAAG in the rat nervous system. J. Neurochem. 1994, 62, 275–281. [Google Scholar] [CrossRef]
- Zhong, C.; Zhao, X.; Van, K.C.; Bzdega, T.; Smyth, A.; Zhou, J.; Kozikowski, A.P.; Jiang, J.; O’Connor, W.T.; Berman, R.F.; et al. NAAG peptidase inhibitor increases dialysate NAAG and reduces glutamate, aspartate and GABA levels in the dorsal hippocampus following fluid percussion injury in the rat. J. Neurochem. 2006, 97, 1015–1025. [Google Scholar] [CrossRef]
- Zuo, D.; Bzdega, T.; Olszewski, R.T.; Moffett, J.R.; Neale, J.H. Effects of N-acetylaspartylglutamate (NAAG) peptidase inhibition on release of glutamate and dopamine in prefrontal cortex and nucleus accumbens in phencyclidine model of schizophrenia. J. Biol. Chem. 2012, 287, 21773–21782. [Google Scholar] [CrossRef]
- Seven, A.B.; Barros-Álvarez, X.; de Lapeyrière, M.; Papasergi-Scott, M.M.; Robertson, M.J.; Zhang, C.; Nwokonko, R.M.; Gao, Y.; Meyerowitz, J.G.; Rocher, J.-P. G-protein activation by a metabotropic glutamate receptor. Nature 2021, 595, 450–454. [Google Scholar] [CrossRef]
- Uno, Y.; Coyle, J.T. Glutamate hypothesis in schizophrenia. Psychiatry Clin. Neurosci. 2019, 73, 204–215. [Google Scholar] [CrossRef]
- Neale, J.H.; Yamamoto, T. N-acetylaspartylglutamate (NAAG) and glutamate carboxypeptidase II: An abundant peptide neurotransmitter-enzyme system with multiple clinical applications. Prog. Neurobiol. 2020, 184, 101722. [Google Scholar] [CrossRef]
- Wroblewska, B.; Wroblewski, J.T.; Pshenichkin, S.; Surin, A.; Sullivan, S.E.; Neale, J.H. N-acetylaspartylglutamate selectively activates mGluR3 receptors in transfected cells. J. Neurochem. 1997, 69, 174–181. [Google Scholar] [CrossRef]
- Trombley, P.Q.; Westbrook, G.L. Excitatory synaptic transmission in cultures of rat olfactory bulb. J. Neurophysiol. 1990, 64, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Valivullah, H.M.; Lancaster, J.; Sweetnam, P.M.; Neale, J.H. Interactions between N-acetylaspartylglutamate and AMPA, kainate, and NMDA binding sites. J. Neurochem. 1994, 63, 1714–1719. [Google Scholar] [CrossRef] [PubMed]
- Fricker, A.C.; Mok, M.H.; de la Flor, R.; Shah, A.J.; Woolley, M.; Dawson, L.A.; Kew, J.N. Effects of N-acetylaspartylglutamate (NAAG) at group II mGluRs and NMDAR. Neuropharmacology 2009, 56, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Neale, J.H. N-acetylaspartylglutamate is an agonist at mGluR(3) in vivo and in vitro. J. Neurochem. 2011, 119, 891–895. [Google Scholar] [CrossRef]
- Mazzitelli, M.; Palazzo, E.; Maione, S.; Neugebauer, V. Group II metabotropic glutamate receptors: Role in pain mechanisms and pain modulation. Front. Mol. Neurosci. 2018, 11, 383. [Google Scholar] [CrossRef]
- Corti, C.; Crepaldi, L.; Mion, S.; Roth, A.L.; Xuereb, J.H.; Ferraguti, F. Altered dimerization of metabotropic glutamate receptor 3 in schizophrenia. Biol. Psychiatry 2007, 62, 747–755. [Google Scholar] [CrossRef]
- Tamaru, Y.; Nomura, S.; Mizuno, N.; Shigemoto, R. Distribution of metabotropic glutamate receptor mGluR3 in the mouse CNS: Differential location relative to pre- and postsynaptic sites. Neuroscience 2001, 106, 481–503. [Google Scholar] [CrossRef]
- Jin, L.E.; Wang, M.; Galvin, V.C.; Lightbourne, T.C.; Conn, P.J.; Arnsten, A.F.; Paspalas, C.D. mGluR2 versus mGluR3 metabotropic glutamate receptors in primate dorsolateral prefrontal cortex: Postsynaptic mGluR3 strengthen working memory networks. Cereb. Cortex 2018, 28, 974–987. [Google Scholar] [CrossRef]
- Mudo, G.; Trovato-Salinaro, A.; Caniglia, G.; Cheng, Q.; Condorelli, D.F. Cellular localization of mGluR3 and mGluR5 mRNAs in normal and injured rat brain. Brain Res. 2007, 1149, 1–13. [Google Scholar] [CrossRef]
- Khacho, P.; Wang, B.; Ahlskog, N.; Hristova, E.; Bergeron, R. Differential effects of N-acetyl-aspartyl-glutamate on synaptic and extrasynaptic NMDA receptors are subunit- and pH-dependent in the CA1 region of the mouse hippocampus. Neurobiol. Dis. 2015, 82, 580–592. [Google Scholar] [CrossRef]
- Nordengen, K.; Morland, C.; Slusher, B.; Gundersen, V. Dendritic localization and exocytosis of NAAG in the rat hippocampus. Cereb. Cortex 2020, 30, 1422–1435. [Google Scholar] [CrossRef] [PubMed]
- Monn, J.A.; Valli, M.J.; Massey, S.M.; Hansen, M.M.; Kress, T.J.; Wepsiec, J.P.; Harkness, A.R.; Grutsch, J.L.; Wright, R.A.; Johnson, B.G. Synthesis, pharmacological characterization, and molecular modeling of heterobicyclic amino acids related to (+)-2-aminobicyclo [3.1.0] hexane-2, 6-dicarboxylic acid (LY354740): Identification of two new potent, selective, and systemically active agonists for group II metabotropic glutamate receptors. J. Med. Chem. 1999, 42, 1027–1040. [Google Scholar]
- Zollinger, M.; Brauchli-Theotokis, J.; Gutteck-Amsler, U.; Do, K.Q.; Streit, P.; Cuenod, M. Release of N-acetylaspartylglutamate from slices of rat cerebellum, striatum, and spinal cord, and the effect of climbing fiber deprivation. J. Neurochem. 1994, 63, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Pittaluga, A.; Barbeito, L.; Serval, V.; Godeheu, G.; Artaud, F.; Glowinski, J.; Cheramy, A. Depolarization-evoked release of N-acetyl-L-aspartyl-L-glutamate from rat brain synaptosomes. Eur. J. Pharmacol. 1988, 158, 263–266. [Google Scholar] [CrossRef]
- Volgyi, K.; Gulyassy, P.; Todorov, M.I.; Puska, G.; Badics, K.; Hlatky, D.; Kekesi, K.A.; Nyitrai, G.; Czurko, A.; Drahos, L.; et al. Chronic Cerebral Hypoperfusion Induced Synaptic Proteome Changes in the rat Cerebral Cortex. Mol. Neurobiol. 2018, 55, 4253–4266. [Google Scholar] [CrossRef]
- Moffett, J.R.; Namboodiri, M.A.; Neale, J.H. Enhanced carbodiimide fixation for immunohistochemistry: Application to the comparative distributions of N-acetylaspartylglutamate and N-acetylaspartate immunoreactivities in rat brain. J. Histochem. Cytochem. Off. J. Histochem. Soc. 1993, 41, 559–570. [Google Scholar] [CrossRef]
- Williamson, L.C.; Neale, J.H. Ultrastructural localization of N-acetylaspartylglutamate in synaptic vesicles of retinal neurons. Brain Res. 1988, 456, 375–381. [Google Scholar] [CrossRef]
- Walder, K.K.; Ryan, S.B.; Bzdega, T.; Olszewski, R.T.; Neale, J.H.; Lindgren, C.A. Immunohistological and electrophysiological evidence that N-acetylaspartylglutamate is a co-transmitter at the vertebrate neuromuscular junction. Eur. J. Neurosci. 2013, 37, 118–129. [Google Scholar] [CrossRef]
- Lodder-Gadaczek, J.; Gieselmann, V.; Eckhardt, M. Vesicular uptake of N-acetylaspartylglutamate is catalysed by sialin (SLC17A5). Biochem. J. 2013, 454, 31–38. [Google Scholar] [CrossRef]
- Becker, I.; Wang-Eckhardt, L.; Lodder-Gadaczek, J.; Wang, Y.; Grünewald, A.; Eckhardt, M. Mice deficient in the NAAG synthetase II gene Rimkla are impaired in a novel object recognition task. J. Neurochem. 2021, 157, 2008–2023. [Google Scholar] [CrossRef]
- Zink, C.F.; Barker, P.B.; Sawa, A.; Weinberger, D.R.; Wang, M.; Quillian, H.; Ulrich, W.S.; Chen, Q.; Jaffe, A.E.; Kleinman, J.E. Association of missense mutation in FOLH1 with decreased NAAG levels and impaired working memory circuitry and cognition. Am. J. Psychiatry 2020, 177, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Zheng, Y.; Wang, X. Self-Regulation of Cerebral Metabolism and Its Neuroprotective Effect After Hypoxic-Ischemic Injury: Evidence From 1H-MRS. Front. Neuroanat. 2021, 15, 45. [Google Scholar] [CrossRef] [PubMed]
- Šácha, P.; Zámečník, J.; Bařinka, C.; Hlouchova, K.; Vicha, A.; Mlčochová, P.; Hilgert, I.; Eckschlager, T.; Konvalinka, J. Expression of glutamate carboxypeptidase II in human brain. Neuroscience 2007, 144, 1361–1372. [Google Scholar] [CrossRef] [PubMed]
- Datta, D.; Leslie, S.N.; Woo, E.; Amancharla, N.; Elmansy, A.; Lepe, M.; Mecca, A.P.; Slusher, B.S.; Nairn, A.C.; Arnsten, A.F. Glutamate Carboxypeptidase II in Aging Rat Prefrontal Cortex Impairs Working Memory Performance. Front. Aging Neurosci. 2021, 13, 760270. [Google Scholar] [CrossRef] [PubMed]
- Crupi, R.; Impellizzeri, D.; Cuzzocrea, S. Role of metabotropic glutamate receptors in neurological disorders. Front. Mol. Neurosci. 2019, 12, 20. [Google Scholar] [CrossRef]
- Krystal, J.H.; Abi-Saab, W.; Perry, E.; D’Souza, D.C.; Liu, N.; Gueorguieva, R.; McDougall, L.; Hunsberger, T.; Belger, A.; Levine, L. Preliminary evidence of attenuation of the disruptive effects of the NMDA glutamate receptor antagonist, ketamine, on working memory by pretreatment with the group II metabotropic glutamate receptor agonist, LY354740, in healthy human subjects. Psychopharmacology 2005, 179, 303–309. [Google Scholar] [CrossRef]
- Arnsten, A.F.; Wang, M. The Evolutionary Expansion of mGluR3-NAAG-GCPII Signaling: Relevance to Human Intelligence and Cognitive Disorders. Am. J. Psychiatry 2020, 177, 1103–1106. [Google Scholar] [CrossRef]
- Olszewski, R.T.; Janczura, K.J.; Bzdega, T.; Der, E.K.; Venzor, F.; O’Rourke, B.; Hark, T.J.; Craddock, K.E.; Balasubramanian, S.; Moussa, C. NAAG peptidase inhibitors act via mGluR3: Animal models of memory, Alzheimer’s, and ethanol intoxication. Neurochem. Res. 2017, 42, 2646–2657. [Google Scholar] [CrossRef]
- Menshchikov, P.; Ivantsova, A.; Manzhurtsev, A.; Ublinskiy, M.; Yakovlev, A.; Melnikov, I.; Kupriyanov, D.; Akhadov, T.; Semenova, N. Separate N-acetyl aspartyl glutamate, N-acetyl aspartate, aspartate, and glutamate quantification after pediatric mild traumatic brain injury in the acute phase. Magn. Reson. Med. 2020, 84, 2918–2931. [Google Scholar] [CrossRef]
- Jackson, P.F.; Cole, D.C.; Slusher, B.S.; Stetz, S.L.; Ross, L.E.; Donzanti, B.A.; Trainor, D.A. Design, synthesis, and biological activity of a potent inhibitor of the neuropeptidase N-acetylated α-linked acidic dipeptidase. J. Med. Chem. 1996, 39, 619–622. [Google Scholar] [CrossRef]
- Olszewski, R.T.; Bukhari, N.; Zhou, J.; Kozikowski, A.P.; Wroblewski, J.T.; Shamimi-Noori, S.; Wroblewska, B.; Bzdega, T.; Vicini, S.; Barton, F.B. NAAG peptidase inhibition reduces locomotor activity and some stereotypes in the PCP model of schizophrenia via group II mGluR. J. Neurochem. 2004, 89, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Majer, P.; Jackson, P.F.; Delahanty, G.; Grella, B.S.; Ko, Y.-S.; Li, W.; Liu, Q.; Maclin, K.M.; Poláková, J.; Shaffer, K.A. Synthesis and biological evaluation of thiol-based inhibitors of glutamate carboxypeptidase II: Discovery of an orally active GCP II inhibitor. J. Med. Chem. 2003, 46, 1989–1996. [Google Scholar] [CrossRef] [PubMed]
- Dash, R.P.; Tichý, T.s.; Veeravalli, V.; Lam, J.; Alt, J.; Wu, Y.; Tenora, L.s.; Majer, P.; Slusher, B.S.; Rais, R. Enhanced oral bioavailability of 2-(phosphonomethyl)-pentanedioic acid (2-PMPA) from its (5-methyl-2-oxo-1, 3-dioxol-4-yl) methyl (ODOL)-based prodrugs. Mol. Pharm. 2019, 16, 4292–4301. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.-F.; Van, K.C.; Gurkoff, G.G.; Kopriva, C.; Olszewski, R.T.; Song, M.; Sun, S.; Xu, M.; Neale, J.H.; Yuen, P.-W. Post-injury administration of NAAG peptidase inhibitor prodrug, PGI-02776, in experimental TBI. Brain Res. 2011, 1395, 62–73. [Google Scholar] [CrossRef]
- Jaarsma, D.; Veenma-van der Duin, L.; Korf, J. N-acetylaspartate and N-acetylaspartylglutamate levels in Alzheimer’s disease post-mortem brain tissue. J. Neurol. Sci. 1994, 127, 230–233. [Google Scholar] [CrossRef]
- Durand, D.; Carniglia, L.; Turati, J.; Ramírez, D.; Saba, J.; Caruso, C.; Lasaga, M. Amyloid-beta neurotoxicity and clearance are both regulated by glial group II metabotropic glutamate receptors. Neuropharmacology 2017, 123, 274–286. [Google Scholar] [CrossRef]
- Bukke, V.N.; Archana, M.; Villani, R.; Romano, A.D.; Wawrzyniak, A.; Balawender, K.; Orkisz, S.; Beggiato, S.; Serviddio, G.; Cassano, T. The dual role of glutamatergic neurotransmission in Alzheimer’s disease: From pathophysiology to pharmacotherapy. Int. J. Mol. Sci. 2020, 21, 7452. [Google Scholar] [CrossRef]
- Bonsi, P.; Cuomo, D.; Picconi, B.; Sciamanna, G.; Tscherter, A.; Tolu, M.; Bernardi, G.; Calabresi, P.; Pisani, A. Striatal metabotropic glutamate receptors as a target for pharmacotherapy in Parkinson’s disease. Amino Acids 2007, 32, 189–195. [Google Scholar] [CrossRef]
- Wang, J.; Wang, F.; Mai, D.; Qu, S. Molecular mechanisms of glutamate toxicity in Parkinson’s disease. Front. Neurosci. 2020, 14, 1201. [Google Scholar] [CrossRef]
- Golembiowska, K.; Konieczny, J.; Ossowska, K.; Wolfarth, S. The role of striatal metabotropic glutamate receptors in degeneration of dopamine neurons. Amino Acids 2002, 23, 199–205. [Google Scholar] [CrossRef]
- Testa, C.M.; Standaert, D.G.; Young, A.B.; Penney, J.B. Metabotropic glutamate receptor mRNA expression in the basal ganglia of the rat. J. Neurosci. 1994, 14, 3005–3018. [Google Scholar] [CrossRef] [PubMed]
- Samadi, P.; Rajput, A.; Calon, F.; Grégoire, L.; Hornykiewicz, O.; Rajput, A.H.; Di Paolo, T. Metabotropic glutamate receptor II in the brains of Parkinsonian patients. J. Neuropathol. Exp. Neurol. 2009, 68, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Li, X. Study on Effect of Striatal mGluR2/3 in Alleviating Motor Dysfunction in Rat PD Model Treated by Exercise Therapy. Front. Aging Neurosci. 2019, 11, 255. [Google Scholar] [CrossRef] [PubMed]
- Murray, T.K.; Messenger, M.J.; Ward, M.A.; Woodhouse, S.; Osborne, D.J.; Duty, S.; O’Neill, M.J. Evaluation of the mGluR2/3 agonist LY379268 in rodent models of Parkinson’s disease. Pharmacol. Biochem. Behav. 2002, 73, 455–466. [Google Scholar] [CrossRef]
- Johnson, K.A.; Conn, P.J.; Niswender, C.M. Glutamate receptors as therapeutic targets for Parkinson’s disease. CNS Neurol. Disord. Drug Targets 2009, 8, 475–491. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, G.; Busceti, C.L.; Pontarelli, F.; Biagioni, F.; Fornai, F.; Paparelli, A.; Bruno, V.; Ruggieri, S.; Nicoletti, F. Protective role of group-II metabotropic glutamate receptors against nigro-striatal degeneration induced by 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine in mice. Neuropharmacology 2003, 45, 155–166. [Google Scholar] [CrossRef]
- Green, J.L.; Dos Santos, W.F.; Fontana, A.C.K. Role of glutamate excitotoxicity and glutamate transporter EAAT2 in epilepsy: Opportunities for novel therapeutics development. Biochem. Pharmacol. 2021, 193, 114786. [Google Scholar] [CrossRef]
- Sarlo, G.L.; Holton, K.F. Brain concentrations of glutamate and GABA in human epilepsy: A review. Seizure 2021, 91, 213–227. [Google Scholar] [CrossRef]
- Celli, R.; Santolini, I.; Van Luijtelaar, G.; Ngomba, R.T.; Bruno, V.; Nicoletti, F. Targeting metabotropic glutamate receptors in the treatment of epilepsy: Rationale and current status. Expert Opin. Ther. Targets 2019, 23, 341–351. [Google Scholar] [CrossRef]
- Miyamoto, M.; Ishida, M.; Shinozaki, H. Anticonvulsive and neuroprotective actions of a potent agonist (DCG-IV) for group II metabotropic glutamate receptors against intraventricular kainate in the rat. Neuroscience 1997, 77, 131–140. [Google Scholar] [CrossRef]
- Attwell, P.; Kent, N.S.; Jane, D.; Croucher, M.; Bradford, H. Anticonvulsant and glutamate release-inhibiting properties of the highly potent metabotropic glutamate receptor agonist (2S, 2′ R, 3′ R)-2-(2′, 3′-dicarboxycyclopropyl) glycine (DCG-IV). Brain Res. 1998, 805, 138–143. [Google Scholar] [CrossRef]
- Caulder, E.H.; Riegle, M.A.; Godwin, D.W. Activation of group 2 metabotropic glutamate receptors reduces behavioral and electrographic correlates of pilocarpine induced status epilepticus. Epilepsy Res. 2014, 108, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Feng, Y.; Pang, Y.; Xu, J.; Yu, B.; Liu, X. Inhibitory effect of group II mGluR agonist 2R, 4R-APDC on cell proliferation in dentate gyrus in rats with epileptic seizure. Eur. Rev. Med. Pharm. Sci. 2015, 19, 2922–2927. [Google Scholar]
- Zhang, H.; Cilz, N.I.; Yang, C.; Hu, B.; Dong, H.; Lei, S. Depression of neuronal excitability and epileptic activities by group II metabotropic glutamate receptors in the medial entorhinal cortex. Hippocampus 2015, 25, 1299–1313. [Google Scholar] [CrossRef] [PubMed]
- Kłodzińska, A.; Bijak, M.; Chojnacka-Wojcik, E.; Kroczka, B.; Świąder, M.; Czuczwar, S.; Pilc, A. Roles of group II metabotropic glutamate receptors in modulation of seizure activity. Naunyn-Schmiedebergs Arch. Pharmacol. 2000, 361, 283–288. [Google Scholar] [CrossRef]
- Witkin, J.M.; Gasior, M.; Schad, C.; Zapata, A.; Shippenberg, T.; Hartman, T.; Slusher, B.S. NAALADase (GCP II) inhibition prevents cocaine-kindled seizures. Neuropharmacology 2002, 43, 348–356. [Google Scholar] [CrossRef]
- Meyerhoff, J.L.; Carter, R.E.; Yourick, D.L.; Slusher, B.S.; Coyle, J.T. Genetically epilepsy-prone rats have increased brain regional activity of an enzyme which liberates glutamate from N-acetyl-aspartyl-glutamate. Brain Res. 1992, 593, 140–143. [Google Scholar] [CrossRef]
- Otalora, L.F.P.; Moffett, J.R.; Garrido-Sanabria, E.R. Selective vulnerability of hippocampal NAAGergic neurons in experimental temporal lobe epilepsy. Brain Res. 2007, 1144, 219–230. [Google Scholar] [CrossRef]
- Garrido-Sanabria, E.R.; Otalora, L.F.P.; Arshadmansab, M.F.; Herrera, B.; Francisco, S.; Ermolinsky, B.S. Impaired expression and function of group II metabotropic glutamate receptors in pilocarpine-treated chronically epileptic rats. Brain Res. 2008, 1240, 165–176. [Google Scholar] [CrossRef]
- Choi, D.W. Excitotoxicity: Still hammering the ischemic brain in 2020. Front. Neurosci. 2020, 14, 579953. [Google Scholar] [CrossRef]
- Han, Y.; Yuan, M.; Guo, Y.-S.; Shen, X.-Y.; Gao, Z.-K.; Bi, X. Mechanism of Endoplasmic Reticulum Stress in Cerebral Ischemia. Front. Cell. Neurosci. 2021, 15, 704334. [Google Scholar] [CrossRef] [PubMed]
- Jelinek, M.; Jurajda, M.; Duris, K. Oxidative Stress in the Brain: Basic Concepts and Treatment Strategies in Stroke. Antioxidants 2021, 10, 1886. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Chen, H.; Wang, L.; Lenahan, C.; Lian, L.; Ou, Y.; He, Y. Mitochondrial Dynamics: A Potential Therapeutic Target for Ischemic Stroke. Front. Aging Neurosci. 2021, 13, 721428. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Gao, S.; Tu, S.; Lenahan, C.; Shao, A.; Sheng, J. Pathophysiology and therapeutic potential of NADPH oxidases in ischemic stroke-induced oxidative stress. Oxid. Med. Cell. Longev. 2021, 2021, 6631805. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Shao, L.; Ma, L. Cerebral edema formation after stroke: Emphasis on blood–brain barrier and the lymphatic drainage system of the brain. Front. Cell. Neurosci. 2021, 15, 716825. [Google Scholar] [CrossRef] [PubMed]
- García-Pupo, L.; Van San, E.; Delgado-Hernández, R.; Berghe, T.V.; Berghe, W.V. Emerging immune and cell death mechanisms in stroke: Saponins as therapeutic candidates. Brain Behav. Immun. Health 2020, 9, 100152. [Google Scholar] [CrossRef] [PubMed]
- Slusher, B.S.; Vornov, J.J.; Thomas, A.G.; Hurn, P.D.; Harukuni, I.; Bhardwaj, A.; Traystman, R.J.; Robinson, M.B.; Britton, P.; Lu, X.C.; et al. Selective inhibition of NAALADase, which converts NAAG to glutamate, reduces ischemic brain injury. Nat. Med. 1999, 5, 1396–1402. [Google Scholar] [CrossRef]
- Tortella, F.C.; Lin, Y.; Ved, H.; Slusher, B.S.; Dave, J.R. Neuroprotection produced by the NAALADase inhibitor 2-PMPA in rat cerebellar neurons. Eur. J. Pharmacol. 2000, 402, 31–37. [Google Scholar] [CrossRef]
- Bacich, D.J.; Wozniak, K.M.; Lu, X.C.; O’Keefe, D.S.; Callizot, N.; Heston, W.D.; Slusher, B.S. Mice lacking glutamate carboxypeptidase II are protected from peripheral neuropathy and ischemic brain injury. J. Neurochem. 2005, 95, 314–323. [Google Scholar] [CrossRef]
- Zhang, Z.; Bassam, B.; Thomas, A.G.; Williams, M.; Liu, J.; Nance, E.; Rojas, C.; Slusher, B.S.; Kannan, S. Maternal inflammation leads to impaired glutamate homeostasis and up-regulation of glutamate carboxypeptidase II in activated microglia in the fetal/newborn rabbit brain. Neurobiol. Dis. 2016, 94, 116–128. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, Z.; Wu, L.; Qiu, Y.; Lin, Y. Suppression of Glutamate Carboxypeptidase II Ameliorates Neuronal Apoptosis from Ischemic Brain Injury. J. Stroke Cerebrovasc. Dis. Off. J. Natl. Stroke Assoc. 2016, 25, 1599–1605. [Google Scholar] [CrossRef] [PubMed]
- Cabeza, O.A.; Zhang, Z.; Khoury, E.S.; Sheldon, R.; Sharma, A.; Zhang, F.; Slusher, B.; Kannan, R.; Kannan, S.; Ferriero, D. Neuroprotective effects of a dendrimer-based glutamate carboxypeptidase inhibitor on superoxide dismutase transgenic mice after neonatal hypoxic-ischemic brain injury. Neurobiol. Dis. 2021, 148, 105201. [Google Scholar] [CrossRef] [PubMed]
- Bratek, E.; Ziembowicz, A.; Bronisz, A.; Salinska, E. The activation of group II metabotropic glutamate receptors protects neonatal rat brains from oxidative stress injury after hypoxia-ischemia. PLoS ONE 2018, 13, e0200933. [Google Scholar] [CrossRef] [PubMed]
- Bratek-Gerej, E.; Bronisz, A.; Ziembowicz, A.; Salinska, E. Pretreatment with mGluR2 or mGluR3 Agonists Reduces Apoptosis Induced by Hypoxia-Ischemia in Neonatal Rat Brains. Oxidative Med. Cell. Longev. 2021, 2021, 8848015. [Google Scholar] [CrossRef]
- Bagri, K.; Kumar, P.; Deshmukh, R. Neurobiology of traumatic brain injury. Brain Inj. 2021, 35, 1113–1120. [Google Scholar] [CrossRef]
- Risbrough, V.; Vaughn, M.; Friend, S. Role of inflammation in TBI-associated risk for neuropsychiatric disorders: State of the evidence and where do we go from here. Biol. Psychiatry 2021, in press. [Google Scholar] [CrossRef]
- Velayudhan, P.S.; Schwab, N.; Hazrati, L.-N.; Wheeler, A.L. Temporal patterns of microglial activation in white matter following experimental mild traumatic brain injury: A systematic literature review. Acta Neuropathol. Commun. 2021, 9, 197. [Google Scholar] [CrossRef]
- Di Pietro, V.; Yakoub, K.M.; Caruso, G.; Lazzarino, G.; Signoretti, S.; Barbey, A.K.; Tavazzi, B.; Lazzarino, G.; Belli, A.; Amorini, A.M. Antioxidant therapies in traumatic brain injury. Antioxidants 2020, 9, 260. [Google Scholar] [CrossRef]
- Kim, S.; Han, S.C.; Gallan, A.J.; Hayes, J.P. Neurometabolic indicators of mitochondrial dysfunction in repetitive mild traumatic brain injury. Concussion 2017, 2, CNC45. [Google Scholar] [CrossRef]
- Kalimon, O.J.; Sullivan, P.G. Sex Differences in Mitochondrial Function Following a Controlled Cortical Impact Traumatic Brain Injury in Rodents. Front. Mol. Neurosci. 2021, 14, 753946. [Google Scholar] [CrossRef]
- Mira, R.G.; Lira, M.; Cerpa, W. Traumatic brain injury: Mechanisms of glial response. Front. Physiol. 2021, 12, 740939. [Google Scholar] [CrossRef] [PubMed]
- Veeramuthu, V.; Seow, P.; Narayanan, V.; Wong, J.H.D.; Tan, L.K.; Hernowo, A.T.; Ramli, N. Neurometabolites alteration in the acute phase of mild Traumatic Brain Injury (mTBI): An in vivo proton magnetic resonance spectroscopy (1H-MRS) Study. Acad. Radiol. 2018, 25, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Zhao, X.; Sarva, J.; Kozikowski, A.; Neale, J.H.; Lyeth, B.G. NAAG peptidase inhibitor reduces acute neuronal degeneration and astrocyte damage following lateral fluid percussion TBI in rats. J. Neurotrauma 2005, 22, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.-f.; Gurkoff, G.G.; Van, K.C.; Song, M.; Lowe, D.A.; Zhou, J.; Lyeth, B.G. NAAG peptidase inhibitor reduces cellular damage in a model of TBI with secondary hypoxia. Brain Res. 2012, 1469, 144–152. [Google Scholar] [CrossRef]
- Gurkoff, G.G.; Feng, J.-F.; Van, K.C.; Izadi, A.; Ghiasvand, R.; Shahlaie, K.; Song, M.; Lowe, D.A.; Zhou, J.; Lyeth, B.G. NAAG peptidase inhibitor improves motor function and reduces cognitive dysfunction in a model of TBI with secondary hypoxia. Brain Res. 2013, 1515, 98–107. [Google Scholar] [CrossRef]
- Cao, Y.; Gao, Y.; Xu, S.; Bao, J.; Lin, Y.; Luo, X.; Wang, Y.; Luo, Q.; Jiang, J.; Neale, J.H. Glutamate carboxypeptidase II gene knockout attenuates oxidative stress and cortical apoptosis after traumatic brain injury. BMC Neurosci. 2016, 17, 15. [Google Scholar] [CrossRef]
- Gao, Y.; Xu, S.; Cui, Z.; Zhang, M.; Lin, Y.; Cai, L.; Wang, Z.; Luo, X.; Zheng, Y.; Wang, Y. Mice lacking glutamate carboxypeptidase II develop normally, but are less susceptible to traumatic brain injury. J. Neurochem. 2015, 134, 340–353. [Google Scholar] [CrossRef]
- Li, M.; Liu, X.; Yue, H.; Xiong, W.; Gu, J.; Xu, M. Transplantation of N-acetyl aspartyl-glutamate synthetase-activated neural stem cells after experimental traumatic brain injury significantly improves neurological recovery. Cell. Physiol. Biochem. 2013, 32, 1776–1789. [Google Scholar] [CrossRef]
- Edinoff, A.N.; Fitz-Gerald, J.S.; Holland, K.A.A.; Reed, J.G.; Murnane, S.E.; Minter, S.G.; Kaye, A.J.; Cornett, E.M.; Imani, F.; Khademi, S.-H. Adjuvant drugs for peripheral nerve blocks: The role of NMDA antagonists, neostigmine, epinephrine, and sodium bicarbonate. Anesthesiol. Pain Med. 2021, 11, e117146. [Google Scholar] [CrossRef]
- Zhuo, M. Ionotropic glutamate receptors contribute to pain transmission and chronic pain. Neuropharmacology 2017, 112, 228–234. [Google Scholar] [CrossRef]
- Boxall, S.; Berthele, A.; Laurie, D.; Sommer, B.; Zieglgänsberger, W.; Urban, L.; Tölle, T. Enhanced expression of metabotropic glutamate receptor 3 messenger RNA in the rat spinal cord during ultraviolet irradiation induced peripheral inflammation. Neuroscience 1997, 82, 591–602. [Google Scholar] [CrossRef]
- Cangro, C.B.; Namboodiri, M.A.; Sklar, L.A.; Corigliano-Murphy, A.; Neale, J.H. Immunohistochemistry and biosynthesis of N-acetylaspartylglutamate in spinal sensory ganglia. J. Neurochem. 1987, 49, 1579–1588. [Google Scholar] [CrossRef] [PubMed]
- Carozzi, V.A.; Canta, A.; Oggioni, N.; Ceresa, C.; Marmiroli, P.; Konvalinka, J.; Zoia, C.; Bossi, M.; Ferrarese, C.; Tredici, G. Expression and distribution of ‘high affinity’ glutamate transporters GLT1, GLAST, EAAC1 and of GCPII in the rat peripheral nervous system. J. Anat. 2008, 213, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Laird, J.; Mason, G.; Webb, J.; Hill, R.; Hargreaves, R. Effects of a partial agonist and a full antagonist acting at the glycine site of the NMDA receptor on inflammation-induced mechanical hyperalgesia in rats. Br. J. Pharmacol. 1996, 117, 1487–1492. [Google Scholar] [CrossRef]
- Taniguchi, K.; Shinjo, K.; Mizutani, M.; Shimada, K.; Ishikawa, T.; Menniti, F.S.; Nagahisa, A. Antinociceptive activity of CP-101,606, an NMDA receptor NR2B subunit antagonist. Br. J. Pharmacol. 1997, 122, 809–812. [Google Scholar] [CrossRef] [PubMed]
- Zahn, P.K.; Brennan, T.J. Lack of effect of intrathecally administered N-methyl-D-aspartate receptor antagonists in a rat model for postoperative pain. J. Am. Soc. Anesthesiol. 1998, 88, 143–156. [Google Scholar] [CrossRef]
- Nozaki-Taguchi, N.; Yaksh, T.L. Pharmacology of Spinal Glutamatergic Receptors in Post–Thermal Injury–evoked Tactile Allodynia and Thermal Hyperalgesia. J. Am. Soc. Anesthesiol. 2002, 96, 617–626. [Google Scholar] [CrossRef]
- Nagel, J.; Belozertseva, I.; Greco, S.; Kashkin, V.; Malyshkin, A.; Jirgensons, A.; Shekunova, E.; Eilbacher, B.; Bespalov, A.; Danysz, W. Effects of NAAG peptidase inhibitor 2-PMPA in model chronic pain–relation to brain concentration. Neuropharmacology 2006, 51, 1163–1171. [Google Scholar] [CrossRef]
- Yamamoto, T.; Nozaki-Taguchi, N.; Sakashita, Y. Spinal N-acetyl-α-linked acidic dipeptidase (NAALADase) inhibition attenuates mechanical allodynia induced by paw carrageenan injection in the rat. Brain Res. 2001, 909, 138–144. [Google Scholar] [CrossRef]
- Yamamoto, T.; Nozaki-Taguchi, N.; Sakashita, Y.; Inagaki, T. Inhibition of spinal N-acetylated-α-linked acidic dipeptidase produces an antinociceptive effect in the rat formalin test. Neuroscience 2001, 102, 473–479. [Google Scholar] [CrossRef]
- Sharpe, E.; Kingston, A.; Lodge, D.; Monn, J.; Headley, P. Systemic pre-treatment with a group II mGlu agonist, LY379268, reduces hyperalgesia in vivo. Br. J. Pharmacol. 2002, 135, 1255–1262. [Google Scholar] [CrossRef][Green Version]
- Kozikowski, A.P.; Zhang, J.; Nan, F.; Petukhov, P.A.; Grajkowska, E.; Wroblewski, J.T.; Yamamoto, T.; Bzdega, T.; Wroblewska, B.; Neale, J.H. Synthesis of urea-based inhibitors as active site probes of glutamate carboxypeptidase II: Efficacy as analgesic agents. J. Med. Chem. 2004, 47, 1729–1738. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Hirasawa, S.; Wroblewska, B.; Grajkowska, E.; Zhou, J.; Kozikowski, A.; Wroblewski, J.; Neale, J.H. Antinociceptive effects of N-acetylaspartylglutamate (NAAG) peptidase inhibitors ZJ-11, ZJ-17 and ZJ-43 in the rat formalin test and in the rat neuropathic pain model. Eur. J. Neurosci. 2004, 20, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Vornov, J.J.; Wozniak, K.M.; Wu, Y.; Rojas, C.; Rais, R.; Slusher, B.S. Pharmacokinetics and pharmacodynamics of the glutamate carboxypeptidase II inhibitor 2-MPPA show prolonged alleviation of neuropathic pain through an indirect mechanism. J. Pharmacol. Exp. Ther. 2013, 346, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Neef, J.; Palacios, D.S. Progress in mechanistically novel treatments for schizophrenia. RSC Med. Chem. 2021, 12, 1459–1475. [Google Scholar] [CrossRef] [PubMed]
- Egerton, A.; Grace, A.A.; Stone, J.; Bossong, M.G.; Sand, M.; McGuire, P. Glutamate in schizophrenia: Neurodevelopmental perspectives and drug development. Schizophr. Res. 2020, 223, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Stahl, S.M. Beyond the dopamine hypothesis of schizophrenia to three neural networks of psychosis: Dopamine, serotonin, and glutamate. CNS Spectr. 2018, 23, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, G.P.; Neill, J.C. Modelling the cognitive and neuropathological features of schizophrenia with phencyclidine. J. Psychopharmacol. 2016, 30, 1141–1144. [Google Scholar] [CrossRef]
- Kantrowitz, J.T.; Woods, S.W.; Petkova, E.; Cornblatt, B.; Corcoran, C.M.; Chen, H.; Silipo, G.; Javitt, D.C. D-serine for the treatment of negative symptoms in individuals at clinical high risk of schizophrenia: A pilot, double-blind, placebo-controlled, randomised parallel group mechanistic proof-of-concept trial. Lancet Psychiatry 2015, 2, 403–412. [Google Scholar] [CrossRef]
- Dallérac, G.; Li, X.; Lecouflet, P.; Morisot, N.; Sacchi, S.; Asselot, R.; Pham, T.H.; Potier, B.; Watson, D.J.; Schmidt, S. Dopaminergic neuromodulation of prefrontal cortex activity requires the NMDA receptor coagonist d-serine. Proc. Natl. Acad. Sci. USA 2021, 118, e2023750118. [Google Scholar] [CrossRef]
- Romei, C.; Raiteri, M.; Raiteri, L. Glycine release is regulated by metabotropic glutamate receptors sensitive to mGluR2/3 ligands and activated by N-acetylaspartylglutamate (NAAG). Neuropharmacology 2013, 66, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Saini, S.; Mancuso, S.; Mostaid, M.S.; Liu, C.; Pantelis, C.; Everall, I.; Bousman, C. Meta-analysis supports GWAS-implicated link between GRM3 and schizophrenia risk. Transl. Psychiatry 2017, 7, e1196. [Google Scholar] [CrossRef] [PubMed]
- Egan, M.F.; Straub, R.E.; Goldberg, T.E.; Yakub, I.; Callicott, J.H.; Hariri, A.R.; Mattay, V.S.; Bertolino, A.; Hyde, T.M.; Shannon-Weickert, C. Variation in GRM3 affects cognition, prefrontal glutamate, and risk for schizophrenia. Proc. Natl. Acad. Sci. USA 2004, 101, 12604–12609. [Google Scholar] [CrossRef] [PubMed]
- Cartmell, J.; Monn, J.A.; Schoepp, D.D. The metabotropic glutamate 2/3 receptor agonists LY354740 and LY379268 selectively attenuate phencyclidine versus d-amphetamine motor behaviors in rats. J. Pharmacol. Exp. Ther. 1999, 291, 161–170. [Google Scholar]
- Rorick-Kehn, L.M.; Johnson, B.G.; Knitowski, K.M.; Salhoff, C.R.; Witkin, J.M.; Perry, K.W.; Griffey, K.I.; Tizzano, J.P.; Monn, J.A.; McKinzie, D.L. In vivo pharmacological characterization of the structurally novel, potent, selective mGlu2/3 receptor agonist LY404039 in animal models of psychiatric disorders. Psychopharmacology 2007, 193, 121–136. [Google Scholar] [CrossRef]
- Ghose, S.; Gleason, K.A.; Potts, B.W.; Lewis-Amezcua, K.; Tamminga, C.A. Differential expression of metabotropic glutamate receptors 2 and 3 in schizophrenia: A mechanism for antipsychotic drug action? Am. J. Psychiatry 2009, 166, 812–820. [Google Scholar] [CrossRef]
- Ghose, S.; Chin, R.; Gallegos, A.; Roberts, R.; Coyle, J.; Tamminga, C. Localization of NAAG-related gene expression deficits to the anterior hippocampus in schizophrenia. Schizophr. Res. 2009, 111, 131–137. [Google Scholar] [CrossRef][Green Version]
- Neale, J.H.; Olszewski, R. A role for N-acetylaspartylglutamate (NAAG) and mGluR3 in cognition. Neurobiol. Learn. Mem. 2019, 158, 9–13. [Google Scholar] [CrossRef]
- Olszewski, R.T.; Bzdega, T.; Neale, J.H. mGluR3 and not mGluR2 receptors mediate the efficacy of NAAG peptidase inhibitor in validated model of schizophrenia. Schizophr. Res. 2012, 136, 160–161. [Google Scholar] [CrossRef]
- Swain, Y.; Gewirtz, J.C.; Harris, A.C. Behavioral predictors of individual differences in opioid addiction vulnerability as measured using iv self-administration in rats. Drug Alcohol Depend. 2021, 221, 108561. [Google Scholar] [CrossRef]
- Ma, B.; Mei, D.; Wang, F.; Liu, Y.; Zhou, W. Cognitive enhancers as a treatment for heroin relapse and addiction. Pharmacol. Res. 2019, 141, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Van der Post, J.; De Visser, S.; De Kam, M.; Woelfler, M.; Hilt, D.; Vornov, J.; Burak, E.; Bortey, E.; Slusher, B.; Limsakun, T. The central nervous system effects, pharmacokinetics and safety of the NAALADase-inhibitor GPI 5693. Br. J. Clin. Pharmacol. 2005, 60, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Vornov, J.J.; Peters, D.; Nedelcovych, M.; Hollinger, K.; Rais, R.; Slusher, B.S. Looking for drugs in all the wrong places: Use of GCPII inhibitors outside the brain. Neurochem. Res. 2019, 45, 1256–1267. [Google Scholar] [CrossRef] [PubMed]
- Date, A.A.; Rais, R.; Babu, T.; Ortiz, J.; Kanvinde, P.; Thomas, A.G.; Zimmermann, S.C.; Gadiano, A.J.; Halpert, G.; Slusher, B.S. Local enema treatment to inhibit FOLH1/GCPII as a novel therapy for inflammatory bowel disease. J. Control. Release 2017, 263, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Kirsch, B.J.; Asaka, R.; Nabi, K.; Quinones, A.; Tan, J.; Antonio, M.J.; Camelo, F.; Li, T.; Nguyen, S. Uncovering the role of N-acetyl-aspartyl-glutamate as a glutamate reservoir in cancer. Cell Rep. 2019, 27, 491–501.e496. [Google Scholar] [CrossRef]
- Asaka, R.; Le, A. Dual role of N-acetyl-aspartyl-glutamate metabolism in cancer monitor and therapy. Mol. Cell. Oncol. 2019, 6, e1627273. [Google Scholar] [CrossRef]
- Guo, S.; Gu, Y.; Qu, J.; Le, A. Bridging the Metabolic Parallels Between Neurological Diseases and Cancer. In The Heterogeneity of Cancer Metabolism; Springer: Cham, Switzerland, 2021; pp. 229–248. [Google Scholar]
- Nedelcovych, M.; Dash, R.P.; Tenora, L.s.; Zimmermann, S.C.; Gadiano, A.J.; Garrett, C.; Alt, J.; Hollinger, K.R.; Pommier, E.; Jančařík, A. Enhanced brain delivery of 2-(phosphonomethyl) pentanedioic acid following intranasal administration of its γ-substituted ester prodrugs. Mol. Pharm. 2017, 14, 3248–3257. [Google Scholar] [CrossRef]
- Rais, R.; Wozniak, K.; Wu, Y.; Niwa, M.; Stathis, M.; Alt, J.; Giroux, M.; Sawa, A.; Rojas, C.; Slusher, B.S. Selective CNS uptake of the GCP-II inhibitor 2-PMPA following intranasal administration. PLoS ONE 2015, 10, e0131861. [Google Scholar] [CrossRef][Green Version]



{kind=link}
{kind=link}
{kind=link}
| Inhibitor | IC50 | Ki | Measuring System | Crosses BBB | References |
|---|---|---|---|---|---|
| 2-PMPA: 2-(phosphonomethyl)pentanedioic acid) | - | 275 pM | Synaptosomal protein preparation; detection of [3H]Glu produced from [3H]NAAG | No | Jackson et al., 1996 [40] |
| 4.1 nM | 1.4 nM | Chinese hamster ovary (CHO) cell transfected with the cloned human GCPII; fluorescence detection of produced glutamate. | Olszewski et al., 2004 [41] | ||
| 2-MPPA: 2-(3-Mercaptopropyl)pentanedioic acid | 30 nM | 90 nM | Purified human recombinant GCPII; detection of [3H]Glu produced from [3H]NAAG | No | Majer et al., 2003 [42] |
| Tetra-ODOL-prodrug for 2-PMPA | Enhanced oral bioavalability; effective after conversion to 2-PMPA | Peroral bioavailability testes in CD-1 mica and beagle dogs | Yes | Dash at el., 2019 [43] | |
| ZJ43: (S)-2-[3-[(S)-1-carboxy-3-methylbutyl]ureido]pentanedioic acid | 2.4 nM | 0.8 nM (GCPIII: 23 nM) | Chinese hamster ovary (CHO) cell transfected with the cloned human GCPII or GCPIII; fluorescence detection of produced glutamate. | Yes | Olszewski et al., 2004 [41] |
| PGI02749: Mono-ester prodrug of ZJ43 | Weak effect per se. Effective after conversion to ZJ43 | Chinese hamster ovary (CHO) cell transfected with the cloned human GCPII or GCPIII; fluorescence detection of produced glutamate. | Yes | Feng et al., 2011 [44] | |
| PGI-02776: Di-ester prodrug of ZJ43 | Moderate effect per se (IC50 ~100 µM). Effective after conversion to ZJ43 | Chinese hamster ovary (CHO) cell transfected with the cloned human GCPII or GCPIII; fluorescence detection of produced glutamate. | Yes | Feng et al., 2011 [44] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morland, C.; Nordengen, K. N-Acetyl-Aspartyl-Glutamate in Brain Health and Disease. Int. J. Mol. Sci. 2022, 23, 1268. https://doi.org/10.3390/ijms23031268
Morland C, Nordengen K. N-Acetyl-Aspartyl-Glutamate in Brain Health and Disease. International Journal of Molecular Sciences. 2022; 23(3):1268. https://doi.org/10.3390/ijms23031268
Chicago/Turabian StyleMorland, Cecilie, and Kaja Nordengen. 2022. "N-Acetyl-Aspartyl-Glutamate in Brain Health and Disease" International Journal of Molecular Sciences 23, no. 3: 1268. https://doi.org/10.3390/ijms23031268
APA StyleMorland, C., & Nordengen, K. (2022). N-Acetyl-Aspartyl-Glutamate in Brain Health and Disease. International Journal of Molecular Sciences, 23(3), 1268. https://doi.org/10.3390/ijms23031268