
A MicroRNA Next-Generation-Sequencing Discovery Assay (miND) for Genome-Scale Analysis and Absolute Quantitation of Circulating MicroRNA Biomarkers

 , , , , , ,
, , , , , ,
Abstract
1. Introduction
2. Results
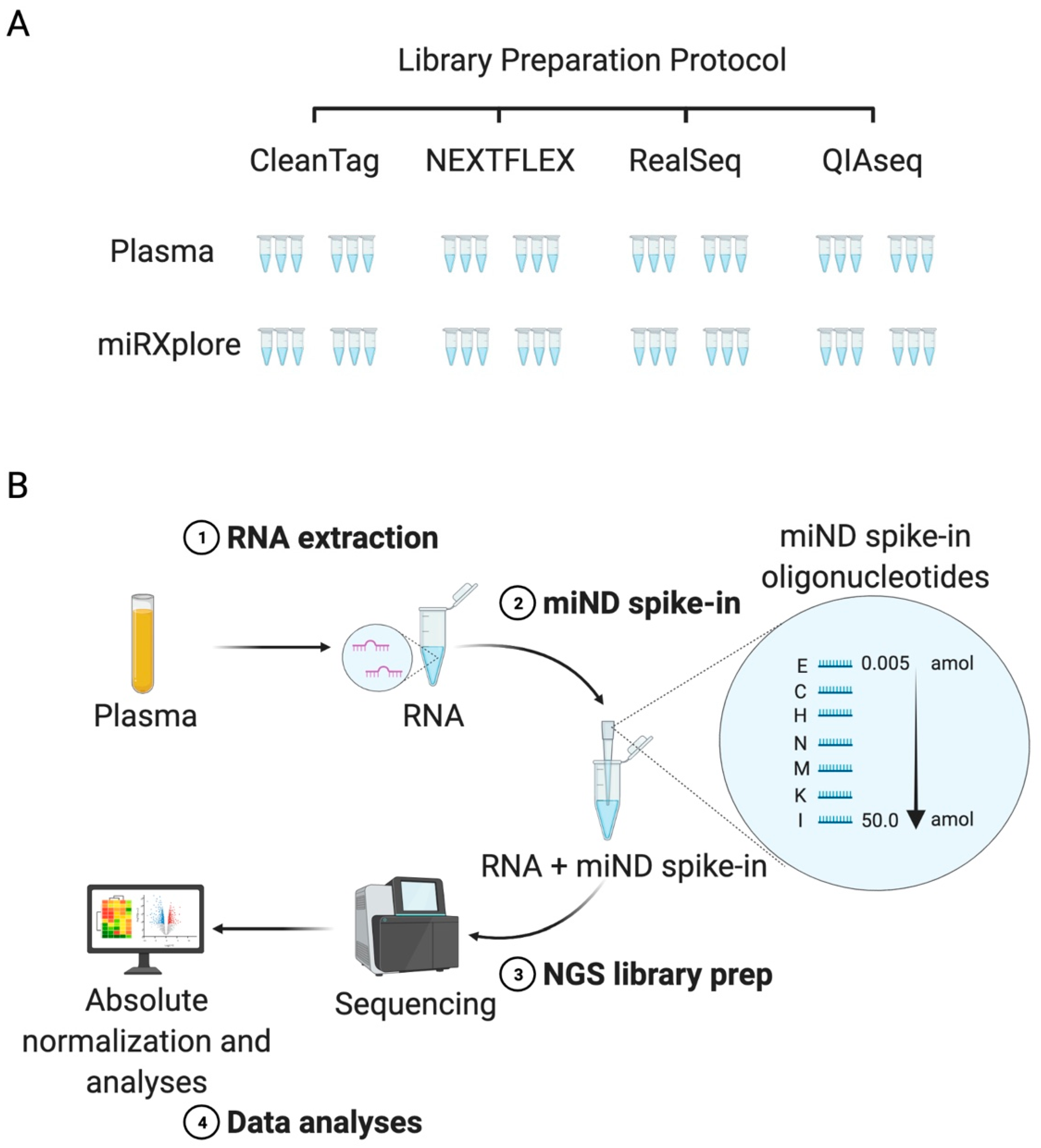
2.1. Study Design, Selection of Commercial Kits, and Reference Material
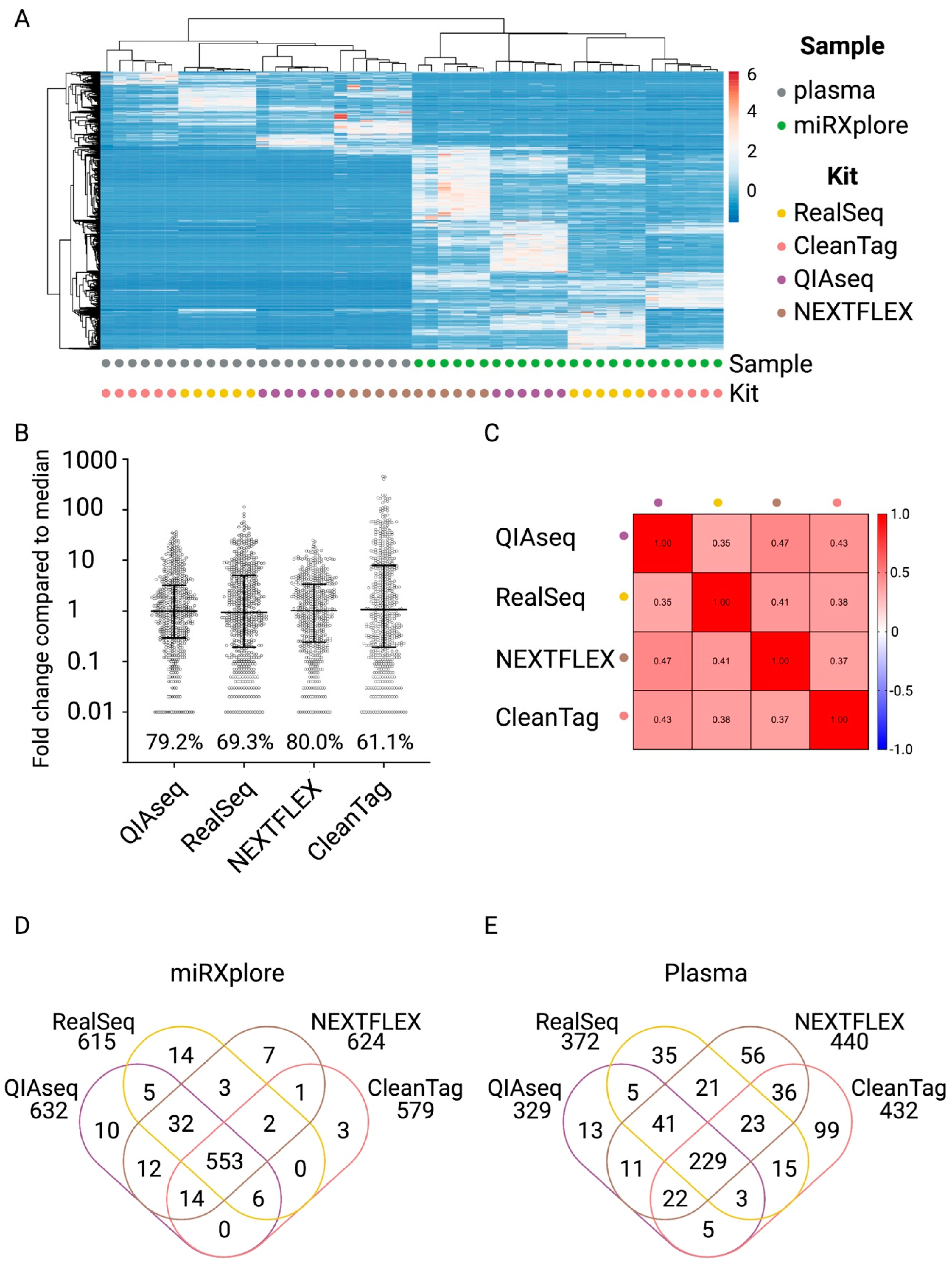
2.2. Characterization of Four Small RNA Sequencing Protocols and Selection of the Protocol for the miND Assay
2.3. Design and Testing of the miND Spike-In for Quality Control and Absolute Normalisation of Small RNA Sequencing Data
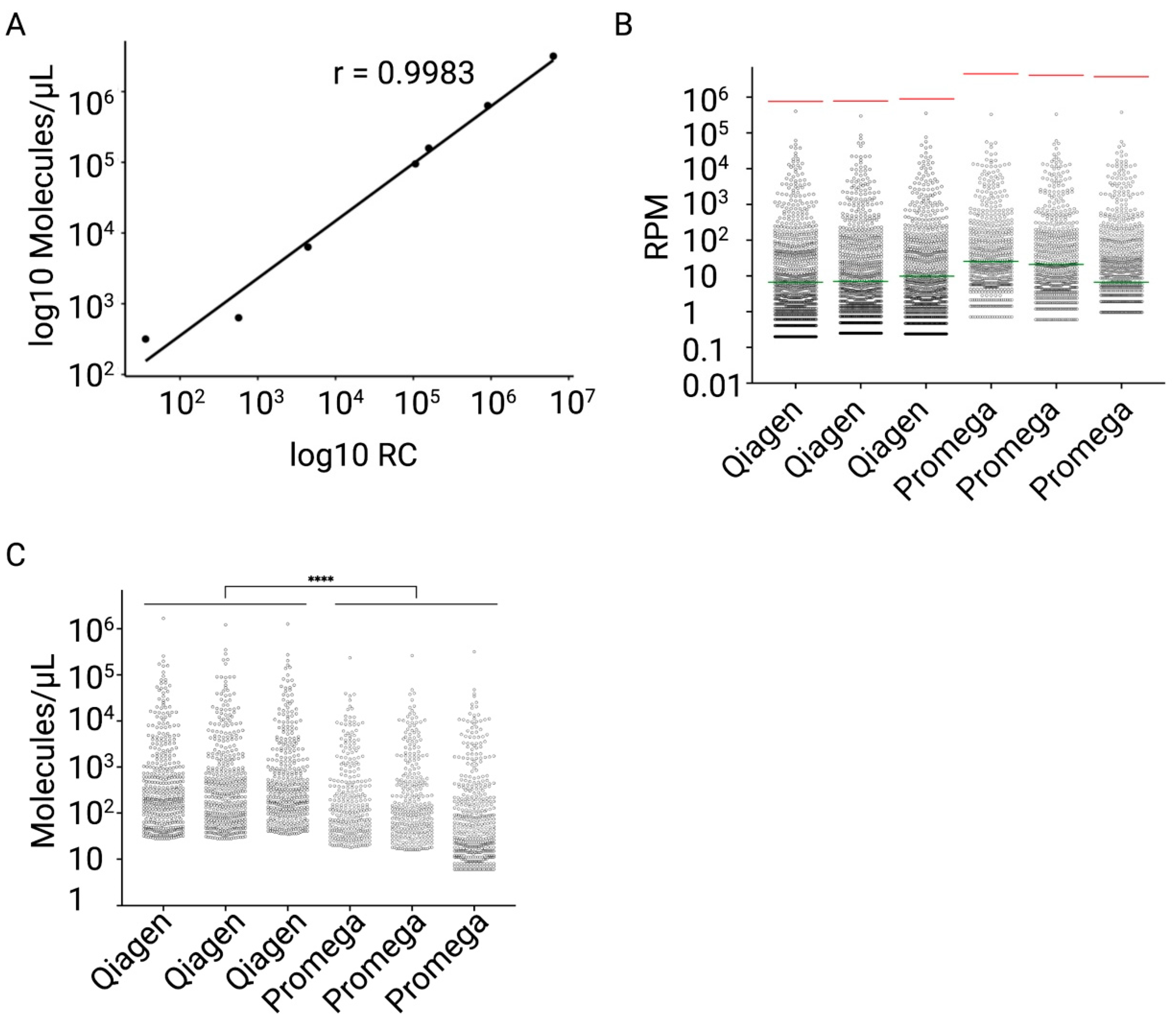
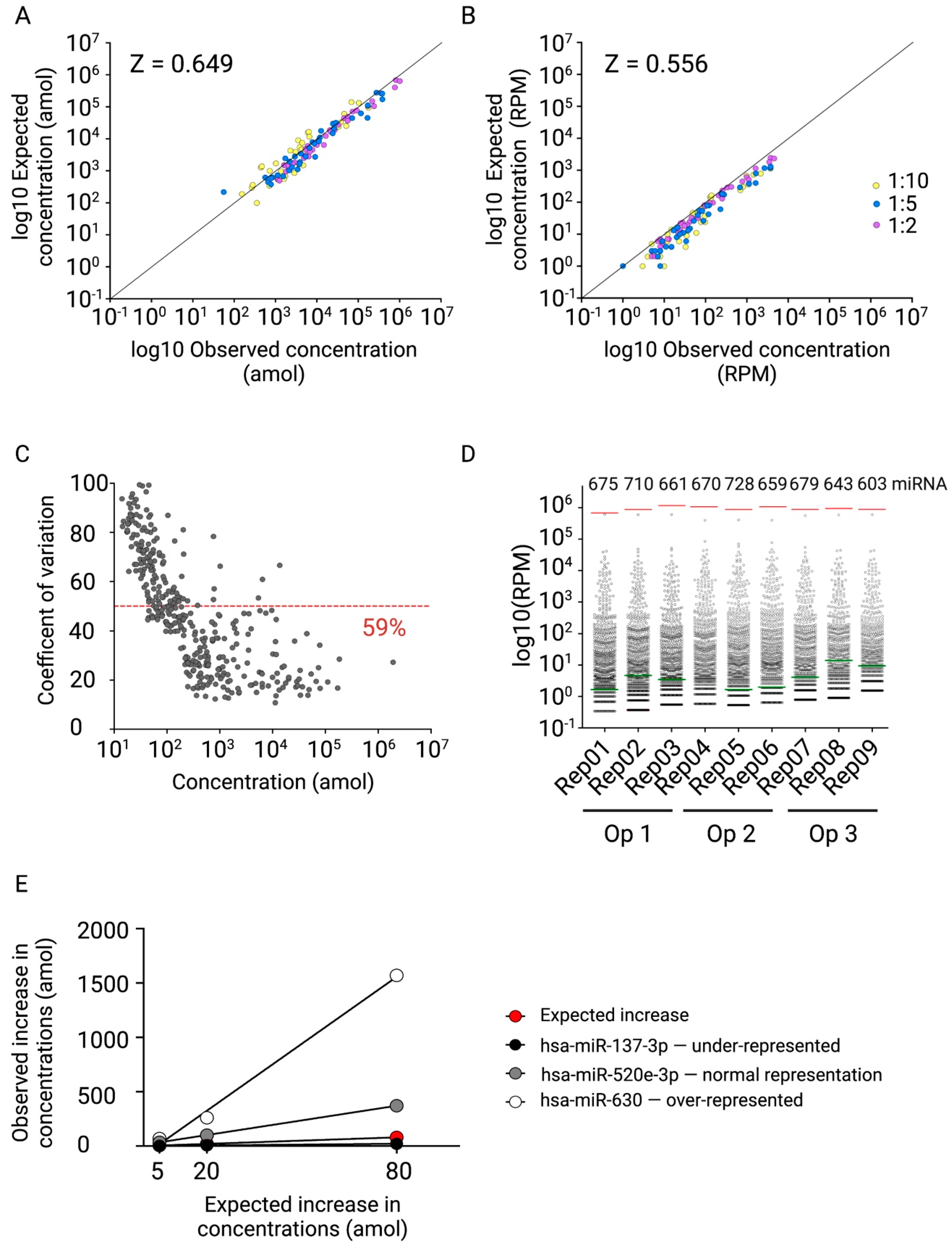
2.4. Fit-for-Purpose Validation of the Established NGS Protocol
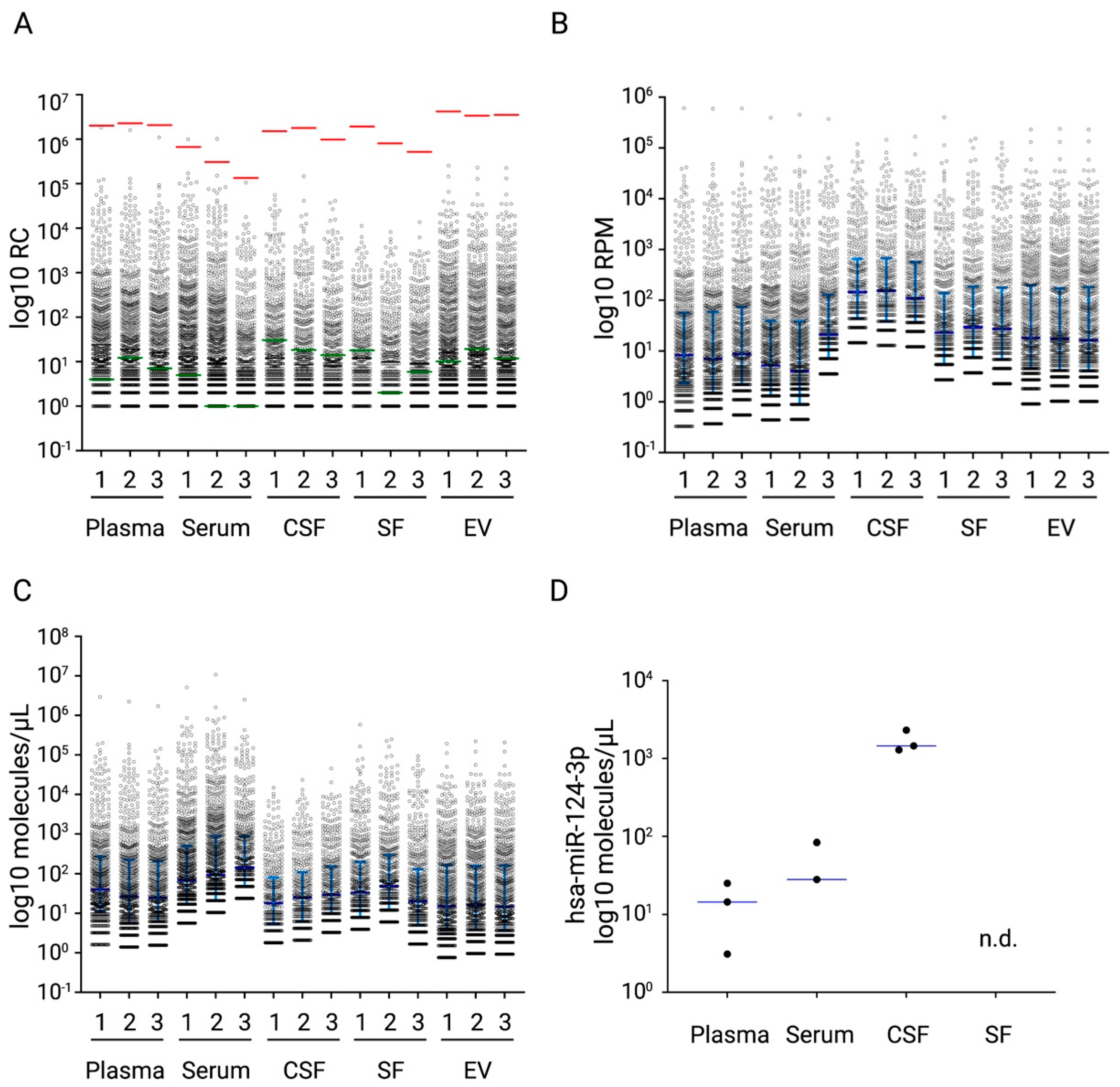
2.5. NGS Analyses of Diverse Biological Samples with the miND Spike-In Assay
3. Discussion
The Value of Spike-Ins for Small RNA Sequencing
4. Materials and Methods
4.1. Samples
4.2. Design of the miND Spike-Ins
4.3. RNA Extraction
4.4. NGS Library Preparation
4.5. RT-qPCR
4.6. Bioinformatic Analyses
4.7. Data Analyses and Statistical Methods
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| APAP | acetaminophen-induced liver injury |
| CSF | cerebrospinal fluid |
| EV | extracellular vesicles |
| FDR | false discovery rate |
| IQR | inter-quartile range |
| NGS | Next generation sequencing |
| NHV | Normal healthy volunteers |
| PPP | platelet-poor plasma |
| RPM | reads per million genome-matching reads |
| SF | synovial fluid |
| UMI | Unique Molecular Indices |
References
- Weber, J.A.; Baxter, D.H.; Zhang, S.; Huang, D.Y.; How Huang, K.; Jen Lee, M.; Galas, D.J.; Wang, K. The microRNA spectrum in 12 body fluids. Clin. Chem. 2010, 56, 1733–1741. [Google Scholar] [CrossRef] [PubMed]
- Godoy, P.M.; Bhakta, N.R.; Barczak, A.J.; Cakmak, H.; Fisher, S.; MacKenzie, T.C.; Patel, T.; Price, R.W.; Smith, J.F.; Woodruff, P.G.; et al. Large differences in small RNA composition between human biofluids. Cell Rep. 2018, 25, 1346–1358. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, C.; Khamina, K.; Theofilatos, K.; Diendorfer, A.B.; Burnap, S.A.; Nabeebaccus, A.; Fish, M.; McPhail, M.J.W.; O’Gallagher, K.; Schmidt, L.E.; et al. Association of cardiometabolic microRNAs with COVID-19 severity and mortality. Cardiovasc. Res. 2021, cvab338. [Google Scholar] [CrossRef] [PubMed]
- Corsten, M.F.; Dennert, R.; Jochems, S.; Kuznetsova, T.; Devaux, Y.; Hofstra, L.; Wagner, D.R.; Staessen, J.A.; Heymans, S.; Schroen, B. Circulating MicroRNA-208b and MicroRNA-499 reflect myocardial damage in cardiovascular disease. Circ. Cardiovasc. Genet. 2010, 3, 499–506. [Google Scholar] [CrossRef]
- Kingsley, S.M.K.; Bhat, B.V. Role of microRNAs in sepsis. Inflamm. Res. 2017, 66, 553–569. [Google Scholar] [CrossRef]
- Nahand, J.S.; Taghizadeh-Boroujeni, S.; Karimzadeh, M.; Borran, S.; Pourhanifeh, M.H.; Moghoofei, M.; Bokharaei-Salim, F.; Karampoor, S.; Jafari, A.; Asemi, Z.; et al. MicroRNAs: New prognostic, diagnostic, and therapeutic biomarkers in cervical cancer. J. Cell. Physiol. 2019, 234, 17064–17099. [Google Scholar] [CrossRef]
- Macha, M.; Seshacharyulu, P.; Krishn, S.R.; Pai, P.; Rachagani, S.; Jain, M.; Batra, S. MicroRNAs (miRNAs) as biomarker(s) for prognosis and diagnosis of gastrointestinal (GI) cancers. Curr. Pharm. Des. 2014, 20, 5287–5297. [Google Scholar] [CrossRef]
- Larrea, E.; Sole, C.; Manterola, L.; Goicoechea, I.; Armesto, M.; Arestin, M.; Caffarel, M.M.; Araujo, A.M.; Araiz, M.; Fernandez-Mercado, M.; et al. New concepts in cancer biomarkers: Circulating miRNAs in liquid biopsies. Int. J. Mol. Sci. 2016, 17, 627. [Google Scholar] [CrossRef]
- Wang, J.; Chen, J.; Sen, S. MicroRNA as biomarkers and diagnostics: MicroRNAs as biomarkers for diagnostics. J. Cell. Physiol. 2016, 231, 25–30. [Google Scholar] [CrossRef]
- Howell, L.; Ireland, L.; Park, B.K.; Goldring, C.E. MiR-122 and other microRNAs as potential circulating biomarkers of drug-induced liver injury. Expert Rev. Mol. Diagn. 2018, 18, 47–54. [Google Scholar] [CrossRef]
- Laterza, O.F.; Lim, L.; Garrett-Engele, P.W.; Vlasakova, K.; Muniappa, N.; Tanaka, W.K.; Johnson, J.M.; Sina, J.F.; Fare, T.L.; Sistare, F.D.; et al. Plasma MicroRNAs as sensitive and specific biomarkers of tissue injury. Clin. Chem. 2009, 55, 1977–1983. [Google Scholar] [CrossRef] [PubMed]
- Erdos, Z.; Barnum, J.E.; Wang, E.; DeMaula, C.; Dey, P.M.; Forest, T.; Bailey, W.J.; Glaab, W.E. Evaluation of the relative performance of pancreas-specific MicroRNAs in rat plasma as biomarkers of pancreas injury. Toxicol. Sci. 2020, 173, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Schofield, A.L.; Brown, J.P.; Brown, J.; Wilczynska, A.; Bell, C.; Glaab, W.E.; Hackl, M.; Howell, L.; Lee, S.; Dear, J.W.; et al. Systems analysis of miRNA biomarkers to inform drug safety. Arch. Toxicol. 2021, 95, 3475–3495. [Google Scholar] [CrossRef] [PubMed]
- Schraml, E.; Hackl, M.; Grillari, J. MicroRNAs and toxicology: A love marriage. Toxicol. Rep. 2017, 4, 634–636. [Google Scholar] [CrossRef] [PubMed]
- Buschmann, D.; Haberberger, A.; Kirchner, B.; Spornraft, M.; Riedmaier, I.; Schelling, G.; Pfaffl, M.W. Toward reliable biomarker signatures in the age of liquid biopsies—How to standardize the small RNA-Seq workflow. Nucleic Acids Res. 2016, 44, 5995–6018. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Landgraf, P.; Rusu, M.; Sheridan, R.; Sewer, A.; Iovino, N.; Aravin, A.; Pfeffer, S.; Rice, A.; Kamphorst, A.O.; Landthaler, M.; et al. A Mammalian microRNA expression atlas based on small RNA library sequencing. Cell 2007, 129, 1401–1414. [Google Scholar] [CrossRef]
- De Rie, D.; Abugessaisa, I.; Alam, T.; Arner, E.; Arner, P.; Ashoor, H.; Åström, G.; Babina, M.; Bertin, N.; Burroughs, A.M.; et al. An integrated expression atlas of miRNAs and their promoters in human and mouse. Nat. Biotechnol. 2017, 35, 872–878. [Google Scholar] [CrossRef]
- Murillo, O.D.; Thistlethwaite, W.; Rozowsky, J.; Subramanian, S.L.; Lucero, R.; Shah, N.; Jackson, A.R.; Srinivasan, S.; Chung, A.; Laurent, C.D.; et al. exRNA Atlas analysis reveals distinct extracellular RNA cargo types and their carriers present across human biofluids. Cell 2019, 177, 463–477.e15. [Google Scholar] [CrossRef]
- Wright, C.; Rajpurohit, A.; Burke, E.E.; Williams, C.; Collado-Torres, L.; Kimos, M.; Brandon, N.J.; Cross, A.J.; Jaffe, A.E.; Weinberger, D.R.; et al. Comprehensive assessment of multiple biases in small RNA sequencing reveals significant differences in the performance of widely used methods. BMC Genom. 2019, 20, 513. [Google Scholar] [CrossRef]
- Fuchs, R.T.; Sun, Z.; Zhuang, F.; Robb, G.B. Bias in ligation-based small RNA sequencing library construction is determined by adaptor and RNA structure. PLoS ONE 2015, 10, e0126049. [Google Scholar] [CrossRef] [PubMed]
- Raabe, C.A.; Tang, T.-H.; Brosius, J.; Rozhdestvensky, T.S. Biases in small RNA deep sequencing data. Nucleic Acids Res. 2014, 42, 1414–1426. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.U.; Pfaffl, M.W.; Ulbrich, S.E. Normalization strategies for microRNA profiling experiments: A “normal” way to a hidden layer of complexity? Biotechnol. Lett. 2010, 32, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Wu, P.-H.; Beane, T.; Zamore, P.D.; Weng, Z. Elimination of PCR duplicates in RNA-seq and small RNA-seq using unique molecular identifiers. BMC Genom. 2018, 19, 531. [Google Scholar] [CrossRef]
- Barberán-Soler, S.; Vo, J.M.; Hogans, R.E.; Dallas, A.; Johnston, B.H.; Kazakov, S.A. Decreasing miRNA sequencing bias using a single adapter and circularization approach. Genome Biol. 2018, 19, 105. [Google Scholar] [CrossRef]
- Mussbacher, M.; Krammer, T.L.; Heber, S.; Schrottmaier, W.C.; Zeibig, S.; Holthoff, H.-P.; Pereyra, D.; Starlinger, P.; Hackl, M.; Assinger, A. Impact of anticoagulation and sample processing on the quantification of human blood-derived microRNA signatures. Cells 2020, 9, 1915. [Google Scholar] [CrossRef]
- Lutzmayer, S.; Enugutti, B.; Nodine, M.D. Novel small RNA spike-in oligonucleotides enable absolute normalization of small RNA-Seq data. Sci. Rep. 2017, 7, 5913. [Google Scholar] [CrossRef]
- Wong, R.K.Y.; MacMahon, M.; Woodside, J.V.; Simpson, D.A. A comparison of RNA extraction and sequencing protocols for detection of small RNAs in plasma. BMC Genom. 2019, 20, 446. [Google Scholar] [CrossRef]
- Campbell, J.D.; Liu, G.; Luo, L.; Xiao, J.; Gerrein, J.; Juan-Guardela, B.; Tedrow, J.; Alekseyev, Y.O.; Yang, I.V.; Correll, M.; et al. Assessment of microRNA differential expression and detection in multiplexed small RNA sequencing data. RNA 2015, 21, 164–171. [Google Scholar] [CrossRef]
- Krauskopf, J.; De Kok, T.M.; Schomaker, S.J.; Gosink, M.; Burt, D.A.; Chandler, P.; Warner, R.L.; Johnson, K.J.; Caiment, F.; Kleinjans, J.C.; et al. Serum microRNA signatures as “liquid biopsies” for interrogating hepatotoxic mechanisms and liver pathogenesis in human. PLoS ONE 2017, 12, e0177928. [Google Scholar] [CrossRef]
- The External RNA Controls Consortium. The External RNA Controls Consortium: A progress report. Nat. Methods 2005, 2, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Munro, S.A.; Lund, S.P.; Pine, P.S.; Binder, H.; Clevert, D.-A.; Conesa, A.; Dopazo, J.; Fasold, M.; Hochreiter, S.; Hong, H.; et al. Assessing technical performance in differential gene expression experiments with external spike-in RNA control ratio mixtures. Nat. Commun. 2014, 5, 5125. [Google Scholar] [CrossRef] [PubMed]
- Plotnikova, A.; Kellner, M.J.; Schon, M.A.; Mosiolek, M.; Nodine, M.D. MicroRNA Dynamics and Functions during Arabidopsis Embryogenesis. Plant Cell 2019, 31, 2929–2946. [Google Scholar] [CrossRef]
- Chomczynski, P.; Wilfinger, W.W.; Eghbalnia, H.R.; Kennedy, A.; Rymaszewski, M.; Mackey, K. Inter-individual differences in RNA levels in human peripheral blood. PLoS ONE 2016, 11, e0148260. [Google Scholar] [CrossRef] [PubMed]
- Baldrich, P.; Tamim, S.; Mathioni, S.; Meyers, B. Ligation bias is a major contributor to nonstoichiometric abundances of secondary siRNAs and impacts analyses of microRNAs. bioRxiv 2020. [Google Scholar] [CrossRef]
- Buchtele, N.; Kovacevic, K.D.; Brostjan, C.; Schwameis, M.; Hayden, H.; Derhaschnig, U.; Firbas, C. Differential Osteoprotegerin Kinetics after Stimulation with Desmopressin and Lipopolysaccharides In Vivo. Thromb Haemost 2020, 120, 1108–1115. [Google Scholar] [CrossRef]
- Andrews, S. Babraham Bioinformatics—FastQC a Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 13 December 2021).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Girón, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2018, 46, D754–D761. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S. The microRNA registry. Nucleic Acids Res. 2004, 32, D109–D111. [Google Scholar] [CrossRef] [PubMed]
- The RNAcentral Consortium. RNAcentral: A hub of information for non-coding RNA sequences. Nucleic Acids Res. 2019, 47, D221–D229. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Seqtk. Available online: https://github.com/lh3/seqtk (accessed on 11 September 2021).
- Excel Microsoft Office. Available online: https://office.microsoft.com/excel (accessed on 13 December 2021).
- GraphPad Software. Available online: https://www.graphpad.com (accessed on 13 December 2021).
- Biorender.com. Available online: https://biorender.com (accessed on 13 December 2021).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oligo | Sequence (5′–3′) | Molar Amount (amol) |
|---|---|---|
| I | (N)(N)(N)(N)ACGAUCGGCUCUA(N)(N)(N)(N) | 50 |
| K | (N)(N)(N)(N)UGAACGUCCGUAC(N)(N)(N)(N) | 10 |
| M | (N)(N)(N)(N)UCUCGCGCGCGUU(N)(N)(N)(N) | 2.5 |
| N | (N)(N)(N)(N)CGAGUAAUGAACG(N)(N)(N)(N) | 1.5 |
| H | (N)(N)(N)(N)GCUACACACGUCG(N)(N)(N)(N) | 0.1 |
| C | (N)(N)(N)(N)UAUUCGCGGUGAC(N)(N)(N)(N) | 0.01 |
| E | (N)(N)(N)(N)ACCUCCGUUUACG(N)(N)(N)(N) | 0.005 |
| Biofluid | Sample | Median (Q3–Q1) | IQR | Number of Detected MicroRNAs | The 5 Most Abundant MicroRNAs on Average |
|---|---|---|---|---|---|
| Plasma | 1 | 6 (41.3–1.7) | 39.6 | 668 | miR-451a, miR-16-5p, miR-486-5p, miR-92a-3p, miR-103a-3p |
| 2 | 4.1 (33.6–0.8) | 32.8 | 707 | ||
| 3 | 3.8 (31.2–0.9) | 30.3 | 658 | ||
| Serum | 1 | 10.2 (76.7–2.6) | 74.1 | 900 | miR-451a, miR-16-5p, miR-92a-3p, miR-486-5p, miR-19b-3p |
| 2 | 14.1 (134.6–3.2) | 131.4 | 925 | ||
| 3 | 21.5 (131.1–7.2) | 123.9 | 600 | ||
| Synovial Fluid | 1 | 9.9 (30.0–1.2) | 57.6 | 548 | miR-21-5p, miR-23a-3p, miR-451a, miR-221-3p, miR-223-3p |
| 2 | 14.4 (45.3–1.8) | 87.0 | 530 | ||
| 3 | 6.0 (19.5–0.8) | 37.5 | 552 | ||
| Cerebrospinal Fluid | 1 | 2.7 (12.2–0.9) | 11.3 | 387 | miR-21-5p, miR-204-5p, miR-145-5p, miR-99a-5p, miR-221-3p |
| 2 | 3.8 (16.4–0.9) | 15.5 | 388 | ||
| 3 | 4.5 (23.1–1.5) | 21.6 | 467 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khamina, K.; Diendorfer, A.B.; Skalicky, S.; Weigl, M.; Pultar, M.; Krammer, T.L.; Fournier, C.A.; Schofield, A.L.; Otto, C.; Smith, A.T.; et al. A MicroRNA Next-Generation-Sequencing Discovery Assay (miND) for Genome-Scale Analysis and Absolute Quantitation of Circulating MicroRNA Biomarkers. Int. J. Mol. Sci. 2022, 23, 1226. https://doi.org/10.3390/ijms23031226
Khamina K, Diendorfer AB, Skalicky S, Weigl M, Pultar M, Krammer TL, Fournier CA, Schofield AL, Otto C, Smith AT, et al. A MicroRNA Next-Generation-Sequencing Discovery Assay (miND) for Genome-Scale Analysis and Absolute Quantitation of Circulating MicroRNA Biomarkers. International Journal of Molecular Sciences. 2022; 23(3):1226. https://doi.org/10.3390/ijms23031226
Chicago/Turabian StyleKhamina, Kseniya, Andreas B. Diendorfer, Susanna Skalicky, Moritz Weigl, Marianne Pultar, Teresa L. Krammer, Catharine Aquino Fournier, Amy L. Schofield, Carolin Otto, Aaron Thomas Smith, and et al. 2022. "A MicroRNA Next-Generation-Sequencing Discovery Assay (miND) for Genome-Scale Analysis and Absolute Quantitation of Circulating MicroRNA Biomarkers" International Journal of Molecular Sciences 23, no. 3: 1226. https://doi.org/10.3390/ijms23031226
APA StyleKhamina, K., Diendorfer, A. B., Skalicky, S., Weigl, M., Pultar, M., Krammer, T. L., Fournier, C. A., Schofield, A. L., Otto, C., Smith, A. T., Buchtele, N., Schoergenhofer, C., Jilma, B., Frank, B. J. H., Hofstaetter, J. G., Grillari, R., Grillari, J., Ruprecht, K., Goldring, C. E., ... Hackl, M. (2022). A MicroRNA Next-Generation-Sequencing Discovery Assay (miND) for Genome-Scale Analysis and Absolute Quantitation of Circulating MicroRNA Biomarkers. International Journal of Molecular Sciences, 23(3), 1226. https://doi.org/10.3390/ijms23031226