
Detecting Bacterial–Human Lateral Gene Transfer in Chronic Lymphocytic Leukemia

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
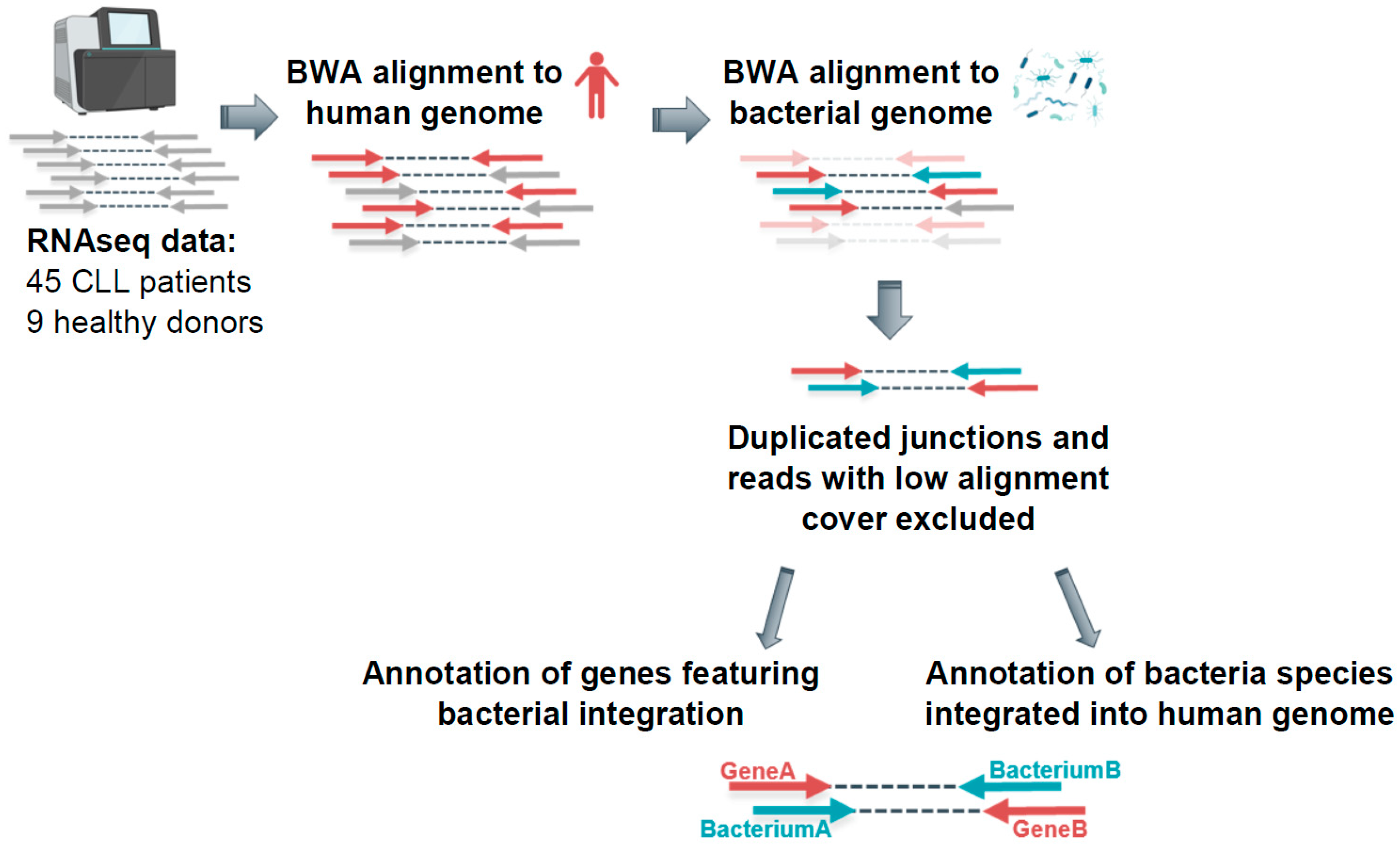
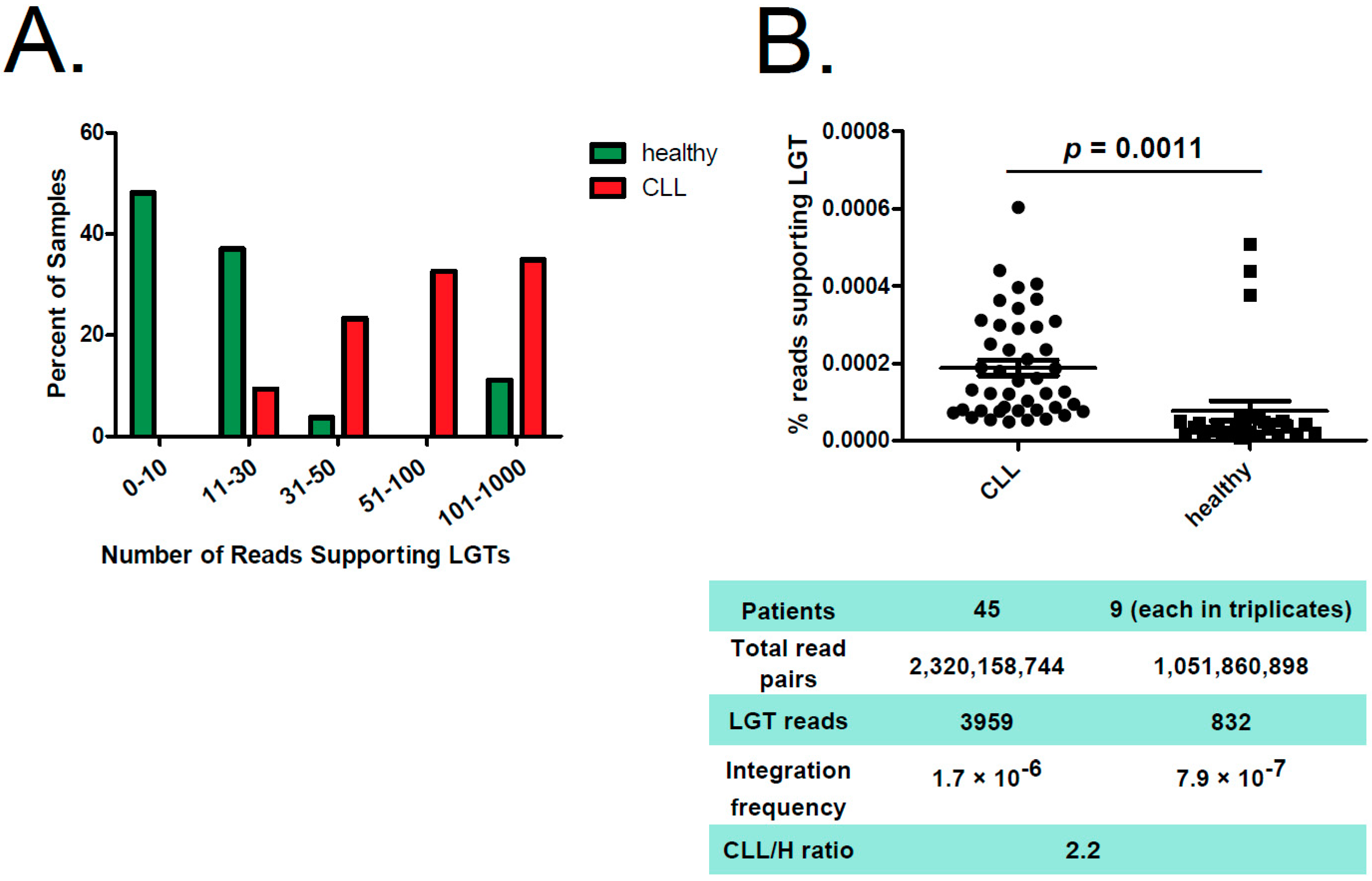
2.1. Detecting Bacterial Integrations into the Human Genome in CLL Patients
2.2. Reads Supporting Bacterial–Human LGT were Enriched in CLL versus Non-Leukemic B Cells
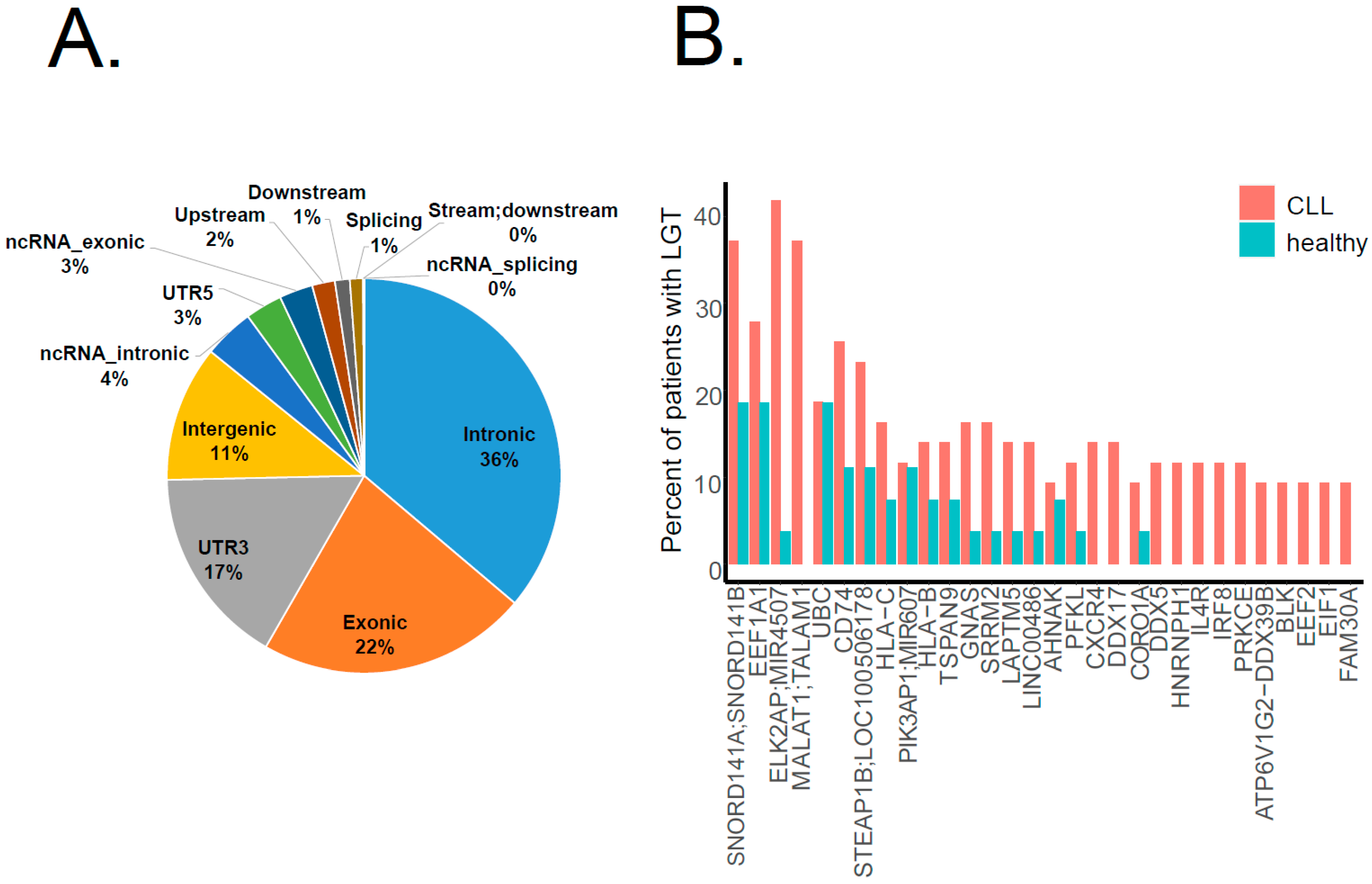
2.3. Bacterial Integrations Occurred Recurrently in Several Genes
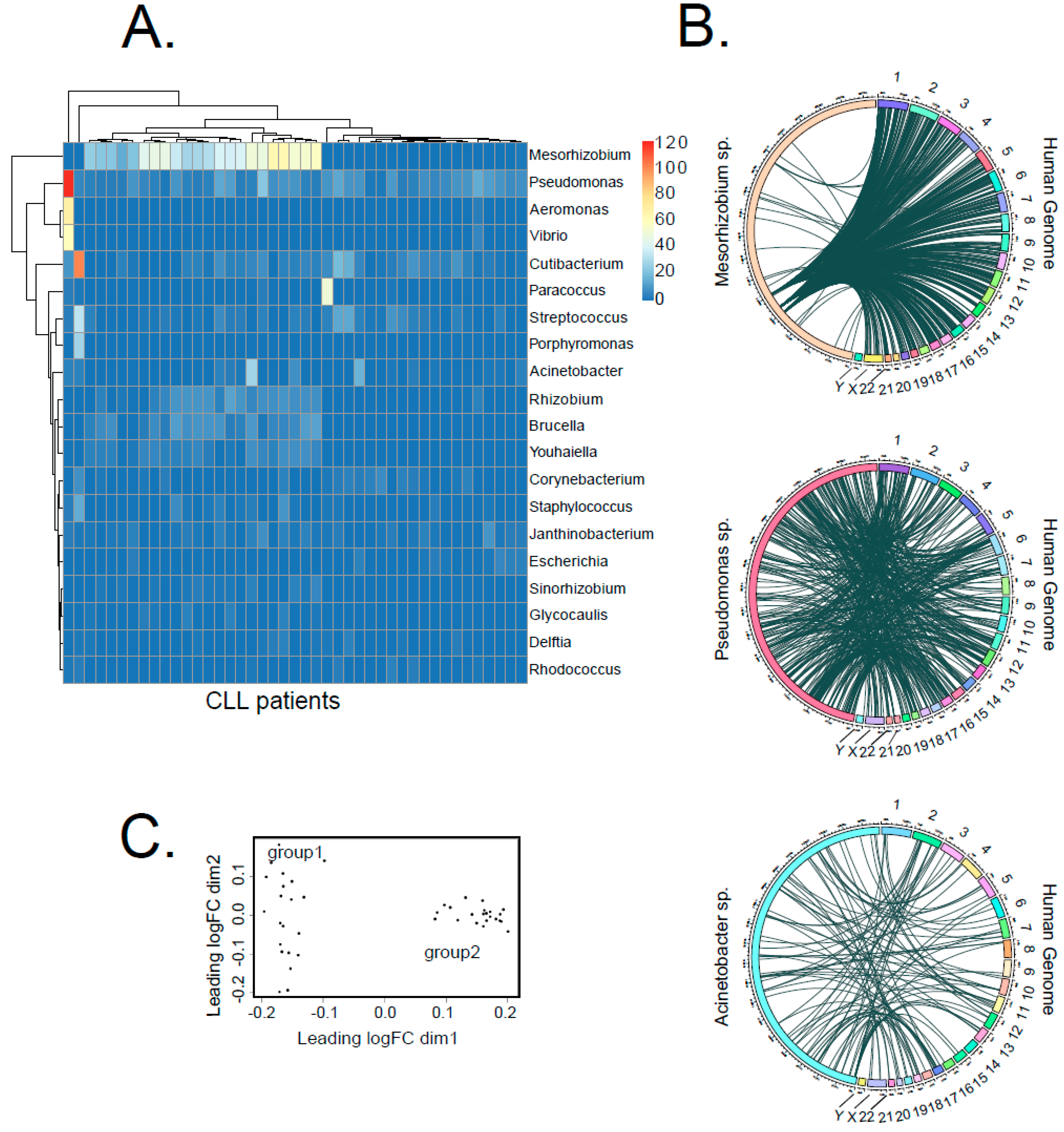
2.4. Some Bacteria Genera Appear to Integrate into Human Genome with Higher Likelihood
2.5. Correlation of LGT Events with Clinically Relevant Parameters
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pleyer, L.; Egle, A.; Hartmann, T.N.; Greil, R. Molecular and Cellular Mechanisms of CLL: Novel Therapeutic Approaches. Nat. Rev. Clin. Oncol. 2009, 6, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Landau, D.A.; Carter, S.L.; Stojanov, P.; McKenna, A.; Stevenson, K.; Lawrence, M.S.; Sougnez, C.; Stewart, C.; Sivachenko, A.; Wang, L.; et al. Evolution and Impact of Subclonal Mutations in Chronic Lymphocytic Leukemia. Cell 2013, 152, 714–726. [Google Scholar] [CrossRef] [Green Version]
- Landau, D.A.; Tausch, E.; Taylor-Weiner, A.N.; Stewart, C.; Reiter, J.G.; Bahlo, J.; Kluth, S.; Bozic, I.; Lawrence, M.; Böttcher, S.; et al. Mutations Driving CLL and Their Evolution in Progression and Relapse. Nature 2015, 526, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Chai-Adisaksopha, C.; Brown, J.R. FCR Achieves Long-Term Durable Remissions in Patients with IGHV-Mutated CLL. Blood 2017, 130, 2278–2282. [Google Scholar] [CrossRef]
- Visentin, A.; Facco, M.; Gurrieri, C.; Pagnin, E.; Martini, V.; Imbergamo, S.; Frezzato, F.; Trimarco, V.; Severin, F.; Raggi, F.; et al. Prognostic and Predictive Effect of IGHV Mutational Status and Load in Chronic Lymphocytic Leukemia: Focus on FCR and BR Treatments. Clin. Lymphoma Myeloma Leuk. 2019, 19, 678–685.e4. [Google Scholar] [CrossRef] [PubMed]
- Kreuzer, K.-A.; Furman, R.R.; Stilgenbauer, S.; Dubowy, R.L.; Kim, Y.; Munugalavadla, V.; Lilienweiss, E.; Reinhardt, H.C.; Cramer, P.; Eichhorst, B.; et al. The Impact of Complex Karyotype on the Overall Survival of Patients with Relapsed Chronic Lymphocytic Leukemia Treated with Idelalisib plus Rituximab. Leukemia 2020, 34, 296–300. [Google Scholar] [CrossRef] [Green Version]
- Hancks, D.C.; Kazazian, H.H. Active Human Retrotransposons: Variation and Disease. Curr. Opin. Genet. Dev. 2012, 22, 191–203. [Google Scholar] [CrossRef] [Green Version]
- Ju, Y.S.; Tubio, J.M.C.; Mifsud, W.; Fu, B.; Davies, H.R.; Ramakrishna, M.; Li, Y.; Yates, L.; Gundem, G.; Tarpey, P.S.; et al. Frequent Somatic Transfer of Mitochondrial DNA into the Nuclear Genome of Human Cancer Cells. Genome Res. 2015, 25, 814–824. [Google Scholar] [CrossRef] [Green Version]
- Pett, M.; Coleman, N. Integration of High-Risk Human Papillomavirus: A Key Event in Cervical Carcinogenesis? J. Pathol. 2007, 212, 356–367. [Google Scholar] [CrossRef]
- Sung, W.-K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-Wide Survey of Recurrent HBV Integration in Hepatocellular Carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.M.; Sieber, K.B.; Dunning Hotopp, J.C. A Review of Bacteria-Animal Lateral Gene Transfer May Inform Our Understanding of Diseases like Cancer. PLoS Genet. 2013, 9, e1003877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global Burden of Cancer Attributable to Infections in 2018: A Worldwide Incidence Analysis. Lancet Glob. Health 2020, 8, e180–e190. [Google Scholar] [CrossRef] [Green Version]
- Schwabe, R.F.; Wang, T.C. Bacteria Deliver a Genotoxic Hit. Science 2012, 338, 52–53. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.C.; Perez-Chanona, E.; Mühlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.-J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal Inflammation Targets Cancer-Inducing Activity of the Microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, D.R.; Sieber, K.B.; Robinson, K.M.; White, J.R.; Ganesan, A.; Nourbakhsh, S.; Hotopp, J.C.D. Bacteria-Human Somatic Cell Lateral Gene Transfer Is Enriched in Cancer Samples. PLoS Comput. Biol. 2013, 9, e1003107. [Google Scholar] [CrossRef]
- Sun, Y.; Ma, L. New Insights into Long Non-Coding RNA MALAT1 in Cancer and Metastasis. Cancers 2019, 11, 216. [Google Scholar] [CrossRef] [Green Version]
- Jima, D.D.; Zhang, J.; Jacobs, C.; Richards, K.L.; Dunphy, C.H.; Choi, W.W.L.; Yan Au, W.; Srivastava, G.; Czader, M.B.; Rizzieri, D.A.; et al. Deep Sequencing of the Small RNA Transcriptome of Normal and Malignant Human B Cells Identifies Hundreds of Novel MicroRNAs. Blood 2010, 116, e118–e127. [Google Scholar] [CrossRef] [Green Version]
- Stein, R.; Mattes, M.J.; Cardillo, T.M.; Hansen, H.J.; Chang, C.-H.; Burton, J.; Govindan, S.; Goldenberg, D.M. CD74: A New Candidate Target for the Immunotherapy of B-Cell Neoplasms. Clin. Cancer Res. 2007, 13, 5556s–5563s. [Google Scholar] [CrossRef] [Green Version]
- Starlets, D.; Gore, Y.; Binsky, I.; Haran, M.; Harpaz, N.; Shvidel, L.; Becker-Herman, S.; Berrebi, A.; Shachar, I. Cell-Surface CD74 Initiates a Signaling Cascade Leading to Cell Proliferation and Survival. Blood 2006, 107, 4807–4816. [Google Scholar] [CrossRef]
- Morishita, Y.; Shimizu, T. Promoting Effect of Intestinal Pseudomonas Aeruginosa on Gastric Tumorigenesis in Rats with N-Methyl-N′-Nitro-N-Nitrosoguanidine. Cancer Lett. 1983, 17, 347–352. [Google Scholar] [CrossRef]
- Avilés-Jiménez, F.; Guitron, A.; Segura-López, F.; Méndez-Tenorio, A.; Iwai, S.; Hernández-Guerrero, A.; Torres, J. Microbiota Studies in the Bile Duct Strongly Suggest a Role for Helicobacter Pylori in Extrahepatic Cholangiocarcinoma. Clin. Microbiol. Infect. 2016, 22, 178.e11–178.e22. [Google Scholar] [CrossRef] [Green Version]
- Fournier, G.P.; Andam, C.P.; Gogarten, J.P. Ancient Horizontal Gene Transfer and the Last Common Ancestors. BMC Evol. Biol. 2015, 15, 70. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.T.; Seifert, H.S. Opportunity and Means: Horizontal Gene Transfer from the Human Host to a Bacterial Pathogen. MBio 2011, 2, e00005-11. [Google Scholar] [CrossRef] [Green Version]
- Crisp, A.; Boschetti, C.; Perry, M.; Tunnacliffe, A.; Micklem, G. Expression of Multiple Horizontally Acquired Genes Is a Hallmark of Both Vertebrate and Invertebrate Genomes. Genome Biol. 2015, 16, 50. [Google Scholar] [CrossRef] [Green Version]
- Sieber, K.B.; Bromley, R.E.; Dunning Hotopp, J.C. Lateral Gene Transfer between Prokaryotes and Eukaryotes. Exp. Cell Res. 2017, 358, 421–426. [Google Scholar] [CrossRef]
- Gassner, F.J.; Schubert, M.; Rebhandl, S.; Spandl, K.; Zaborsky, N.; Catakovic, K.; Blaimer, S.; Hebenstreit, D.; Greil, R.; Geisberger, R. Imprecision and DNA Break Repair Biased towards Incompatible End Joining in Leukemia. Mol. Cancer Res. 2018, 16, 428–438. [Google Scholar] [CrossRef] [Green Version]
- Blauwkamp, T.A.; Thair, S.; Rosen, M.J.; Blair, L.; Lindner, M.S.; Vilfan, I.D.; Kawli, T.; Christians, F.C.; Venkatasubrahmanyam, S.; Wall, G.D.; et al. Analytical and Clinical Validation of a Microbial Cell-Free DNA Sequencing Test for Infectious Disease. Nat. Microbiol. 2019, 4, 663–674. [Google Scholar] [CrossRef]
- Zozaya-Valdés, E.; Wong, S.Q.; Raleigh, J.; Hatzimihalis, A.; Ftouni, S.; Papenfuss, A.T.; Sandhu, S.; Dawson, M.A.; Dawson, S.-J. Detection of Cell-Free Microbial DNA Using a Contaminant-Controlled Analysis Framework. Genome Biol. 2021, 22, 187. [Google Scholar] [CrossRef] [PubMed]
- Bessman, N.J.; Sonnenberg, G.F. Emerging Roles for Antigen Presentation in Establishing Host-Microbiome Symbiosis. Immunol. Rev. 2016, 272, 139–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gassner, F.J.; Zaborsky, N.; Buchumenski, I.; Levanon, E.Y.; Gatterbauer, M.; Schubert, M.; Rauscher, S.; Hebenstreit, D.; Nadeu, F.; Campo, E.; et al. RNA Editing Contributes to Epitranscriptome Diversity in Chronic Lymphocytic Leukemia. Leukemia 2021, 35, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, P.G.; Jares, P.; Rico, D.; Gomez-Lopez, G.; Martinez-Trillos, A.; Villamor, N.; Ecker, S.; Gonzalez-Perez, A.; Knowles, D.G.; Monlong, J.; et al. Transcriptome Characterization by RNA Sequencing Identifies a Major Molecular and Clinical Subdivision in Chronic Lymphocytic Leukemia. Genome Res. 2014, 24, 212–226. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akimova, E.; Gassner, F.J.; Greil, R.; Zaborsky, N.; Geisberger, R. Detecting Bacterial–Human Lateral Gene Transfer in Chronic Lymphocytic Leukemia. Int. J. Mol. Sci. 2022, 23, 1094. https://doi.org/10.3390/ijms23031094
Akimova E, Gassner FJ, Greil R, Zaborsky N, Geisberger R. Detecting Bacterial–Human Lateral Gene Transfer in Chronic Lymphocytic Leukemia. International Journal of Molecular Sciences. 2022; 23(3):1094. https://doi.org/10.3390/ijms23031094
Chicago/Turabian StyleAkimova, Ekaterina, Franz Josef Gassner, Richard Greil, Nadja Zaborsky, and Roland Geisberger. 2022. "Detecting Bacterial–Human Lateral Gene Transfer in Chronic Lymphocytic Leukemia" International Journal of Molecular Sciences 23, no. 3: 1094. https://doi.org/10.3390/ijms23031094
APA StyleAkimova, E., Gassner, F. J., Greil, R., Zaborsky, N., & Geisberger, R. (2022). Detecting Bacterial–Human Lateral Gene Transfer in Chronic Lymphocytic Leukemia. International Journal of Molecular Sciences, 23(3), 1094. https://doi.org/10.3390/ijms23031094